

Abstract
Whether signal transduction pathways regulate epigenetic states in response to environmental cues remains poorly understood. We demonstrate here that Smad3, signaling downstream of transforming growth factor β, interacts with the zinc finger domain of CCCTC-binding factor (CTCF), a nuclear protein known to act as “the master weaver of the genome.” This interaction occurs via the Mad homology 1 domain of Smad3. Although Smad2 and Smad4 fail to interact, an alternatively spliced form of Smad2 lacking exon 3 interacts with CTCF. CTCF does not perturb well established transforming growth factor β gene responses. However, Smads and CTCF co-localize to the H19 imprinting control region (ICR), which emerges as an insulator in cis and regulator of transcription and replication in trans via direct CTCF binding to the ICR. Smad recruitment to the ICR requires intact CTCF binding to this locus. Smad2/3 binding to the ICR requires Smad4, which potentially provides stability to the complex. Because the CTCF-Smad complex is not essential for the chromatin insulator function of the H19 ICR, we propose that it can play a role in chromatin cross-talk organized by the H19 ICR.
Keywords: Epigenetics, Insulin-like Growth Factor (IGF), Signal Transduction, Smad Transcription Factor, Transforming Growth Factor β (TGFβ)
Introduction
Genomic imprinting is manifested by the translation of gametic marks into the parent of origin-dependent gene expression patterns. The neighboring Igf2 (insulin-like growth factor 2) and H19 genes are generally considered the paradigms of genomic imprinting because their expression is monoallelic from opposite parental alleles and governed by shared enhancers (1, 2). The repression of the maternal Igf2 and paternal H19 alleles depends on a differentially methylated ICR5 in the 5′ region of the H19 gene (3). This feature is mediated by the only currently known mammalian insulator, the zinc finger protein CTCF (4, 5). CTCF has a central domain comprised by 11 zinc fingers, flanked by long N- and C-terminal domains. Although CTCF interacts with only the unmethylated, maternal H19 ICR allele, it also protects this region from de novo methylation. CTCF bound to the H19 ICR has been implicated in both local and long range interactions between chromatin fibers both in cis and in trans (6–9). This evidence implicates CTCF in the control of diverse biological processes.
Transforming growth factor β (TGFβ) is a secreted cytokine with vital functions during embryogenesis, adult tissue homeostasis, and disease pathogenesis, such as with cancer (10, 11). TGFβ signals via Smad proteins (Smad2 and Smad3) that are phosphorylated by the cell surface TGFβ type I receptor and rapidly move to the nucleus in association with the common mediator Smad4, where they regulate transcription (12). Smad3, upon phosphorylation by the TGFβ type I receptor and entry to the nucleus, binds efficiently to the DNA sequence 5′-CAGACA-3′, also known as the Smad-binding element (SBE) (13). Despite the established role of TGFβ and its family members in developmental processes, connections between TGFβ signaling and control of the epigenome have not been made.
Here we describe a novel cross-talk between CTCF and the Smad pathway of TGFβ. CTCF forms complexes with Smads, and together they are recruited to the H19 ICR. Smad recruitment requires prior CTCF binding to the ICR. Although TGFβ signaling regulates expression of Igf2, this feature involves only the already active paternal allele. Although this observation rules out an effect on chromatin insulator function, we propose that TGFβ signaling may influence cross-talk between chromatin fibers. This proposal is in keeping with the co-adaptor function of the Smad complex and hence its ability to establish interactions within and between chromosomes.
EXPERIMENTAL PROCEDURES
Cells, Mice, Viruses, Ligands, Antibodies, and Plasmids
Mouse mammary epithelial NMuMG, human mammary carcinoma MDA-MB-468, human hepatocellular carcinoma HepG2, and human embryonic kidney 293T (HEK293T) cells were purchased from American Type Culture Collection and cultured as described previously (14). Primary embryonic fibroblasts and primary cells from newborn liver were derived from mice of the following crosses: 142* × SD7 (♀mut × ♂wt) and SD7 × 142* (♀wt × ♂mut). Strain 142* harbors mutations in three of four CTCF-binding sites in the H19 ICR on chromosome 7 and has previously been described (15). Strain SD7 is congenic; it is derived from Mus musculus domesticus and carries the distal end of chromosome 7 of Mus musculus spretus (16). Progeny of the SD7 × 142* cross have wild type maternally inherited H19 ICR and are designated as wt. Progeny of the reciprocal cross have a mutant maternally inherited H19 ICR and are designated as mut. Ethical approval was obtained from the Animal Ethics Committee in Uppsala, Sweden. Adenoviruses expressing LacZ and FLAG-tagged wild type Smad4 have been described earlier (14).
Recombinant mature TGFβ1 was purchased from PeproTech EC Ltd. The following antibodies were used for chromatin immunoprecipitation: against CTCF (antibody 612149) from BD Transduction Laboratories, against Smad2 (antibody S-20) from Santa Cruz Biotechnology, against Smad3 (antibody 51-1500) from Zymed Laboratories Inc., and against Smad4 (antibody sc-7154) from Santa Cruz Biotechnology. The antibodies used for co-immunoprecipitation were mouse monoclonal anti-FLAG (M2) purchased from Sigma, the mouse monoclonal anti-Myc (antibody 9E10), and rabbit anti-Smad3 (antibody 51-1500) from Zymed Laboratories Inc.. The antibodies used for immunoblotting of total cell lysates or of DNA affinity precipitation assays were, in addition to those listed above: mouse anti-GST (antibody sc-138) from Santa Cruz Biotechnology, rabbit anti-phospho-Smad3 (C-terminal, antibody C25A9) from Cell Signaling Technology (antibody 9520S), and mouse anti-glyceraldehyde-3-phosphate dehydrogenase (antibody AM4300) from Ambion.
The mammalian expression vectors pcDNA3 encoding C-terminally hemagglutinin-tagged constitutively active (T204D) TGFβ type I receptor (ALK5 (activin receptor-like kinase 5)), N-terminally Myc6-tagged Smad2, Smad3, and Smad4 have been described previously (17). The empty GST vector (pGEX-4T1), the GST fusion vectors with full-length Smad2, Smad3 and Smad4, Smad3 deletions, ΔMH1 and ΔMH2, and MH1 domain swaps between Smad2 and Smad3, GST-Smad3+GAG, +TID, GST-Smad2-ΔGAG, -ΔTID, -ΔGAGΔTID have been described previously (17, 18). The vectors encoding pcDNA3 FLAG-tagged CTCF and GST fusions of CTCF domains, N-terminal, C-terminal, zinc finger (Zn 1–11), and shorter zinc finger domains (Zn 1–4 and Zn 1–7) were kind gifts from J. Leers and R. Renkawitz (University of Gissen, Gissen, Germany) (19). The promoter-luciferase constructs pCAGA12-MLP-luc, pCMV-β-galactosidase, and p800-PAI-1-luc have been described (20).
