

Abstract
Hyperglycemia, a key factor in insulin resistance and diabetic pathology, is associated with cellular oxidative stress that promotes oxidative protein modifications. We report that protein nitration is responsive to changes in glucose concentrations in 3T3-L1 adipocytes. Alterations in the extent of tyrosine nitration as well as the cellular nitroproteome profile correlated tightly with changing glucose concentrations. The target proteins we identified are involved in fatty acid binding, cell signaling, protein folding, energy metabolism, antioxidant capacity, and membrane permeability. The nitration of adipocyte fatty acid binding protein (FABP4) at Tyr19 decreases, similar to phosphorylation, the binding of palmitic acid to the fatty acid-free protein. This potentially alters intracellular fatty acid transport, nuclear translocation of FABP4, and agonism of PPAR gamma. Our results suggest that protein tyrosine nitration may be a factor in obesity, insulin resistance, and the pathogenesis of diabetes.
Keywords: Adipocytes, Nitric oxide, Oxidative stress, Protein tyrosine nitration, Glucose, Obesity, Insulin resistance, Diabetes
Introduction
The prevalence of obesity and one of its foremost comorbidities, type 2 diabetes [1], is increasing worldwide in epidemic proportions [2–4]. A key link between both metabolic diseases is the chronic subacute inflammatory status that is characteristic for obesity. Together other factors like endoplasmic reticulum (ER) stress and chronic inflammation may impair the insulin-stimulated glucose uptake in insulin-sensing tissues like liver, muscles, and adipose tissue [5]. The resulting insulin resistance, a primary condition in obesity, is a crucial step in the pathogenesis of type 2 diabetes and the lead cause of impaired glucose tolerance (IGT) [6]. However, limited glycemic control is maintained by compensatory increases in β-cells insulin secretion, resulting in hyperinsulinemia [7]. The full transition to type 2 diabetes is triggered by β-cell failure [1].
Regulation and control of the systemic metabolic homeostasis and energy storage by interorgan communication networks are critical for this process. Adipocytes of the white adipose tissue are an important part of this network due to their endocrine and secretory function as well as the capacity to store and release lipids [8]. Alterations in the metabolic state of hypertrophic adipocytes and the recruitment of immune cells like macrophages, especially in the obese visceral adipose tissue, are now thought to play an important regulatory role in the obesity-associated pathological processes [1,8,9]. This includes the accumulation and redistribution of potentially toxic metabolic byproducts like nonesterified fatty acids as well as the altered release of peptide hormones (adipokines) and expression of proinflammatory cytokines (e.g., interleukins IL-1 and IL-6; tumor necrosis factor-α, TNFα; interferon-γ, IFNγ) [1,6,8–11]. These factors are detrimental for insulin signaling and glucose homeostasis in liver, skeletal muscle, and adipose tissue itself [1,8]. They also affect the expression of insulin and metabolic enzymes in β-cells [9]. Thus, the regulation of the metabolic state of adipocytes is highly relevant for the onset of insulin resistance and type 2 diabetes.
At physiological levels, nitric oxide (NO) acts as a signaling molecule regulating energy homeostasis in adipose tissue by stimulating glucose uptake and insulin-responsive glucose transporter protein-4 (GLUT4) translocation along with increasing glucose and fatty acid metabolism [10,11]. In adipocytes NO is generated by endothelial (eNOS) and inducible (iNOS) NO synthase [12]. Adipogenic differentiation and obesity increase the expression of iNOS leading to an augmented generation of NO. Since insulin increases NO generation in human preadipocytes [12], one of the contributing factors might be the increased insulin secretion by β-cells due to insulin resistance. Glycemic dysregulation leading to a proinflammatory response and the augmentation of reactive oxygen species (ROS) [13] could further modulate NO bioavailability in adipocytes. Conditions characterized by the simultaneous generation of increased amounts of NO and ROS like superoxide are prone to oxidative protein modifications, particularly protein tyrosine nitration [14].
Protein tyrosine nitration can be part of a transient adaptive response based on regulated nitration/denitration or have detrimental effects on excessive and potentially accumulative modification due to overwhelmed cellular response mechanisms [14–17]. Thus, protein nitration in adipocytes could be a crucial factor in adipose dysfunction and therefore obesity-related pathologies. However, the effects of elevated glucose or lipid levels on protein tyrosine nitration in adipocytes have not been studied. In the present study, we therefore identified the target proteins for tyrosine nitration in 3T3-L1 adipocytes under different hyperglycemic conditions. The results provide insights into the cellular effects of protein nitration in adipocytes.
Materials and methods
Cell culture
Mouse 3T3-L1 cells (ATCC, CRL-173) were used as a model for white adipose tissue adipocytes. Cells were grown at 37 °C under isobaric conditions (5% CO2, 95% air) in humidified atmosphere using Corning CellBIND culture material. Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 5 mM D-glucose (normoglycemic standard), 10% fetal bovine serum, 2 mM L-glutamine, 100 units/ml penicillin, and 100 μg/ml streptomycin was used to propagate the cells. The media were changed every day and cells were maintained at <60% confluence prior to diffentiation. For adipocyte differentiation 3T3-L1 cells were grown to confluency (Day 0), and then stimulated for 2 days with 1 μM dexamethasone, 0.5 mM isobutyl-methylxanthine, and 2 μg/ml insulin (Day 2), followed by another 2 days with 2 μg/ml insulin alone (Day 4). By Day 4 media were changed to regular DMEM and fully differentiated phenotype, including the accumulation of lipid droplets, reached at Days 8 to 10, which was monitored by light microscopy. At this time experiments were performed. Differentiated 3T3-L1 cells express adipokines like leptin and adiponectin [18].
Conditions of elevated glucose levels
To simulate in vivo conditions of normal fasting glucose (NFG), which is now linked to plasma glucose levels of less than 5.2 mM, cells were constantly cultured at 5 mM D-glucose and media changed every 24 h. As IFG is related to glucose levels between 5.6 and 6.9 mM and IGT is associated with a postprandial hyperglycemia marked by D-glucose levels of 7.8–11 mM [6,19], 3T3-L1 cells were exposed to 6.5, 8, and 11 mM D-glucose in DMEM in the presence of 2 μg/ml insulin to support glucose uptake. To exclude the influence of osmotic changes, variable amounts of L-glucose were added resulting in 11 mM total glucose. The time of exposure was 24 or 12 h with intermittent phases of 5 and 11 mM glucose. The intermittent exposure was used to simulate physiological fluctuations in glucose levels according to the fact that postprandial blood glucose level regularly peaks approximately 30–120 min after the start of a meal [6].
Cell lysis
After removing culture media cells were washed three times with PBS and lysed by adding lysis buffer (7.8 M urea, 2.2 M thiourea, and 1% Triton X-100). For two-dimensional electrophoreses 1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS), 1% dithiothreitol (DTT), and 1% IPG ampholytes (Bio-Lyte 3/10) were added immediately before isoelectric focusing [15,20].