Transfections and Infections
Calcium phosphate DNA co-precipitation, transient transfections of HEK293T or HepG2 cells, and adenoviral transient infections of MDA-MB-468 cells were performed as described earlier (14, 17). For short interfering RNA (siRNA)-mediated knockdown of endogenous CTCF, NMuMG or HepG2 cells at 80% confluency were transfected with CTCF siRNA (Dharmacon Inc.) using Dharmafect reagent (Dharmacon Inc.) according to the manufacturer's instructions. siRNAs were transfected at a concentration of 5–20 nm in 6-well plates for 48 h, and in some experiments, after 24 h, the treatment with siRNA was repeated (double treatment). For stimulation experiments with TGFβ1, the cells were starved by replacing the medium with Dulbecco's modified Eagle's medium containing 1% serum for 8 h minimum before the end of the siRNA treatment. The CTCF siRNA pool was from Dharmacon Inc. (L-20165-00-0020, human CTCF, NM_006565) and contained four siRNAs: ON-targetplus SMARTpool siRNA J-20165-07, GAUGAAGACUGAAGUAAUG; ON-targetplus SMARTpool siRNA J-20165-08, GGAGAAACGAAGAAGAGUA; ON-targetplus SMARTpool siRNA J-20165-09, GAAGAUGCCUGCCACUUAC; and ON-targetplus SMARTpool siRNA J-20165-10, GAACAGCCCAUAAACAUAG. The nonsilencing siRNA controls used in this study were either siScrambled or siLuciferase from Dharmacon Inc.
Co-immunoprecipitations and DNA Affinity Precipitations
Associations between transfected FLAG-tagged CTCF and Myc6-tagged Smad2, Smad3, or Smad4 in HEK293T cells and between endogenous CTCF and Smad3 in HepG2 cells were monitored as described previously by co-immunoprecipitation assays in total cell lysates (17). DNA affinity precipitations using a concatamerized Smad-binding element (4×CAGA DNA) and transfected FLAG-CTCF and endogenous phospho-Smad3 in HepG2 cells were performed exactly as described (21). The sequence of the double-stranded 4× CAGA oligonucleotide is: 5′-CAGACAGTCAGACAGTCAGACAGTCAGACAGT-3′ (sense strand) and 5′-ACTGTCTGACTGTCTGACTGTCTGACTGTCTG-3′ (antisense strand).
In Vitro Translation
The FLAG-tagged CTCF cDNA from pCDNA3-FLAG-CTCF was subjected to in vitro transcription and translation using the TnT quick coupled transcription/translation system, following the supplier's instructions (Promega). Briefly, 1 μg of plasmid was incubated for 1 h at 30 °C in a reaction mix containing the rabbit reticulocyte lysate, T7 RNA polymerase, nucleotides, salts, recombinant RNasin ribonuclease inhibitor, and 20 μCi of [35S]methionine/cysteine (PerkinElmer Life Sciences). A control without plasmid in the reaction mix was used to monitor for unspecific translation products.
GST Pulldown Assays
Cell extracts from NMuMG cells or reticulocyte lysates after in vitro translation were incubated in the presence of GST-Sepharose beads to wash away the unspecific binding by incubating on a rotating wheel for 2 h at 4 °C. After washing with lysis buffer containing increasing amounts of NaCl, the beads were washed with lysis buffer, and the final pellet was directly resuspended in the sample buffer, loaded on acrylamide gel, and subjected to electrophoresis. The amount and the quality of GST fusion proteins incubated with the lysates were monitored on a gel stained with Coomassie Brilliant Blue. GST pulldown from transfected NMuMG cell extracts was analyzed by anti-FLAG immunoblotting. GST pulldown from in vitro translated plasmids was analyzed after drying the acrylamide gel and phosphorimaging (with a FUJIFILM FLA-3000 unit and associated software) to visualize the radioactively labeled proteins.
Chromatin Immunoprecipitation (ChIP)
Approximately 106 subconfluent NMuMG cells or the corresponding number of mouse embryonic fibroblasts (MEFs) with or without 4 h of stimulation with 5 ng/ml TGFβ1 were cross-linked at 37 °C for 10 min using 1% paraformaldehyde. The cross-linking was quenched with 0.125 m glycine, and the cells were washed twice with ice-cold phosphate-buffered saline containing protease inhibitors. After centrifugation, the cells were resuspended in 200 μl of SDS lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris-HCl, pH 8.1) and incubated on ice for 10 min. The chromatin was sheared by sonication (with a Branson Digital Sonifier) to an average size of 1 kb as described previously (22) and precleared by incubating with Sepharose A or G 4 Fast Flow from Amersham Biosciences with slow rotation overnight at 4 °C. The Sepharose beads were previously washed three times in 15 mm Tris-HCl, pH 7.5, 1 mm EDTA, 150 mm NaCl, 0.05% Triton X-100, 1 mg/ml bovine serum albumin, and 1 mg/ml herring sperm DNA from Invitrogen. At this step, a fraction of the precleared chromatin was kept as input material, and each ChIP reaction (∼10 μg of chromatin) was incubated with 1 μg of antibody at 4 °C for 4 h. After this, 60 μl of Sepharose A/G 4 Fast Flow beads were added to each reaction. The chromatin antibody-Sepharose bead complexes were washed as follows: 30 min in low salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, pH 8.0, 20 mm Tris-HCl, pH 8.0, 150 mm NaCl), 15 min in high salt wash buffer (0.1% SDS, 1% Triton X-100, 2 mm EDTA, pH 8.0, 20 mm Tris-HCl, pH 8.0, 500 mm NaCl), 15 min in LiCl wash buffer (0.25 m LiCl, 1% Igepal-CA630, 1% sodium deoxycholate, 1 mm EDTA, pH 8.0, 10 mm Tris-HCl, pH 8.0), and twice for 20 min in TE buffer (10 mm Tris-HCl, pH 8.0, 1 mm EDTA, pH 8.0). The DNA-protein complexes were eluted with 2× 250 μl of freshly made elution buffer (1% SDS and 0.1 m NaHCO3). To reverse the cross-links, 20 μl of 5 m NaCl was added to each of the samples, which were incubated at 65 °C overnight. The proteins were degraded by proteinase K (Amersham Biosciences), and DNA was extracted by phenol/chloroform/isoamyl alcohol extraction, purified, and resuspended in water. All of the ChIP experiments were repeated three times or more.
Real Time PCR Analysis of ChIP Samples
The DNA obtained from the chromatin immunoprecipitation was analyzed together with a sample to which no antibody had been added and a dilution of the input material () using quantitative (Q) real time PCR. Q-PCR primers and TaqMan probes (Table 1) were designed to amplify and analyze the wild type and mutated H19 ICR. All of the Q-PCR analyses were done in triplicate and repeated a minimum of three times. All of the reactions were performed using iCycler iQTM0 170–8740 and iQ Supermix (Bio-Rad), with the following cycling conditions: 95 °C for 3 min and 40 cycles of 95 °C for 10 s and 65 °C for 45 s.
TABLE 1.