Two-dimensional gel electrophoresis
Two-dimensional gel electrophoresis was performed with the IEF/Criterion gel system (Bio-Rad, Hercules, CA) [16,20]. The first dimension used lysis buffer (above) and 11-cm nonlinear pH 3–10 immobilized pH gradient (IPG) strips. IPG strips were rehydrated with sample at 50 V/14 h, and then isoelectric focusing was performed by a linear increase to 250 V over 20 min followed by a linear increase to 8000 V over 170 min and then held at 8000 V until a total of 45 kV h is reached. For the second dimension, the IPG strips were equilibrated for 12 min in 50 mM Tris/HCl, pH 8.8, 6 M urea, 30% glycerol, 2% SDS, 1% DTT, and bromophenol blue, and then 15 min in 50 mM Tris/HCl, pH 8.8, 6 M urea, 30% glycerol, 2% SDS, 2% iodoacetamide, and bromophenol blue. The strips then were embedded in 1% (wt/vol) agarose on the top of 12.5% acrylamide gels containing 4% stacking gel (Criterion gel). The second dimension SDS/PAGE was performed essentially according to Laemmli. After completion acrylamide gels were soaked 20 min in transfer buffer (25 mM Tris/HCl, 192 mM glycine, pH 8.3, and 20% methanol) and then partially electro-transferred to a Hybond-P PVDF membrane (Bio-Rad) using a semidry transfer apparatus. The gels then were stained with colloidal Coo-massie blue (GelCode blue stain).
Western analysis
PVDF Membranes were blocked for 60 min by using blocking buffer (25 mM Tris, 150 mM NaCl, pH 7.5, 0.2% Tween 20, and 1.5% BSA). Membranes were then probed for 60 min at 25 °C with a monoclonal antibody against 3-nitrotyrosine (1:5000; clone 1A6, Upstate Biotechnology) in blocking buffer. The membranes were then washed four times in washing buffer (20 mM Tris, 150 mM NaCl, pH 7.5, and 0.2% Tween 20), probed 60 min at 25 °C with a goat anti-mouse antibody (horseradish peroxidase conjugate, 1:3.000, Bio-Rad), and finally washed again four times in washing buffer. Immunopositive spots were visualized by chemiluminescence using ECL-Plus reagent (Amersham Biosciences, Little Chalfont, Buckinghamshire, England) according to the manufacturer. The nitrotyrosine immunoreactivity results were verified by reduction of nitrotyrosine to aminotyrosine with sodium hydrosulfite followed by determination of remaining nitrotyrosine immunoreactivity using anti-nitrotyrosine antibody [16,20].
Protein identification by MALDI-TOF mass spectrometry
In-gel spots matching spots immunopositive for 3-nitrotyrosine on immunoblots were subjected to in-gel tryptic digestion. The tryptic peptide mixtures were analyzed by matrix-assisted laser desorption ionization/time-of-flight mass spectrometry (MALDI-TOF/TOF PE Biosystems Model 4800) as primarily described in detail [20,21]. Measured peptide masses were used to search the Swiss-Prot, TrEMBL, and NCBI sequence databases using Mascot (http://www.matrixscience.com). However, these proteins should be considered putatively nitrated until the nitrated sites have been identified by sequence analysis. All searches were performed with a mass tolerance of 0.005% error (50 ppm). Only proteins that reproducibly showed positive nitrotyrosine immunoreactivity in all samples from each experimental condition were included in the analysis.
FABP4-GST expression
GST (glutathione S transferase)-tagged FABP4 was expressed in BL21 Escherichia coli cells transformed with the plasmid pGEX3-FABP4. Cells were grown overnight at 37 °C in LB media containing 100 μM ampicilin. Ten milliliters of this culture was added to 1 liter of TB media containing 100 μM ampicillin and grown until OD 600 reached 0.8. IPTG was then added to a final concentration of 1 mM and left for 18 h at room temperature with shaking. Cells were harvested by centrifugation. The bacterial pellet was resuspended in potassium phosphate buffer (100 mM potassium phosphate, 150 mM NaCl, pH 7.0) containing protease inhibitors (5 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstain, and 24 μg/ml Pefabloc SC), 1 mg/ml lysozyme, and 5 units/ml deoxyribonuclease 1. Cells were then sonicated on ice and centrifuged at 16,000 g for 30 min at 4°C. The supernatant was loaded onto a column containing 5 ml glutathione-Sepharose preequilibrated with potassium phosphate buffer. After washing with 100 ml of potassium phosphate buffer, the protein was eluted with potassium phosphate buffer containing 20 mM reduced glutathione. FABP4 was then dialyzed with phosphate buffer overnight at 4°C and concentrated with Amicon ultra-4 centrifugal filter devices (Millipore) with a 3-kDA molecular weight cutoff. Protein yield was 6.5 mg per liter of bacterial culture.
FABP4 purification
The FABP4-GST protein construct, which contains a thrombin cleavage site between the GST and the mouse FABP4, was incubated with 10 U/mg thrombin (7.5 U/λ) for 20 min at 37°C in phosphate buffer. The reaction was stopped with serine protease inhibitors (5 μg/ml aprotinin, 24 μg/ml Pefabloc SC). FABP4 was separated from GST and thrombin by FPLC at 4°C using a Sephacryl S-200 column and potassium phosphate buffer at a flow rate of 0.5 ml/min. Purification was verified by SDS-PAGE and solely FABP4 containing eluate concentrated to 1 mg/ml with Amicon ultra-4 centrifugal filter devices with 3 kDA molecular weight cutoff.
Biotinylation of FABP4
GST-free FABP4 was reacted with EZ-link Sulfo-NHS-LC-Biotin (Pierce) at a molar ratio of 1:20 in potassium phosphate buffer, pH 7.0, for 2h at 4°C and 1h at room temperature according to the manufacturer.
Nitration of FABP4
GST-free FABP4 and biotinylated FABP4 were nitrated with hydrogen peroxide-free peroxynitrite (Calbiochem) at molar ratios of 1:10 and 1:50 as reported recently [22]. Briefly, FABP4 was exposed to peroxynitrite for 30 s at 25 °C in potassium phosphate buffer, pH 7, with 25 mM sodium bicarbonate by rapid mixing. Thereby equal amounts of 0.1 M NaOH were added (with and without peroxynitrite), which changed the pH of the protein solution from 7 to 7.1. This excludes any nitration of tyrosine residues by acidic reactions of decomposition products like nitrate. Nitration in the presence of fatty acid was done at a molar ratio of 1:1 between FABP4 and palmitic acid. After nitration the protein solutions were stored on ice until used. Aliquots were taken for Western analysis. The concentration of the peroxynitrite stock solution was UV spectrophotometrically determined in 0.1 M sodium hydroxide using ε =1670 M−1cm−1 at 302 nm.