Q-PCR primer and probe sequences
| Clone 704 forward primer | 5′-GAT TAC ATC AGT TAG GGG TC-3′ |
| Clone 704 reverse primer | 5′-GCC ACC ACC TGT ATT GGC AG-3′ |
| KvDMR (Kcnq1) forward primer | 5′-GTG TGC TTT TCT CTG CAT GG-3′ |
| KvDMR (Kcnq1) reverse primer | 5′-CAG GAC AAA CAC TGA GGA GG-3′ |
| H19 ICR CTCF-binding site 3 forward primer | 5′-GCT GTT ATG TGC AAC AAG GG-3′ |
| H19 ICR CTCF-binding site 3 reverse primer | 5′-AAG TTG GCA GCA TTT GGG C-3′ |
| TaqMan probe complementary to wild type H19 ICR | 5′-TAC CGC GCG GTG GCA GCA TAC TCC TAA AT-3′ |
| TaqMan probe complementary to 142* H19 ICR | 5′-CGG ATG CTA CCG CGC GAT ATC AGC ATA CT-3′ |
| Xist forward primer | 5′-GTC GCC AAC CTA ATG CAG AAG-3′ |
| Xist reverse primer | 5′-AAA CGA GCA AAC ATG GCT GG-3′ |
Luciferase Assays
HepG2 cells were transiently transfected with TGFβ/Smad-responsive promoter-reporter pCAGA12-MLP-luc and p800-PAI-1-luc constructs in the presence of mock (pCDNA3) or pCDNA3-FLAG-CTCF plasmids for 24 h prior to stimulation with TGFβ1 for another 16 h. pCMV-galactosidase was co-transfected as control for normalization. Luciferase reporter assays were performed with the enhanced luciferase assay kit from BD PharMingen, Inc., according to the protocol of the manufacturer. Normalized promoter activity data are plotted in bar graphs that represent the average values from triplicate determinations with standard deviations. Each independent experiment was repeated at least twice.
mRNA Expression Analysis
Total NMuMG or HepG2 RNA after transient siRNA transfection and stimulation with 5 ng/ml TGFβ1 for 4 h was isolated using the RNeasy kit from Qiagen. Real time Q-PCR analysis of the total RNA for specific expression of Snail1, PAI-1, and Smad7 mRNA was performed as described previously (14). The primers used for PCR amplification were: human PAI-1, sense, 5′-GAGACAGGCAGCTCGGATTC-3′, and antisense, 5′-GGCCTCCCAAAGTGCATTAC-3′; human GAPDH, sense, 5′-GGAGTCAACGGATTTGGTCGTA-3′, and antisense, 5′-GGCAACAATATCCACTTTACCA-3′; human SMAD7, sense, 5′-ACCCGATGGATTTTCTCAAACC-3′, and antisense, 5′-GCCAGATAATTCGTTCCCCCT-3′; mouse Snail1, sense, 5′-CCACTGCAACCGTGCTTTT-3′, and antisense, 5′-CACATCCGAGTGGGTTTGG-3′; mouse Pai-1, sense, 5′-GGCAGATCCAAGATGCTATGG-3′, and antisense, 5′-TCATTCTTGTTCCACGGCC-3′; and mouse Gapdh, sense, 5′-TGTGTCCGTCGTGGATCTGA-3′, and antisense, 5′-CCTGCTTCACCACCTTCTTGA-3′. The levels of H19 and Igf2 mRNA were analyzed using TaqMan gene expression probes Mm00469706_g1 (H19) and Mm00439565_g1 (Igf2) purchased from Applied Biosystems and performed according to the manufacturer's recommendations using MEF 142* × SD7 and SD7 × 142* mRNA.
The 4-h stimulated samples were then further studied for allele-specific expression of H19 and Igf2 as described previously (15) and visualized using the Bioanalyzer 5100 from Agilent. In more detail, diagnostic BglII restriction digestions of the amplified cDNA take advantage of a mouse strain-specific polymorphism on the H19 transcribed sequence. BglII digestion of H19 cDNA encoded by the chromosome carrying the mutations (mut) on the CTCF-binding sites (M. musculus domesticus strain) is expected to give rise to a single 521-bp DNA fragment, whereas cDNA encoded from the chromosome carrying the wild type CTCF-binding sites (M. musculus spretus strain) is expected to give rise to two DNA fragments of 384 and 137 bp. BsaAI digestion of Igf2 cDNA encoded by the chromosome carrying the mutations (mut) on the CTCF-binding sites (M. musculus domesticus strain) is expected to give rise to a single 602-bp DNA fragment, whereas cDNA encoded from the chromosome carrying the wild type CTCF-binding sites (M. musculus spretus strain) is expected to give rise to two DNA fragments of 473 and 129 bp.
RESULTS
Smad3 Directly Interacts with CTCF
A previous report suggested the possibility that Smad proteins might form physical complexes with CTCF on the β-amyloid gene enhancer (23). We tested the generality of this hypothesis by expressing CTCF and the three Smads of the TGFβ pathway (Smad2, Smad3, and Smad4) in human embryonic kidney cells and performing co-immunoprecipitation assays (Fig. 1A). A sustained TGFβ stimulus was provided to the cells by co-transfecting a constitutively active form of the TGFβ type I receptor (ALK5T204D). CTCF co-precipitated with Smad3, but not with Smad2 or Smad4, even in the absence of receptor activation, and such activation weakly enhanced the CTCF-Smad complex. These data were confirmed by co-immunoprecipitation assays of endogenous CTCF and Smad3 in human hepatocarcinoma HepG2 cells (Fig. 1B) and in embryonic kidney cells (data not shown). We could also detect the endogenous CTCF-Smad3 complex in these cells even in the absence of stimulation with TGFβ (Fig. 1B). Because CTCF is integrated into the chromatin, these results suggest that a certain pool of Smad3 always exists in the nucleus and is capable of making contacts with factors that are tightly bound to DNA, such as CTCF.
FIGURE 1.

Smad3 interacts with the zinc finger domain of CTCF. A, immunoprecipitation (IP) of FLAG-CTCF followed by immunoblotting (IB) for the indicated proteins after transfection of HEK293T cells with the indicated plasmids. Total cell lysates (TCL) demonstrate the input Smad levels. An asterisk indicates a nonspecific protein. B, co-immunoprecipitation of endogenous CTCF with endogenous Smad3 from HepG2 cells in the absence of TGFβ stimulation. Immunoblots of total cell lysates are shown as input, and flag represents immunoprecipitation with a nonspecific antibody. C, GST pulldown assay of transfected Myc-Smad3 with the indicated GST fragments of CTCF. An aliquot of transfected HEK293T cells shows the Smad3 expression used as input, and Ponceau staining shows the recombinant CTCF fragments. An asterisk indicates a nonspecific protein. Molecular size markers (in kDa) are shown in the bottom panel. D, schematic diagram of Smad2, its alternatively spliced isoform Smad2Δex3, Smad3, and Smad4. The MH1, linker, and MH2 domains are highlighted, along with the C-terminal di-serine motif that is phosphorylated by the TGFβ type I receptor. Within the MH1 domain, features of the proteins discussed in this paper are emphasized: the N-terminal Smad2-specific peptide insert GAG, the exon 3 corresponding peptide insert TID, and the juxtaposed DNA-binding domain (DBD) of all Smads. Splicing of exon 3 is indicated, and the lack of the GAG insert from the sequence of Smad3 is also shown.