Identification of FABP4 nitration sites by LC-MS/MS
Nonnitrated and nitrated FABP4 were subjected to SDS/PAGE according to Laemmli and the resulting protein bands excised and treated with trypsin [22]. The resulting extracts were mixed 1:11% (v/v) acetic acid and 20 μl was transferred to the autosampler of the ThermoScientific LTQ ion trap mass spectrometer system. For analysis a volume of 10 μl was injected onto a 8 cm×75 μm Phenomenex Jupiter C18 reversed-phase capillary chromatography column using an Eksigent splitless nanoflow LC system. The peptides were eluted from the column by an acetonitrile/0.1% (v/v) formic acid gradient at 250 nl/min. The microelectrospray ion source was operated at 2.5 kV. Full-scan mass spectra were acquired to determine peptide molecular weights and product ion spectra to determine amino acid sequences in successive instrument scans. The data were analyzed by using all collisionally induced dissociation spectra (CID) collected in the experiment to search the NCBI sequence databases using Mascot. Each identification was verified by manual inspection of several matching spectra. The potential nitration sites were interrogated by manual inspection of the spectra and through the use of the program Sequest.
Fatty acid binding to FABP4
Nitrated and nonnitrated biotinylated FABP4 were incubated with equimolar amounts of [9,10-3H]palmitic acid (50 Ci/mmol; Sigma; final ethanol concentration 1% v/v) in potassium phosphate buffer for 2 h at room temperature. Then the biotinylated FABP4 with bound palmitic acid was separated using UltraLink Immobilized NeutrAvidin Protein (Pierce) according to the manufacturer. Washed Immobilized NeutrAvidin Protein with bound FABP4 was quantatively transferred to scintillation vials and activity measured using a Beckman Coulter LS6500 Multipurpose scintillation counter.
Statistics and data analysis
Presented 2D blots are representative for three independent experiments. All other data are presented as mean±SEM. Unpaired two-tailed Student’s t tests were performed using the Prism software package version 4 (GraphPad). Differences were considered significant at P<0.02.
Results
Glucose concentration-dependent protein tyrosine nitration
Our established proteomic method [16,20] was used to examine the effects of glucose on the nitroproteome of differentiated mouse 3T3-L1 adipocytes to reveal the potential involvement of NO-mediated oxidative protein modifications in the pathology of obesity and insulin resistance. To avoid premature oxidative stress, excessive triglyceride accumulation, and deregulated basal glucose uptake as well as the onset of insulin resistance, preadipocytes were differentiated under normoglycemic conditions, hence in the presence of 5 mM glucose [13]. The increase in glucose concentration from 5 mM to physiologically/pathologically relevant concentrations of 6.5, 8, and 11 mM resulted in a general and concentration-dependent increase in 3-nitrotyrosine immunoreactivity (Figs. 1A–D). These results are consistent with previous studies showing increased glucose-mediated generation of nitrating reagents fueled by elevated generation of NO and ROS like superoxide in adipocytes [13] and retinal Müller cells [23,24]. However, this regimen also caused alterations of the nitroproteome that are highly target selective as a subset of the protein targets had a decrease or loss of immunoreactivity with increasing glucose concentrations rather than an increase.
Fig. 1.
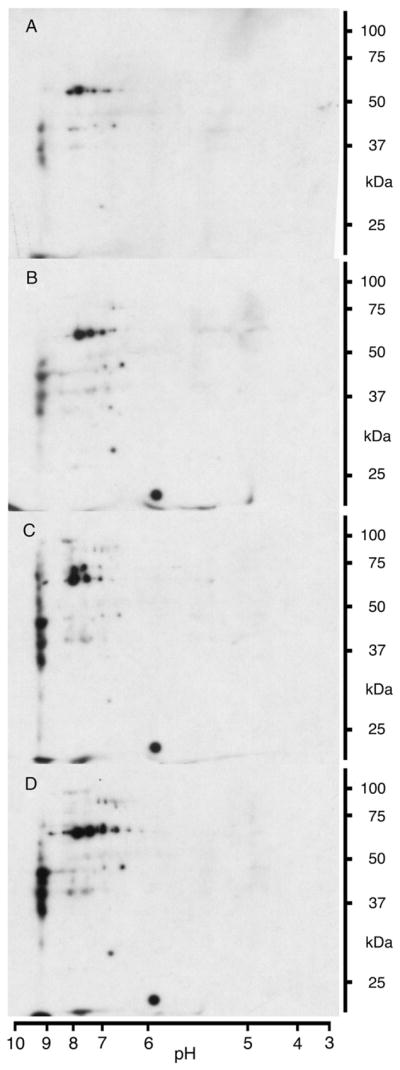
Glucose concentration-dependent increase in protein tyrosine nitration. Representative 2D blots for 3-nitrotyrosine immunoreactivity of 3T3-L1 adipocytes sampled after 24 h of exposure to 5 (A), 6.5 (B), 8 (C), and 11 (D) mM glucose. Cells have been cultured at 5 mM glucose prior to the experiment.
Effect of fluctuating glucose concentrations on protein tyrosine nitration
To test how physiological glucose concentration fluctuations influence protein nitration in 3T3-L1 adipocytes, physiological conditions were mimicked by switching the cell culture four to five times between media containing 5 or 11 mM glucose over a period of 12 h (Figs. 2A–C). This approach revealed that fluctuating glucose levels result in a pronounced increase in protein tyrosine nitration after repeated 2-h 11 mM glucose phases (Fig. 2C). Despite the widely similar alterations in the 2D nitroproteome profiles, the increase in 3-nitrotyrosine immunoreactivity resulting from 12 h of intermittent exposure to 11 mM glucose notably exceeded the one resulting from 24 h of continuous exposure (compare Figs. 1D and 2C). However, a comparison with cells grown at 5 mM glucose showed that a period of 4 h at 5 mM glucose following two intermittent 2-h 11 mM glucose phases resulted in the return to the base tyrosine nitration level (compare Figs. 2B with A and C). This points toward cellular mechanisms that remove nitrated proteins by increased turnover [25] and/or denitration of modified target proteins [15,17]. In case of a regulated denitration, protein nitration could be part of a cellular response to minimize glucotoxicity that includes NO-dependent glucose uptake [26].
Fig. 2.
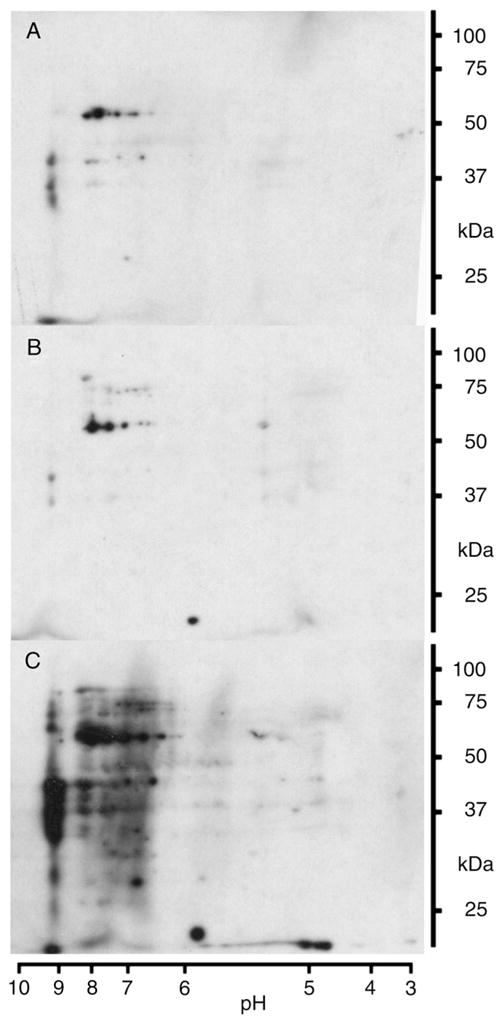
Increase in protein tyrosine nitration by fluctuating glucose levels. Representative 2D blots for 3-nitrotyrosine immunoreactivity of 3T3-L1 adipocytes sampled after exposure to specified conditions. Cells have been cultured at 5 mM glucose prior to the experiment. Conditions for glucose exposure were: (A) 12 h 5 mM with media change after 2, 4, 6, and 10 h; (B) 2 h 11 mM, 2 h 5 mM, 2 h 11 mM, and 4 h 5 mM; (C) 2 h 11 mM, 2 h 5 mM, 2 h 11 mM, 4 h 5 mM, and 2 h 11 mM.
Identities of the nitrated proteins
The identification of the tyrosine nitrated proteins is a prerequisite for the approximation of the potential (patho)physiological impact of their modification. Thus, following 2D SDS-PAGE, immunoblotting, and in-gel tryptic digestion, product peptides from nitrotyrosine-immunoreactive protein spots were subjected to mass spectrometric analysis. The modified proteins identified in samples from 3T3-L1 adipocytes that were exposed to 11 mM glucose are listed in Table 1. They participate in a variety of physiological processes essential for the maintenance of normal metabolic homeostasis and paracrine function. The affected processes comprise glycolysis, fatty acid binding, mitochondrial metabolism, mitochondrial permeability, protein folding, protein translation, antioxidant defense, and signal transduction.
Table 1.
Identification of nitrated proteins
| Symbol | Protein | % Sequence coverage | Peptide matches |
|---|---|---|---|
| AC2 | Aconitase 2, mitochondrial | 27 | 16 |
| FABP4 | Adipocyte fatty acid binding protein (aP2/FABP4) | 38 | 6 |
| ALDA | Aldolase A | 52 | 12 |
| CAT | Catalase | 17 | 7 |
| G3PDH | Glyceraldehyde-3-phosphate dehydrogenase (G3PDH) | 15 | 3 |
| RNP | Heterogeneous nuclear ribonucleoprotein A2/B1 (hnRNP-A2/B1) | 23 | 6 |
| HSP60 | Heat shock protein 60 (HSP60) | 26 | 8 |
| IVDH | Isovaleryl-CoA dehydrogenase | 19 | 7 |
| MDH | Malate dehydrogenase | 36 | 9 |
| NDKB | Nucleoside-diphosphate kinase B (NDK-B) | 59 | 9 |
| PGK | Phosphoglycerate kinase | 24 | 8 |
| ER60 | Protein disulfide isomerase A3 (ERp60, Erp57, ER-60) | 25 | 11 |
| PK | Pyruvate kinase | 31 | 12 |
| GDI | Rho GDP-dissociation inhibitor 2 (RhoGDI2) | 50 | 7 |
| TP | Trifunctional protein, mitochondrial, α-chain | 12 | 7 |
| VDAC | Voltage-dependent anion-selective channel protein 1 (VDAC-1) | 40 | 7 |
Proteins immunopositive for 3-nitrotyrosine from 3T3-L1 adipocytes were identified by mass spectrometry. Values for sequence coverage and peptide matches originate from a Mascot search using MALDI-TOF mass spectrometry peptide map data. Only proteins reproducibly present in the 2D nitroproteome profiles of all samples from each experimental condition using 11 mM glucose are listed.
Nitration of FABP4
FABP4 contains two tyrosine residues and both have been demonstrated to be critical for function. To identify the site of tyrosine modification in FABP4, we used capillary column LC-tandem mass spectrometry. The analysis confirmed the protein identity (NCBI Accession No. 14149635). Successive analysis showed that exposure of FABP4 to peroxynitrite at molar ratios of 1:10 as well as 1:50 leads to nitration of tyrosine 19 (Tyr19) of the peptide LVSSENFDDYMK (Fig. 3). In all cases in which Tyr19 was modified, methionine 20 (Met20) was also found oxidized. This observation is consistent with the preferential oxidation of the methionine prior to the tyrosine nitration under the conditions used. Thereby the increase of the molar ratio from 1:10 to 1:50 led to a several-fold increase in the amount of peptides with oxidized Met20 as well as the comodification in the form of nitrated Tyr19 and oxidized Met20 relative to unmodified peptide. This increase was more pronounced for the double-modified peptide.
Fig. 3.

Identification of tyrosine 19 as a nitration site in FABP4 using capillary column LC-tandem mass spectrometry. Collision-induced dissociation (CID) spectra of the tryptic peptide peptide 10-LVSSENFDDYMK-21 from FABP4 (NCBI Accession No. 14149635). Representative CID spectra of the unmodified peptide (LVSSENFDDYMK), methionine oxidized peptide (LVSSENFDDYMoK), and tyrosine nitrated-methionine oxidized peptide (LVSSENFDDYNO2MoK) clearly showing the site of the modified amino acids Tyr19 and Met20. The interpretation of each spectrum is inset.
Fatty acid binding to nitrated FABP4
To determine the potential functional impact of the nitration of Tyr19 and oxidation of Met20 the affect of peroxynitrite exposure on the fatty acid binding ability of FABP4 was determined. Depending on the molar ratio of FAPB4 and peroxynitrite, the exposure to peroxynitrite was associated with a significant decrease in the binding of palmitic acid to FABP4 (Fig. 4). However, treatment of FABP4 with decomposed peroxynitrate had no effect on the fatty acid binding. This decrease in binding occurs likely due to the alteration of the pKa of the hydroxyl group and therefore the polarity of the tyrosine residue by nitration [27]. In the case of FABP4 the change in polarity probably mimics phosphorylation [23,28], which has been shown to greatly inhibit lipid binding and release [29].
Fig. 4.

Fatty acid binding to FABP4. The binding of [9,10-3H]palmitic acidto nonnitrated and nitrated FABP4 is shown dependent on the applied peroxynitrite concentration. The data are represented as mean±SEM of 4 independent experiments (n=4). Thereby asterisks represent alterations with P<0.02 and were therefore considered significant.