Using a panel of GST fusions to the CTCF domains, we found that Smad3 preferentially interacts with the central CTCF zinc finger domain (Fig. 1C). Further GST pulldown assays confirmed the specificity of interaction between CTCF and Smad3 (Fig. 2A), and use of recombinant proteins confirmed that CTCF interacts directly with Smad3 but not with Smad2 or Smad4 (Fig. 2B). Incubation with both recombinant Smad3 and Smad4 led to a significantly stronger complex with CTCF (Fig. 2A), suggesting that the presence of Smad4 stabilizes or enhances formation of this ternary protein complex. GST pulldown assays with two deletion mutants of Smad3 indicated that CTCF interacts with the N-terminal conserved domain of Smad3 (Fig. 3A), known as Mad homology 1 (MH1 domain; Fig. 1D), which binds to specific DNA sequences and also carries the Smad nuclear localization signal (11).
FIGURE 2.

Direct interaction between Smad3 and CTCF. A, GST pulldown assay of transfected FLAG-CTCF with the indicated GST-Smad proteins. An aliquot of transfected HEK293T cell total cell lysate (TCL) shows the CTCF expression used as input. Anti-GST immunoblot (IB) shows the recombinant GST-Smad proteins, and asterisks indicate degradation fragments. B, GST pulldown assay of in vitro translated, [35S]methionine/cysteine-labeled FLAG-CTCF with the indicated GST-Smad proteins. Autoradiograms show the radioactive CTCF protein, and the GST-Smads used are shown after Coomassie Brilliant Blue staining of the gels. The asterisks indicate degradation fragments. Molecular size markers (in kDa) are shown.
FIGURE 3.

CTCF interacts with the MH1 domain of Smad3, and exon 3 in Smad2 prohibits this interaction. Shown are GST pulldown assays of in vitro translated, [35S]methionine/cysteine-labeled FLAG-CTCF with the indicated GST-Smad3 deletion mutants (A) and GST-Smad2/Smad3 swapping mutants (B). The conditions and presentation are exactly as described for Fig. 2B. In B, M indicates the molecular size marker lane.
The MH1 domains of Smad2 and Smad3 are structurally identical except for the presence of two short amino acid sequence inserts, one named GAG and the second named TID, in Smad2 (Fig. 1D) (24). The TID insert corresponds to exon 3 of Smad2, which can be alternatively spliced, thus forming a natural Smad2 variant that highly resembles Smad3. The presence of the TID insert (exon 3) explains why full-length Smad2 fails to bind to DNA or to the importins that import Smad3 into the nucleus (18, 24). Thus, it is possible that the Smad2-specific GAG and/or TID peptide sequences might interfere with its interaction with CTCF. To test this hypothesis we repeated the GST pulldown assays with a new panel of Smad2/Smad3 MH1 domain swapping mutants (Fig. 3B). Engineering peptide insert TID into the sequence of wild type Smad3 abolished the interaction with CTCF (Fig. 3B). Conversely, deleting only peptide insert TID from Smad2 made this protein capable of binding to CTCF, whereas the presence or absence of the N-terminal peptide insert GAG had no effect (Fig. 3B). Thus, CTCF can specifically form complexes with Smad3 or an alternatively spliced form of Smad2 that lacks exon3.
CTCF Minimally Affects Gene Responses to Smad3
The specificity of interaction between the CTCF zinc finger domain and the Smad3 MH1/DNA-binding domain suggested that CTCF might regulate the function of Smad3 during TGFβ signaling. A classic target of TGFβ/Smad3 signaling is the PAI-1 (plasminogen activator inhibitor 1) gene (25). DNA affinity precipitation experiments showed that phosphorylated Smad3 could be readily detected bound to a SBE DNA sequence derived from the PAI-1 enhancer; co-expression of CTCF did not perturb such binding to DNA (Fig. 4A).
FIGURE 4.

CTCF does not affect binding of Smad3 to DNA. A, DNA affinity precipitation assay with biotinylated 4× CAGA oligonucleotide and cell extracts from transfected HepG2 cells in the absence or presence of 5 ng/ml TGFβ1 for 2 h. The levels of transfected FLAG-CTCF and endogenous phospho-Smad3 are shown in control total cell lysate (TCL) and after the DNA affinity precipitation. An asterisk shows a nonspecific protein. B and C, promoter-luciferases assays in HepG2 cells transfected with the indicated reporter and mock plasmid or two amounts of CTCF plasmid prior to stimulation with 5 ng/ml TGFβ1 for 16 h (black bars). The average values and standard deviations from triplicate determinations are plotted.
Transcriptional reporter assays using the same PAI-1 enhancer SBE element fused to the luciferase cDNA, which is established as a potent Smad3-dependent reporter (25), failed to show any significant impact of CTCF overexpression on the responsiveness of this promoter to TGFβ (Fig. 4B). A longer, 800-bp enhancer-promoter fragment of PAI-1 fused to luciferase also confers strong responsiveness to TGFβ. However, co-expression of CTCF did not significantly change this response (Fig. 4C), in agreement with the DNA binding data of Fig. 4A.
At the endogenous level, potent knockdown of CTCF (Fig. 5A) in the mouse mammary epithelial NMuMG cell line exhibited weak enhancement of the expression of the mouse Pai-1 gene and no measurable effects at all on Snail expression, as revealed by real time Q-PCR (Fig. 5B). Similar results were gathered from a well established human cell model for TGFβ/Smad responses, human hepatocellular carcinoma HepG2 cells, where efficient knockdown of endogenous CTCF (Fig. 5C) weakly enhanced expression of human PAI-1 and did not at all perturb the expression pattern of the SMAD7 gene (Fig. 5D). These results suggested that the functional importance of the Smad3-CTCF interaction may involve cellular processes other than mainstream Smad3-dependent gene responses. However, it is also possible that CTCF might exert a weak repressive effect on a subset of TGFβ-responsive genes, such as PAI-1.
FIGURE 5.

Endogenous CTCF minimally regulates Smad3-dependent gene responses. A and C, immunoblot of endogenous CTCF and control glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein from NMuMG (A) or HepG2 (C) cells transfected with the indicated siRNAs prior to stimulation with 5 ng/ml TGFβ1 for 8 h. B and D, real time Q-PCR assays for mouse Pai-1 and Snail (B) or human PAI-1 and SMAD7 (D) mRNA expression in the same cells as shown in A and C, respectively. siScr. (siScrambled) represents a nonspecific scrambled short interfering RNA sequence. The average values and standard deviations from triplicate determinations are shown.