Discussion
Continuous consumption of surplus lipids and carbohydrates is a major causative factor for obesity. The disproportionate or excessive gain of white adipose tissue mass affects the systemic energy balance as well as glucose and lipid metabolism [8], which is correlated with the development of comorbidities like type 2 diabetes, hepatosteatosis, and atherosclerosis [5]. Thereby different molecular factors like nonesterified fatty acids and cytokines, originating from the dysregulation of metabolic and secretory/endocrine functions of adipocytes as well as the chronic subacute inflammation, contribute to an inadequate control of blood glucose due to insulin resistance. Acute or chronically elevated glucose can cause toxicity that correlates with oxidative stress, favoring oxidative protein modifications like protein nitration [14]. Insulin resistance is also associated with an increased generation of reactive species like peroxynitrite [30]. The various potential (patho)physiological effects of protein nitration under these conditions will depend on target proteins and the extent of modification [17]. In this context our study represents the first systematic investigation that correlates glucose and protein tyrosine nitration in adipocytes. Our data show that the extent of cellular tyrosine protein nitration, along with an expansion of the nitroproteome, mirrored rises in the glucose concentration during continuous exposure as well as during the simulation of a more physiologically relevant periodic fluctuation in the glucose concentration. Thus, our study adds greatly to the understanding of the effects of oxidants like peroxynitrite in insulin resistance [30].
The largest functional group of target proteins comprised metabolic enzymes including enzymes of the glycolytic pathway, tricarboxylic acid (TCA) cycle, and fatty acid β-oxidation. Among the proteins identified, the glycolytic pathway was most extensively modified. We have recently shown that nitration of aldolase A inhibits enzyme activity [22]. G3PDH and phosphoglycerate kinase were also found to be sensitive to protein nitration and/or thiol oxidation [31,32]. Thus, the simultaneous nitration of aldolase A, G3PDH, phosphoglycerate kinase, and pyruvate kinase suggests that elevated glucose levels can decrease the glycolytic activity in adipocytes. The metabolism of glucose carbons could be suppressed further, as the mitochondrial TCA cycle enzymes malate dehydrogenase [33] and aconitase [34,35] become inactivated by oxidative modifications. Tyrosine nitration acts probably as markers for these oxidative modifications. These metabolic alterations potentially lead to a decreased generation of ATP, GTP, NADHs and FADH2, a decrease in mitochondrial membrane potential as well as a depletion or quantitative shift of essential metabolic intermediates like oxaloacetate. A depletion or shift in these intermediates has the potential to decrease mitochondrial fatty acid β-oxidation, which might be further affected by the nitration of the α-chain of the trifunctional protein. In combination with the potential impairment of integrity and biogenesis as well as the altered permeability of mitochondria through the nitration of heat shock protein 60 [36–38] and voltage-dependent anion-selective channel protein 1 [39,40], this could result in mitochondrial dysfunction accompanied by altered fuel metabolism and lipid homeostasis. Elevated nonesterified fatty acids and metabolic lipid products transmit stress responses through activation of several kinases, including c-jun NH2-terminal kinase (JNK) and inhibitor of kappa kinase (IκK) [41].
In adipocytes lipid trafficking/metabolism and hormone action are integrated with stress and inflammatory responses by aP2/FABP4, the predominant fatty acid binding protein, which modulates the systemic glucose and lipid metabolism [41,42]. All FABPs (1–9) in mammals share the same general tertiary structure. It consists of a β-barrel around the ligand-binding cavity covered with a helix-turn-helix cap and contains only two tyrosine residues, Tyr19 and Tyr128, which are highly conserved [43]. Both tyrosine residues are functionally important. The carboxylate group of a lipid ligand directly interacts with Tyr128 through hydrogen bonding [43,44] and phosphorylation of Tyr19 by insulin receptor tyrosine kinase [43,45] regulates lipid binding and release [29]. Nitration of Tyr19 mimicking and modulating phosphorylation [23,28] could therefore represent an additional regulatory mechanism especially under stress conditions. This, in turn, could further affect the regulatory interaction of FABP4 with hormone-sensitive lipase [46], nuclear translocation of FABP4, and agonist supply for peroxisome-proliferator activated receptor γ (PPAR-γ) [47–49] as well as the activation of JNK [41,42]. Alterations in PPAR-γ agonism further alter FABP4 expression, lipolysis, fatty acid β-oxidation, and mitochondrial biogenesis [50]. Therefore, the nitration of FABP4 could be an important factor for the adipose tissue and systemic response to high metabolic load in regard of adipocytokines as well as glucose and lipid metabolism.
The (patho)physiological effects resulting from the high metabolic load and inflammatory responses also depend on the status of the ER and cellular antioxidant capacity. In adipocytes under hyperglycemic conditions this capacity is likely weakened by the tyrosine nitration of the antioxidant enzyme catalase, which is associated with inactivation [51]. Moreover, alterations in the agonist stimulation of PPAR-γ could decrease the expression of catalase [52]. The potential effects of the nitration protein disulfide isomerase A3 are unknown, but the oxidative folding of glycoproteins in the ER and/or the interaction with calnexin and calreticulin could be affected [53].
The nitration of Rho GDP-dissociation inhibitor 2 (RhoGDI2) in 3T3-L1 adipocytes has the potential to further alter metabolic stress response. RhoGDI2, whose abundance decreases during differentiation [54], has a relatively narrow specificity for Cdc42, Rac1, and RhoA of the Rho family of small GTP-binding proteins [55]. It contains a tyrosine phosphorylation site at Tyr24 [56], which decreases the ability for complex formation with target proteins. Thus, tyrosine nitration has the potential to alter the physiological function of Cdc42, Rac1, and/or RhoA and therefore stress fiber formation, adipocyte morphology [57,58], and maybe even glucose [59] and GLUT4 trafficking [57,58,60]. Nitration of nucleoside-diphosphate kinase B (NDK-B) could contribute to these effects as it can exist in two distinct pools in 3T3 fibroblasts: one population is transiently translocated to the cell periphery on activation of receptor tyrosine kinases and G-protein-coupled receptors, whereas a second pool binds constitutively to microtubule-associated vesicles [61]. The association with vesicles might provide a regulated source of GTP for GTPases that control intracellular trafficking, while the potential interaction with integrin cytoplasmic domain-associated protein 1α (ICAP-1α) as a consequence of Rac activation might affect cell morphology and behavior during adhesion [61]. NDK-B further binds polyunsaturated fatty acids [62], interacts with heterotrimeric G protein βγ dimers [63], facilitates coat protein complex II assembly [64], and induces c-myc [65] in various cell types. These functions of NDK-B could, if present in adipocytes, provide additional mechanisms by which NDK-B nitration might alter adipocyte biology.