Smad Proteins Bind to Imprinted Loci
Given the strong link between CTCF and the epigenetics of imprinted chromatin domains, we hypothesized that Smads might get recruited to CTCF-bound imprinted loci based on their ability to interact with CTCF. To this end, we performed Q-PCR analysis after ChIP in the mouse mammary epithelial cell system described above (Fig. 6). The ChIP-Q-PCR analysis showed that CTCF, as established before (26–28), could readily be found in association with the chromatin of three imprinted loci, the H19 ICR, the KvDMR, and the X-chromosomal Xist locus (Fig. 6A).
FIGURE 6.

CTCF and Smads co-occupy CTCF-binding loci such as the H19 ICR. ChIP-Q-PCR analysis of four genomic fragments corresponding to three known imprinted loci, H19 ICR, KvDMR, and Xist, and a control intergenic region, clone 704. ChIP was performed in NMuMG cells stimulated with TGFβ1 for 4 h using specific antibodies against CTCF (A), Smad3 (B), and Smad4 (C). Q-PCR data show the average values and standard deviations calculated from triplicate determinations.
Interestingly, after stimulation of these cells with TGFβ1, we could also monitor Smad3 and Smad4 recruited to the H19 ICR and KvDMR loci (Fig. 6, A and B). Smad3 also showed weak recruitment to the Xist locus, which was not altered after TGFβ stimulation (Fig. 6B). Conversely, control chromatin, such as clone 704, which represents an intergenic region with weak CTCF binding, recruited significant levels of Smad3 and Smad4 in a ligand-dependent manner, whereas CTCF binding to this site was measurable but weak (Fig. 6A). These results demonstrate that TGFβ enhances Smad recruitment to a selective group of CTCF-binding sites.
Smad Recruitment to the H19 ICR Requires an Intact Smad4
We previously demonstrated that Smad4 fails to bind directly to CTCF (Figs. 1–3). However, ChIP experiments clearly showed recruitment of Smad4 together with Smad3 and CTCF to imprinted loci (Fig. 6). Furthermore, Smad recruitment was always significantly enhanced by TGFβ stimulation, implying that Smad4 recruitment to the H19 ICR, and other loci, could be mediated via oligomerization with Smad3. To examine whether Smad4 had any functional importance for the recruitment of Smad proteins to the H19 ICR, we performed ChIP experiments in a human breast carcinoma cell line, MDA-MB-468, that is completely devoid of Smad4 because of genomic deletion of the Smad4 locus. In this cell model, TGFβ stimulates and activates Smad2 and Smad3 properly; however, the transcriptional responses of TGFβ are severely crippled because of the lack of Smad4 (14).
ChIP analysis after infection of MDA-MB-468 cells with an adenovirus expressing the control protein LacZ showed that CTCF bound to the H19 ICR, similar to normal cells, and TGFβ had no impact on this recruitment (Fig. 7A). Unexpectedly, no Smad2 and very weak Smad3 binding could be measured after TGFβ stimulation (Fig. 7, B and C), suggesting that Smad4 is required for the effective recruitment of Smad2/3 to the H19 ICR. Smad4 showed no binding as expected, because it is not expressed in these cells (Fig. 7D).
FIGURE 7.

Co-occupation of the H19 ICR by CTCF and Smads requires Smad4. A–D, MDA-MB-468 cells that lack Smad4 were infected with adenoviruses expressing control, LacZ (left bars), or wild type Smad4 (right bars) and were stimulated with TGFβ1 for 4 h (+) or left untreated (−) prior to ChIP with specific antibodies for CTCF (A), Smad2 (B), Smad3 (C), and Smad4 (D). Immunoprecipitated chromatin was amplified with primers specific for the H19 ICR. E and F, input chromatin controls (E) and nonspecific IgG controls (F) are also shown after PCR amplification for the same samples analyzed in A–D.
To confirm the importance of Smad4, MDA-MB-468 cells were reconstituted with wild type Smad4 after adenoviral infection, which rescued recruitment of Smad2, Smad3, and Smad4 to the H19 ICR after TGFβ stimulation (Fig. 7, B–D), although CTCF binding to the locus was not affected by the expression of exogenous Smad4 (Fig. 7A). We conclude that the H19 ICR recruits a native Smad protein complex that also requires the presence of Smad4 in a TGFβ-dependent manner.
The CTCF-Smad Complex Forms Only on H19 ICR Chromatin of the Maternally Inherited Allele
To address whether the epigenetic status of the H19 ICR affected Smad recruitment to this locus, we employed primary mouse hepatocytes and used allele-specific probes for the H19 ICR that distinguish between the maternally inherited and the paternally inherited chromosomes (29). CTCF can only bind to the unmethylated, maternally inherited copy of the H19 ICR, whereas CTCF occupancy is excluded from the methylated paternal copy of the ICR.
Repeating the ChIP analysis in this cell system upon stimulation with TGFβ1 for the same period of time revealed a closely similar pattern of recruitment of all three Smad proteins and CTCF to the H19 ICR (Fig. 8A). Smad2, Smad3, and Smad4 showed highly ligand-dependent recruitment to the maternally inherited copy of the H19 ICR in primary hepatocytes, whereas CTCF recruitment was constitutive, and TGFβ stimulation only weakly affected CTCF binding to the maternally inherited copy of the H19 ICR. In contrast, the paternally inherited copy of the H19 ICR failed to recruit either CTCF or Smad proteins. These results suggest that TGFβ-stimulated recruitment of Smad proteins to the H19 ICR reflects the parent of origin-dependent epigenetic state of this region.
FIGURE 8.

Co-occupation of the H19 ICR by CTCF and Smads requires intact binding sites for CTCF on DNA. A, CTCF and Smads bind only to the maternally inherited allele of the H19 ICR. Primary hepatocytes were stimulated with TGFβ1 for 4 h (black bars) or left untreated (white bars) prior to ChIP with the indicated antibodies. Immunoprecipitated chromatin was amplified with primers specific for the H19 ICR, which can discriminate between the maternally inherited allele (♀) and the paternally inherited allele (♂). Input chromatin and nonspecific IgG controls are also shown after PCR amplification. B, MEFs were stimulated with TGFβ1 for 4 h (black bars) or left untreated (white bars) prior to ChIP. The genotype of the amplified allele is shown as wild type or mutant with respect to the H19 ICR point mutations engineered in the specific sequences where CTCF binds. C, nonspecific IgG controls and input chromatin controls for the same samples analyzed in B are shown. In all of the panels, the Q-PCR data show the average values and standard deviations calculated from triplicate determinations.
Smads Bind the H19 ICR in a CTCF-dependent Manner
CTCF binds directly to its cognate DNA sequences in the H19 ICR (29). Inspection of 250 bp flanking the human and mouse H19 ICR could not predict obvious SBEs in this sequence (data not shown). To examine whether Smad recruitment to the H19 ICR depended on CTCF binding to the ICR, we employed primary hepatocytes from a transgenic mouse that harbors specific point mutations in the CTCF-binding sites of the H19 ICR (15). The mutated H19 ICR allele fails to interact with CTCF in vitro and in vivo (15). To separate the parental alleles, we exploited allele-specific probes that reproducibly discriminated between the wild type and mutant H19 ICR allele (30).