Conclusion
The understanding is incomplete of the molecular mechanisms linking obesity to insulin resistance, type 2 diabetes, and other comorbidities. However, in recent years it has become clear that inflammation and adverse metabolic alterations are key factors. Many of these factors are associated with oxidative stress, and protein tyrosine nitration is an important (patho)physiological mechanism by which oxidative stress is manifested in cells. The physiological functions of the proteins identified in this study implicate tyrosine nitration as a potentially significant mechanism for the development of insulin resistance itself and the associated complications in obesity.
Acknowledgments
The work was supported by National Institute of Health Grant NIH P01 HL076491. The pGEX3-FABP4 plasmid was generously provided by Dr. Noa Noy.
Abbreviations
- FABP4
fatty acid binding protein 4
- RhoGDI2
Rho GDP-dissociation inhibitor 2
- NDK-B
nucleoside-diphosphate kinase B
- GLUT4
insulin-responsive glucose transporter protein-4
- PPAR-γ
peroxisome-proliferator activated receptor γ
- TCA
tricarboxylic acid
- ER
endoplasmic reticulum
- IGT
impaired glucose tolerance
- IL-1β
interleukin-1β
- ROS
reactive oxygen species
- NO
nitric oxide
- NOS
nitric oxide synthase
- SOD
superoxide dismutase
- MALDI
matrix-assisted laser desorption ionization/time-of-flight mass spectrometry
- CID
collision-induced dissociation
References
- 1.Muoio DM, Newgard CB. Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol. 2008 doi: 10.1038/nrm2327. [DOI] [PubMed] [Google Scholar]
- 2.Cara JF, Chaiken RL. Type 2 diabetes and the metabolic syndrome in children and adolescents. Curr Diabetes Rep. 2006;6:241–250. doi: 10.1007/s11892-006-0041-8. [DOI] [PubMed] [Google Scholar]
- 3.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–1053. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 4.Jones KL. Role of obesity in complicating and confusing the diagnosis and treatment of diabetes in children. Pediatrics. 2008;121:361–368. doi: 10.1542/peds.2007-1234. [DOI] [PubMed] [Google Scholar]
- 5.Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9:367–377. doi: 10.1038/nrm2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bock G, Dalla Man C, Campioni M, Chittilapilly E, Basu R, Toffolo G, Cobelli C, Rizza R. Pathogenesis of pre-diabetes: mechanisms of fasting and postprandial hyperglycemia in people with impaired fasting glucose and/or impaired glucose tolerance. Diabetes. 2006;55:3536–3549. doi: 10.2337/db06-0319. [DOI] [PubMed] [Google Scholar]
- 7.Weir GC, Bonner-Weir S. A dominant role for glucose in beta cell compensation of insulin resistance. J Clin Invest. 2007;117:81–83. doi: 10.1172/JCI30862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosen ED, Spiegelman BM. Adipocytes as regulators of energy balance and glucose homeostasis. Nature. 2006;444:847–853. doi: 10.1038/nature05483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao YF, Feng DD, Hernandez M, Chen C. 3T3-L1 adipocytes induce dysfunction of MIN6 insulin-secreting cells via multiple pathways mediated by secretory factors in a co-culture system. Endocrine. 2007;31:52–60. doi: 10.1007/s12020-007-0001-3. [DOI] [PubMed] [Google Scholar]
- 10.Jobgen WS, Fried SK, Fu WJ, Meininger CJ, Wu G. Regulatory role for the arginine-nitric oxide pathway in metabolism of energy substrates. J Nutr Biochem. 2006;17:571–588. doi: 10.1016/j.jnutbio.2005.12.001. [DOI] [PubMed] [Google Scholar]
- 11.Tanaka T, Nakatani K, Morioka K, Urakawa H, Maruyama N, Kitagawa N, Katsuki A, Araki-Sasaki R, Hori Y, Gabazza EC, Yano Y, Wada H, Nobori T, Sumida Y, Adachi Y. Nitric oxide stimulates glucose transport through insulin-independent GLUT4 translocation in 3T3-L1 adipocytes. Eur J Endocrinol/Eur Fed Endocr Soc. 2003;149:61–67. doi: 10.1530/eje.0.1490061. [DOI] [PubMed] [Google Scholar]
- 12.Engeli S, Janke J, Gorzelniak K, Bohnke J, Ghose N, Lindschau C, Luft FC, Sharma AM. Regulation of the nitric oxide system in human adipose tissue. J Lipid Res. 2004;45:1640–1648. doi: 10.1194/jlr.M300322-JLR200. [DOI] [PubMed] [Google Scholar]
- 13.Lin Y, Berg AH, Iyengar P, Lam TK, Giacca A, Combs TP, Rajala MW, Du X, Rollman B, Li W, Hawkins M, Barzilai N, Rhodes CJ, Fantus IG, Brownlee M, Scherer PE. The hyperglycemia-induced inflammatory response in adipocytes: the role of reactive oxygen species. J Biol Chem. 2005;280:4617–4626. doi: 10.1074/jbc.M411863200. [DOI] [PubMed] [Google Scholar]
- 14.Pacher P, Beckman JS, Liaudet L. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 2007;87:315–424. doi: 10.1152/physrev.00029.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koeck T, Fu X, Hazen SL, Crabb JW, Stuehr DJ, Aulak KS. Rapid and selective oxygen-regulated protein tyrosine denitration and nitration in mitochondria. J Biol Chem. 2004;279:27257–27262. doi: 10.1074/jbc.M401586200. [DOI] [PubMed] [Google Scholar]
- 16.Aulak KS, Miyagi M, Yan L, West KA, Massillon D, Crabb JW, Stuehr DJ. Proteomic method identifies proteins nitrated in vivo during inflammatory challenge. Proc Natl Acad Sci U S A. 2001;98:12056–12061. doi: 10.1073/pnas.221269198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Koeck T, Stuehr DJ, Aulak KS. Mitochondria and regulated tyrosine nitration. Biochem Soc Trans. 2005;33:1399–1403. doi: 10.1042/BST0331399. [DOI] [PubMed] [Google Scholar]
- 18.Fischer-Posovszky P, Wabitsch M, Hochberg Z. Endocrinology of adipose tissue—an update. Horm Metab Res. 2007;39:314–321. doi: 10.1055/s-2007-976539. [DOI] [PubMed] [Google Scholar]
- 19.Sorkin JD, Muller DC, Fleg JL, Andres R. The relation of fasting and 2-h postchallenge plasma glucose concentrations to mortality: data from the Baltimore Longitudinal Study of Aging with a critical review of the literature. Diabetes Care. 2005;28:2626–2632. doi: 10.2337/diacare.28.11.2626. [DOI] [PubMed] [Google Scholar]
- 20.Aulak KS, Koeck T, Crabb JW, Stuehr DJ. Proteomic method for identification of tyrosine-nitrated proteins. Methods Mol Biol. 2004;279:151–165. doi: 10.1385/1-59259-807-2:151. [DOI] [PubMed] [Google Scholar]
- 21.Miyagi M, Sakaguchi H, Darrow RM, Yan L, West KA, Aulak KS, Stuehr DJ, Hollyfield JG, Organisciak DT, Crabb JW. Evidence that light modulates protein nitration in rat retina. Mol Cell Proteomics. 2002;1:293–303. doi: 10.1074/mcp.m100034-mcp200. [DOI] [PubMed] [Google Scholar]
- 22.Koeck T, Levison B, Hazen SL, Crabb JW, Stuehr DJ, Aulak KS. Tyrosine nitration impairs mammalian aldolase A activity. Mol Cell Proteomics. 2004;3:548–557. doi: 10.1074/mcp.M300141-MCP200. [DOI] [PubMed] [Google Scholar]
- 23.Zhan X, Du Y, Crabb JS, Gu X, Kern TS, Crabb JW. Targets of tyrosine nitration in diabetic rat retina. Mol Cell Proteomics. 2008;7:864–874. doi: 10.1074/mcp.M700417-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 24.Du Y, Smith MA, Miller CM, Kern TS. Diabetes-induced nitrative stress in the retina, and correction by aminoguanidine. J Neurochem. 2002;80:771–779. doi: 10.1046/j.0022-3042.2001.00737.x. [DOI] [PubMed] [Google Scholar]
- 25.Elfering SL, Haynes VL, Traaseth NJ, Ettl A, Giulivi C. Aspects, mechanism, and biological relevance of mitochondrial protein nitration sustained by mitochondrial nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2004;286:H22–H29. doi: 10.1152/ajpheart.00766.2003. [DOI] [PubMed] [Google Scholar]
- 26.McGrowder D, Ragoobirsingh D, Brown P. Modulation of glucose uptake in adipose tissue by nitric oxide-generating compounds. J Biosci. 2006;31:347–354. doi: 10.1007/BF02704107. [DOI] [PubMed] [Google Scholar]
- 27.Sokolovsky M, Riordan JF, Vallee BL. Tetranitromethane. A reagent for the nitration of tyrosyl residues in proteins. Biochemistry. 1966;5:3582–3589. doi: 10.1021/bi00875a029. [DOI] [PubMed] [Google Scholar]
- 28.Mallozzi C, Di Stasi AM, Minetti M. Nitrotyrosine mimics phosphotyrosine binding to the SH2 domain of the src family tyrosine kinase lyn. FEBS Lett. 2001;503:189–195. doi: 10.1016/s0014-5793(01)02726-0. [DOI] [PubMed] [Google Scholar]
- 29.Buelt MK, Xu Z, Banaszak LJ, Bernlohr DA. Structural and functional characterization of the phosphorylated adipocyte lipid-binding protein (pp 15) Biochemistry. 1992;31:3493–3499. doi: 10.1021/bi00128a025. [DOI] [PubMed] [Google Scholar]
- 30.Duplain H, Sartori C, Dessen P, Jayet PY, Schwab M, Bloch J, Nicod P, Scherrer U. Stimulation of peroxynitrite catalysis improves insulin sensitivity in high fat diet-fed mice. J Physiol. 2008;586:4011–4016. doi: 10.1113/jphysiol.2008.154302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buchczyk DP, Grune T, Sies H, Klotz LO. Modifications of glyceralde-hyde-3-phosphate dehydrogenase induced by increasing concentrations of peroxynitrite: early recognition by 20S proteasome. Biol Chem. 2003;384:237–241. doi: 10.1515/BC.2003.026. [DOI] [PubMed] [Google Scholar]
- 32.Markland FS, Bacharach AD, Weber BH, O’Grady TC, Saunders GC, Umemura N. Chemical modification of yeast 3-phosphoglycerate kinase. J Biol Chem. 1975;250:1301–1310. [PubMed] [Google Scholar]
- 33.Varrone S, Consiglio E, Covelli I. The nature of inhibition of mitochondrial malate dehydrogenase by thyroxine, iodine cyanide and molecular iodine. Eur J Biochem/FEBS. 1970;13:305–312. doi: 10.1111/j.1432-1033.1970.tb00932.x. [DOI] [PubMed] [Google Scholar]
- 34.Tortora V, Quijano C, Freeman B, Radi R, Castro L. Mitochondrial aconitase reaction with nitric oxide, S-nitrosoglutathione, and peroxynitrite: mechanisms and relative contributions to aconitase inactivation. Free Radic Biol Med. 2007;42:1075–1088. doi: 10.1016/j.freeradbiomed.2007.01.007. [DOI] [PubMed] [Google Scholar]
- 35.Han D, Canali R, Garcia J, Aguilera R, Gallaher TK, Cadenas E. Sites and mechanisms of aconitase inactivation by peroxynitrite: modulation by citrate and glutathione. Biochemistry. 2005;44:11986–11996. doi: 10.1021/bi0509393. [DOI] [PubMed] [Google Scholar]
- 36.Khor HK, Fisher MT, Schoneich C. Potential role of methionine sulfoxide in the inactivation of the chaperone GroEL by hypochlorous acid (HOCl) and peroxynitrite (ONOO−) J Biol Chem. 2004;279:19486–19493. doi: 10.1074/jbc.M310045200. [DOI] [PubMed] [Google Scholar]
- 37.Voos W, Rottgers K. Molecular chaperones as essential mediators of mitochondrial biogenesis. Biochim Biophys Acta. 2002;1592:51–62. doi: 10.1016/s0167-4889(02)00264-1. [DOI] [PubMed] [Google Scholar]
- 38.Deocaris CC, Kaul SC, Wadhwa R. On the brotherhood of the mitochondrial chaperones mortalin and heat shock protein 60. Cell Stress Chaperones. 2006;11:116–128. doi: 10.1379/CSC-144R.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.O’Rourke B. Mitochondrial ion channels. Annu Rev Physiol. 2007;69:19–49. doi: 10.1146/annurev.physiol.69.031905.163804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lemasters JJ. Modulation of mitochondrial membrane permeability in pathogenesis, autophagy and control of metabolism. J Gastroenterol Hepatol. 2007;22(Suppl 1):S31–S37. doi: 10.1111/j.1440-1746.2006.04643.x. [DOI] [PubMed] [Google Scholar]
- 41.Erbay E, Cao H, Hotamisligil GS. Adipocyte/macrophage fatty acid binding proteins in metabolic syndrome. Curr Atheroscler Rep. 2007;9:222–229. doi: 10.1007/s11883-007-0023-6. [DOI] [PubMed] [Google Scholar]
- 42.Hotamisligil GS. Inflammation and metabolic disorders. Nature. 2006;444:860–867. doi: 10.1038/nature05485. [DOI] [PubMed] [Google Scholar]