Upon TGFβ1 stimulation for 4 h, ChIP Q-PCR analysis revealed as expected a closely similar pattern of recruitment of Smad proteins and CTCF to the maternally inherited wild type H19 ICR allele (Fig. 8B). Interestingly, when the mutated H19 ICR allele was inherited maternally, we observed a complete loss of recruitment not only of CTCF, as expected, but also of the ligand-activated Smad2, Smad3, and Smad4 complex (Fig. 8B). The basal recruitment of all tested proteins to the mutant H19 ICR was equivalent to that observed in the allele of paternal origin. These data strongly support the conclusion that TGFβ-activated Smads can access the H19 ICR chromatin via the CTCF-binding sites. We also conclude that the parent of origin-dependent epigenetic status of the H19 ICR is an important determinant of this complex.
TGFβ Regulates Expression of H19 and Igf2 from the Corresponding Transcriptionally Active Alleles
We finally examined whether the observed recruitment of Smads and CTCF to the H19 ICR could correlate with an impact of TGFβ on the expression levels of H19 and Igf2 mRNAs. Moreover, we wanted to examine any impact by TGFβ on the manifestation of the imprinted Igf2 expression pattern. To this end, we used MEFs derived from the same transgenic mice that carry the mutations in the CTCF-binding sites of the H19 ICR, as described above. Comparing basal levels of expression of H19 mRNA in cells from mice derived from the two reciprocal crosses, we confirmed that when the CTCF-binding site mutation is on the maternal chromosome, H19 expression was almost background (Fig. 9A). Conversely, when the mutated allele was paternally inherited, H19 expression was clearly measurable (set to value 1; Fig. 9A). Stimulation of the two types of MEFs with TGFβ1 for 4 h dramatically induced H19 expression. However, using a diagnostic BglII restriction site (31), we were able to document that this induction was specific for the maternal H19 allele when it carried the mutated H19 ICR in its 5′-flank (Fig. 9, A and B). We conclude that the wild type H19 ICR allele appears to be able to repress TGFβ-dependent induction of H19 on the maternal chromosome. Moreover, this ability of TGFβ to activate maternal H19 expression must depend on cis regulatory elements other than the H19 ICR.
FIGURE 9.
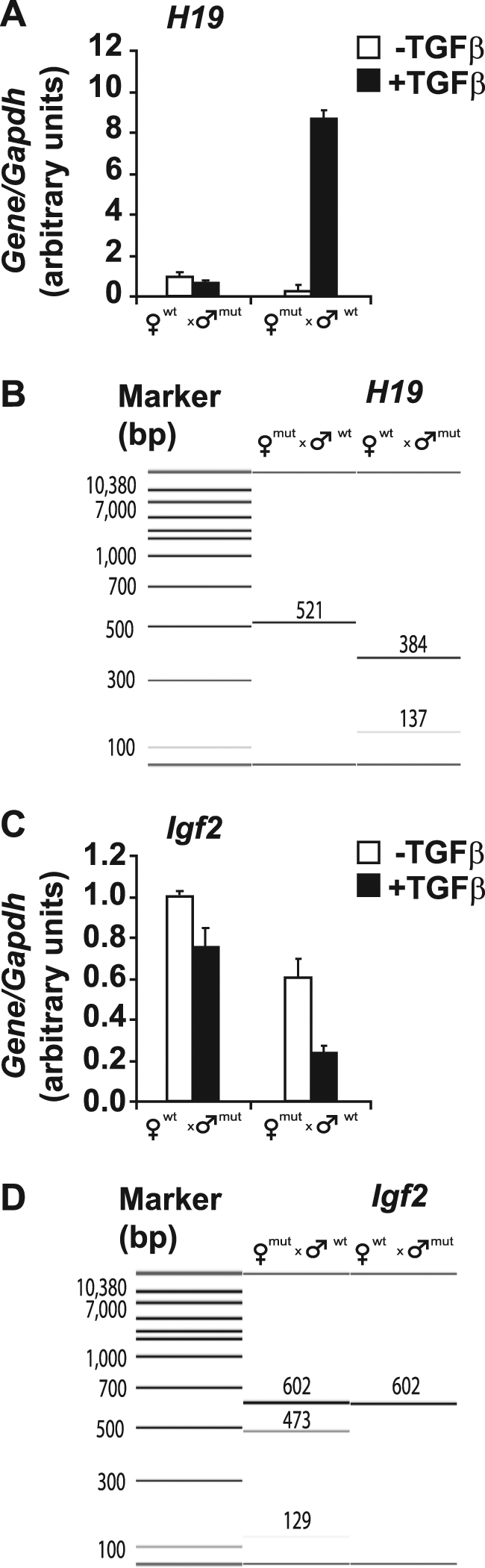
TGFβ regulates expression of the H19 and Igf2 genes. A and C, real time PCR analysis of H19 (A) and Igf2 (C) mRNA expression in MEFs derived from mouse embryos that inherited the mutant CTCF-binding sites from their mother (♀) or their father (♂) and stimulated with TGFβ1 for 0 (−) or 4 h (+). The average relative expression levels normalized to the housekeeping gene Gapdh calculated from triplicate sample determinations are plotted together with their error bars. B and D, cDNA fragments produced after restriction digestion with BglII (H19 cDNA) or BsaAI (Igf2 cDNA) and analyzed on a Bioanalyzer. The cDNAs were the same samples used for real time PCR analysis in A and C and correspond to the 4-h stimulation time point with TGFβ1. Molecular size markers (in bp) are also shown in each panel, and the specific size of each fragment is printed above each corresponding DNA band.
Next, we analyzed Igf2 mRNA expression and its allele-specific origin in response to TGFβ. When analyzing the basal condition, i.e. without TGFβ1 stimulation, we observed that Igf2 expression is 60% in cells with the mutated H19 ICR inherited maternally (142* × SD7) in comparison with cells with the mutated allele inherited paternally (SD7 × 142*) (Fig. 9C). Igf2 is expressed from both alleles in the former case in contrast to the monoallelic expression in the latter case. We confirmed the allele specificity based on a diagnostic BsaAI restriction digestion of the amplified cDNA as established before (31) and taking advantage of a mouse strain-specific BsaAI polymorphism on the Igf2 transcribed sequence (16) (see also Fig. 9D). Because the 142* × SD7 MEFs expressed Igf2 biallelically, we attribute the lower level of Igf2 expression in these cells compared with the SD7 × 142* cells to expression heterogeneity in the MEF culture. Importantly, a 4-h post-TGFβ stimulation significantly repressed Igf2 expression irrespective of its parental origin in the 142* × SD7 cells. Moreover, Igf2 expression was repressed also from the paternal allele in the SD7 × 142* cells without a sign of activity of the maternal Igf2 allele. We conclude that TGFβ signaling enforces a transcriptional control on the H19/Igf2 locus without modifying the allelic usage of either Igf2 or H19 and hence no apparent involvement of the H19 ICR.