- 43.Xu Z, Bernlohr DA, Banaszak LJ. Crystal structure of recombinant murine adipocyte lipid-binding protein. Biochemistry. 1992;31:3484–3492. doi: 10.1021/bi00128a024. [DOI] [PubMed] [Google Scholar]
- 44.Sha RS, Kane CD, Xu Z, Banaszak LJ, Bernlohr DA. Modulation of ligand binding affinity of the adipocyte lipid-binding protein by selective mutation. Analysis in vitro and in situ. J Biol Chem. 1993;268:7885–7892. [PubMed] [Google Scholar]
- 45.Hresko RC, Hoffman RD, Flores-Riveros JR, Lane MD. Insulin receptor tyrosine kinase-catalyzed phosphorylation of 422(aP2) protein. Substrate activation by long-chain fatty acid. J Biol Chem. 1990;265:21075–21085. [PubMed] [Google Scholar]
- 46.Smith AJ, Thompson BR, Sanders MA, Bernlohr DA. Interaction of the adipocyte fatty acid-binding protein with the hormone-sensitive lipase: regulation by fatty acids and phosphorylation. J Biol Chem. 2007;282:32424–32432. doi: 10.1074/jbc.M703730200. [DOI] [PubMed] [Google Scholar]
- 47.Adida A, Spener F. Adipocyte-type fatty acid-binding protein as inter-compartmental shuttle for peroxisome proliferator activated receptor gamma agonists in cultured cell. Biochim Biophys Acta. 2006;1761:172–181. doi: 10.1016/j.bbalip.2006.02.006. [DOI] [PubMed] [Google Scholar]
- 48.Ayers SD, Nedrow KL, Gillilan RE, Noy N. Continuous nucleocytoplasmic shuttling underlies transcriptional activation of PPARgamma by FABP4. Biochemistry. 2007;46:6744–6752. doi: 10.1021/bi700047a. [DOI] [PubMed] [Google Scholar]
- 49.Wolfrum C, Borrmann CM, Borchers T, Spener F. Fatty acids and hypoli-pidemic drugs regulate peroxisome proliferator-activated receptors alpha- and gamma-mediated gene expression via liver fatty acid binding protein: a signaling path to the nucleus. Proc Natl Acad Sci U S A. 2001;98:2323–2328. doi: 10.1073/pnas.051619898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Laplante M, Festuccia WT, Soucy G, Gelinas Y, Lalonde J, Berger JP, Deshaies Y. Mechanisms of the depot specificity of peroxisome proliferator-activated receptor gamma action on adipose tissue metabolism. Diabetes. 2006;55:2771–2778. doi: 10.2337/db06-0551. [DOI] [PubMed] [Google Scholar]
- 51.Ghosh S, Janocha AJ, Aronica MA, Swaidani S, Comhair SA, Xu W, Zheng L, Kaveti S, Kinter M, Hazen SL, Erzurum SC. Nitrotyrosine proteome survey in asthma identifies oxidative mechanism of catalase inactivation. J Immunol. 2006;176:5587–5597. doi: 10.4049/jimmunol.176.9.5587. [DOI] [PubMed] [Google Scholar]
- 52.Okuno Y, Matsuda M, Kobayashi H, Morita K, Suzuki E, Fukuhara A, Komuro R, Shimabukuro M, Shimomura I. Adipose expression of catalase is regulated via a novel remote PPARgamma-responsive region. Biochem Biophys Res Commun. 2008;366:698–704. doi: 10.1016/j.bbrc.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 53.Hatahet F, Ruddock LW. Substrate recognition by the protein disulfide isomerases. FEBS J. 2007;274:5223–5234. doi: 10.1111/j.1742-4658.2007.06058.x. [DOI] [PubMed] [Google Scholar]
- 54.Welsh GI, Griffiths MR, Webster KJ, Page MJ, Tavare JM. Proteome analysis of adipogenesis. Proteomics. 2004;4:1042–1051. doi: 10.1002/pmic.200300675. [DOI] [PubMed] [Google Scholar]
- 55.Takai Y, Sasaki T, Matozaki T. Small GTP-binding proteins. Physiol Rev. 2001;81:153–208. doi: 10.1152/physrev.2001.81.1.153. [DOI] [PubMed] [Google Scholar]
- 56.Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, Zhang H, Zha XM, Polakiewicz RD, Comb MJ. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol. 2005;23:94–101. doi: 10.1038/nbt1046. [DOI] [PubMed] [Google Scholar]
- 57.Bar-Sagi D, Hall A. Ras and Rho GTPases: a family reunion. Cell. 2000;103:227–238. doi: 10.1016/s0092-8674(00)00115-x. [DOI] [PubMed] [Google Scholar]
- 58.DerMardirossian C, Bokoch GM. GDIs: central regulatory molecules in Rho GTPase activation. Trends Cell Biol. 2005;15:356–363. doi: 10.1016/j.tcb.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 59.Usui I, Imamura T, Huang J, Satoh H, Olefsky JM. Cdc42 is a Rho GTPase family member that can mediate insulin signaling to glucose transport in 3T3-L1 adipocytes. J Biol Chem. 2003;278:13765–13774. doi: 10.1074/jbc.M208904200. [DOI] [PubMed] [Google Scholar]
- 60.Hou JC, Shigematsu S, Crawford HC, Anastasiadis PZ, Pessin JE. Dual regulation of Rho and Rac by p120 catenin controls adipocyte plasma membrane trafficking. J Biol Chem. 2006;281:23307–23312. doi: 10.1074/jbc.M603127200. [DOI] [PubMed] [Google Scholar]
- 61.Gallagher BC, Parrott KA, Szabo G, de SOA. Receptor activation regulates cortical, but not vesicular localization of NDP kinase. J Cell Sci. 2003;116:3239–3250. doi: 10.1242/jcs.00630. [DOI] [PubMed] [Google Scholar]
- 62.Brock TG. Capturing proteins that bind polyunsaturated fatty acids: demonstration using arachidonic acid and eicosanoids. Lipids. 2008;43:161–169. doi: 10.1007/s11745-007-3136-3. [DOI] [PubMed] [Google Scholar]
- 63.Wieland T. Interaction of nucleoside diphosphate kinase B with heterotrimeric G protein betagamma dimers: consequences on G protein activation and stability. Naunyn-Schmiedeberg’s Arch Pharmacol. 2007;374:373–383. doi: 10.1007/s00210-006-0126-6. [DOI] [PubMed] [Google Scholar]
- 64.Kapetanovich L, Baughman C, Lee TH. Nm23H2 facilitates coat protein complex II assembly and endoplasmic reticulum export in mammalian cells. Mol Biol Cell. 2005;16:835–848. doi: 10.1091/mbc.E04-09-0785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Arnaud-Dabernat S, Masse K, Smani M, Peuchant E, Landry M, Bourbon PM, Le Floch R, Daniel JY, Larou M. Nm23-M2/NDP kinase B induces endogenous c-myc and nm23-M1/NDP kinase A overexpression in BAF3 cells. Both NDP kinases protect the cells from oxidative stress-induced death. Exp Cell Res. 2004;301:293–304. doi: 10.1016/j.yexcr.2004.07.026. [DOI] [PubMed] [Google Scholar]