DISCUSSION
We show here that CTCF physically interacts with the Smad2/3/4 complex. This interaction brings the Smad complex to CTCF-binding sites that generally map to linker regions between positioned nucleosomes. Most importantly, because CTCF-binding sites are generally sensitive to CpG methylation (32), the CTCF link provides an epigenetic dimension to TGFβ signaling.
The selectivity of interaction of Smad3, but not Smad2 or Smad4, with CTCF (Figs. 1–3) is of interest. This selective interaction requires the MH1 domain of Smad3 (Fig. 10A) and suggests that because Smad3 binds to CTCF via its DNA-binding domain, the interaction may preclude the possibility of concomitant Smad3 binding to DNA and to CTCF (Fig. 10B). In addition, the requirement for Smad4 for the recruitment of Smad2 and Smad3 to the H19 ICR (Fig. 7A) suggests that Smad4 may stabilize the nuclear Smad complex, which is necessary for its proper association to chromatin via CTCF (see also the model of Fig. 10B). Because Smad3 may be engaged with binding to CTCF by means of its MH1 domain and to other Smads via its phosphorylated MH2 domain, Smad4 may provide an additional binding site to DNA via its own available MH1 domain. The functional role of the Smad2 MH1 domain that fails to associate with either CTCF or DNA remains open for future investigation. However, a nuclear complex with the spliced isoform Smad2Δex3 is capable of providing a second CTCF-binding arm to the multimeric complex (Fig. 10B).
FIGURE 10.

The CTCF-Smad complex. A, schematic model of the interacting protein domains between Smad3 and CTCF. The Smad3 MH1 and MH2 domains and the CTCF N- and C-terminal and 11 zinc finger domains are indicated. An arrow points to the interaction between MH1 and zinc finger domains. B, hypothetical model of a chromatin-bound Smad2Δex3/Smad3/Smad4/CTCF complex on the H19 ICR locus. TGFβ promotes the accumulation of a nuclear Smad trimer with two phosphorylated R-Smad subunits and one Smad4 subunit. The Smads bind to each other via their MH2 domains. The existence of the Smad2Δex3/Smad3/Smad4 trimer is supported by the experimental evidence shown here, but its exact stoichiometry remains unclear. CTCF is shown as a monomer for simplicity. However, it is well established that it binds to chromatin as a homo-oligomer. The Smad4 MH1 domain is proposed to tether to SBE DNA, whereas each of Smad2Δex3 and Smad3 MH1 domains with CTCF binds to distinct genomic CTCF-binding sites. A possible functional role of such a complex is the interaction of distant genomic sites within a single chromosome or even between different chromosomes (question mark).
The interaction between CTCF and Smads (Figs. 1–3) and the dependence of CTCF-binding sites for the recruitment of Smads to the maternal H19 ICR allele strongly suggest that Smads do not bind to sequences within the H19 ICR but rather are recruited via CTCF. There is a single precedence for common recruitment of CTCF and Smad proteins to the regulatory region of the APP (amyloid precursor protein) gene (23). APP expression is induced by TGFβ signaling, and CTCF seems to participate in this regulatory mechanism. However, this study has not defined whether Smads bind directly to the APP enhancer or whether they are recruited via binding to CTCF or other common interacting components. In contrast, we were unable to establish robust regulation of well established gene and cell responses to TGFβ in various cell types by CTCF (Figs. 4 and 5). The examples of APP gene regulation and our evidence on PAI-1 gene expression (Fig. 5) do leave open the possibility that CTCF may be involved in regulation of the transcriptional output of a subset of TGFβ-responsive genes.
Moreover, TGFβ signaling did not modify the repressed status of the maternal Igf2 or H19 alleles (Fig. 9), which depends on the CTCF-binding sites within the H19 ICR (15). We thus conclude that TGFβ signaling targets the paternal Igf2 allele independent of the maternal H19 ICR allele. Furthermore, the presence of Smad3 cannot play an essential role in the parent of origin-specific insulator function associated with the H19 ICR. This argument is based on previous work that established that the H19 ICR insulator function is maintained in human choriocarcinoma cells such as JEG-3 (29), despite the fact that these tumor cells suffer a complete deletion of the Smad3 locus (33). Moreover, there is to our knowledge no other indication in the literature that the maternal-specific repression of the Igf2 locus depends on exogenous TGFβ.
Although these data render the functionality of the CTCF-Smad2/3/4 complex enigmatic, we note that CTCF has been identified bound to several thousand sites in the human, mouse, and fly genomes (34–38). Even though most of these sites map at intergenic regions and at chromatin boundaries between transcriptionally silent and active chromatin, a number of the CTCF-binding sites reside in proximity to gene promoters/enhancers. It is thus possible that TGFβ signaling modulates the function of CTCF-binding sites in a context-dependent manner.
Furthermore, because CTCF has been implicated as a master weaver of the genome (5), TGFβ signaling might also modulate the organization of large chromatin domains via the formation of chromatin loops and bridges (4–6). This option is attractive given the ability of the Smad complex to interact with other proteins to potentially stabilize interactions between chromatin fibers involving CTCF.
In conclusion, this study provides the first evidence that CTCF recruits Smad proteins to its binding sites and that this recruitment can be epigenetically controlled. Such cross-talk can be achieved by the domain-specific molecular interaction between CTCF and Smads that we demonstrate here. This work opens the possibility that the functional consequence of such a molecular interaction may mediate control of long range chromatin associations by a major developmental signaling pathway such as TGFβ.
Acknowledgment
We thank Lars van der Heide for useful comments during preparation of the manuscript.
This work was supported by grants from the Ludwig Institute for Cancer Research, Swedish Cancer Society Project 4855-B03-01XAC, and Natural Sciences Foundation of Sweden Project K2004-32XD-14936-01A (to A. M.) and grants from the Swedish Science Research Council, the Swedish Cancer Society, the Swedish Pediatric Cancer Foundation, the Lundberg Foundation, and the HEROIC European Union integrated project (to R. O.).
- ICR
- imprinting control region(s)
- ChIP
- chromatin immunoprecipitation
- CTCF
- CCCTC-binding factor
- GST
- glutathione S-transferase
- MH
- Mad homology
- PAI
- plasminogen activator inhibitor
- SBE
- Smad-binding element
- TGF
- transforming growth factor
- luc
- luciferase
- siRNA
- short interfering RNA
- MEF
- mouse embryonic fibroblast
- Q-PCR
- quantitative PCR.
REFERENCES
- 1.Ohlsson R., Paldi A., Graves J. A. (2001) Trends Genet. 17, 136–141 [DOI] [PubMed] [Google Scholar]
- 2.Verona R. I., Mann M. R., Bartolomei M. S. (2003) Annu. Rev. Cell Dev. Biol. 19, 237–259 [DOI] [PubMed] [Google Scholar]
- 3.Reik W., Dean W., Walter J. (2001) Science 293, 1089–1093 [DOI] [PubMed] [Google Scholar]
- 4.Ohlsson R., Lobanenkov V., Klenova E. (2010) BioEssays 32, 37–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Phillips J. E., Corces V. G. (2009) Cell 137, 1194–1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Göndör A., Ohlsson R. (2009) Nature 461, 212–217 [DOI] [PubMed] [Google Scholar]
- 7.Sandhu K. S., Shi C., Sjölinder M., Zhao Z., Göndör A., Liu L., Tiwari V. K., Guibert S., Emilsson L., Imreh M. P., Ohlsson R. (2009) Genes Dev. 23, 2598–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Splinter E., Heath H., Kooren J., Palstra R. J., Klous P., Grosveld F., Galjart N., de Laat W. (2006) Genes. Dev. 20, 2349–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao Z., Tavoosidana G., Sjölinder M., Göndör A., Mariano P., Wang S., Kanduri C., Lezcano M., Sandhu K. S., Singh U., Pant V., Tiwari V., Kurukuti S., Ohlsson R. (2006) Nat. Genet. 38, 1341–1347 [DOI] [PubMed] [Google Scholar]
- 10.Massagué J. (2008) Cell 134, 215–230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu M. Y., Hill C. S. (2009) Dev. Cell 16, 329–343 [DOI] [PubMed] [Google Scholar]
- 12.Moustakas A., Heldin C. H. (2009) Development 136, 3699–3714 [DOI] [PubMed] [Google Scholar]
- 13.Shi Y., Wang Y. F., Jayaraman L., Yang H., Massagué J., Pavletich N. P. (1998) Cell 94, 585–594 [DOI] [PubMed] [Google Scholar]
- 14.Kowanetz M., Valcourt U., Bergström R., Heldin C. H., Moustakas A. (2004) Mol. Cell. Biol. 24, 4241–4254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pant V., Mariano P., Kanduri C., Mattsson A., Lobanenkov V., Heuchel R., Ohlsson R. (2003) Genes. Dev. 17, 586–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dean W., Bowden L., Aitchison A., Klose J., Moore T., Meneses J. J., Reik W., Feil R. (1998) Development 125, 2273–2282 [DOI] [PubMed] [Google Scholar]
- 17.Morén A., Hellman U., Inada Y., Imamura T., Heldin C. H., Moustakas A. (2003) J. Biol. Chem. 278, 33571–33582 [DOI] [PubMed] [Google Scholar]
- 18.Kurisaki A., Kose S., Yoneda Y., Heldin C. H., Moustakas A. (2001) Mol. Biol. Cell 12, 1079–1091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lutz M., Burke L. J., Barreto G., Goeman F., Greb H., Arnold R., Schultheiss H., Brehm A., Kouzarides T., Lobanenkov V., Renkawitz R. (2000) Nucleic Acids Res. 28, 1707–1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurisaki K., Kurisaki A., Valcourt U., Terentiev A. A., Pardali K., Ten Dijke P., Heldin C. H., Ericsson J., Moustakas A. (2003) Mol. Cell. Biol. 23, 4494–4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Thuault S., Tan E. J., Peinado H., Cano A., Heldin C. H., Moustakas A. (2008) J. Biol. Chem. 283, 33437–33446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schübeler D., Scalzo D., Kooperberg C., van Steensel B., Delrow J., Groudine M. (2002) Nat. Genet. 32, 438–442 [DOI] [PubMed] [Google Scholar]
- 23.Burton T., Liang B., Dibrov A., Amara F. (2002) Biochem. Biophys. Res. Commun. 295, 713–723 [DOI] [PubMed] [Google Scholar]
- 24.Dennler S., Huet S., Gauthier J. M. (1999) Oncogene 18, 1643–1648 [DOI] [PubMed] [Google Scholar]
- 25.Dennler S., Itoh S., Vivien D., ten Dijke P., Huet S., Gauthier J. M. (1998) EMBO J. 17, 3091–3100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chao W., Huynh K. D., Spencer R. J., Davidow L. S., Lee J. T. (2002) Science 295, 345–347 [DOI] [PubMed] [Google Scholar]
- 27.Kanduri C., Fitzpatrick G., Mukhopadhyay R., Kanduri M., Lobanenkov V., Higgins M., Ohlsson R. (2002) J. Biol. Chem. 277, 18106–18110 [DOI] [PubMed] [Google Scholar]
- 28.Mancini-Dinardo D., Steele S. J., Levorse J. M., Ingram R. S., Tilghman S. M. (2006) Genes. Dev. 20, 1268–1282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kanduri C., Pant V., Loukinov D., Pugacheva E., Qi C. F., Wolffe A., Ohlsson R., Lobanenkov V. V. (2000) Curr. Biol. 10, 853–856 [DOI] [PubMed] [Google Scholar]
- 30.Bergström R., Whitehead J., Kurukuti S., Ohlsson R. (2007) Cell Cycle 6, 450–454 [DOI] [PubMed] [Google Scholar]
- 31.Pant V., Kurukuti S., Pugacheva E., Shamsuddin S., Mariano P., Renkawitz R., Klenova E., Lobanenkov V., Ohlsson R. (2004) Mol. Cell. Biol. 24, 3497–3504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mukhopadhyay R., Yu W., Whitehead J., Xu J., Lezcano M., Pack S., Kanduri C., Kanduri M., Ginjala V., Vostrov A., Quitschke W., Chernukhin I., Klenova E., Lobanenkov V., Ohlsson R. (2004) Genome Res. 14, 1594–1602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Xu G., Chakraborty C., Lala P. K. (2002) Biochem. Biophys. Res. Commun. 294, 1079–1086 [DOI] [PubMed] [Google Scholar]
- 34.Barski A., Cuddapah S., Cui K., Roh T. Y., Schones D. E., Wang Z., Wei G., Chepelev I., Zhao K. (2007) Cell 129, 823–837 [DOI] [PubMed] [Google Scholar]
- 35.Kim T. H., Abdullaev Z. K., Smith A. D., Ching K. A., Loukinov D. I., Green R. D., Zhang M. Q., Lobanenkov V. V., Ren B. (2007) Cell 128, 1231–1245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xie X., Mikkelsen T. S., Gnirke A., Lindblad-Toh K., Kellis M., Lander E. S. (2007) Proc. Natl. Acad. Sci. U.S.A. 104, 7145–7150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cuddapah S., Jothi R., Schones D. E., Roh T. Y., Cui K., Zhao K. (2009) Genome Res. 19, 24–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smith S. T., Wickramasinghe P., Olson A., Loukinov D., Lin L., Deng J., Xiong Y., Rux J., Sachidanandam R., Sun H., Lobanenkov V., Zhou J. (2009) Dev. Biol. 328, 518–528 [DOI] [PMC free article] [PubMed] [Google Scholar]