

Abstract
Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate are ubiquitous calcium-mobilizing messengers produced by the same family of multifunctional enzymes, the ADP-ribosyl cyclases. Not all ADP-ribosyl cyclases have been identified, and how production of different messengers is achieved is incompletely understood. Here, we report the cloning and characterization of a novel ADP-ribosyl cyclase (SpARC4) from the sea urchin, a key model organism for the study of calcium-signaling pathways. Like several other members of the ADP-ribosyl cyclase superfamily, SpARC4 is a glycoprotein targeted to the plasma membrane via a glycosylphosphatidylinositol anchor. However, unlike most other members, SpARC4 shows a remarkable preference for producing cyclic ADP-ribose over nicotinic acid adenine dinucleotide phosphate. Mutation of a single residue (tyrosine 142) within a noncanonical active site reversed this striking preference. Our data highlight further diversification of this unusual enzyme family, provide mechanistic insight into multifunctionality, and suggest that different ADP-ribosyl cyclases are fine-tuned to produce specific calcium-mobilizing messengers.
Keywords: Calcium, Calcium Intracellular Release, NAD, Nicotinic Acid, Site-directed Mutagenesis, ADP-ribosyl Cyclase, NAADP, Cyclic ADP-ribose, Sea Urchin
Introduction
Changes in cytosolic calcium levels govern a multitude of cellular processes. Many extracellular stimuli mediate complex changes in calcium through production of second messengers (1). These molecules effect release of calcium from intracellular stores. In addition to inositol 1,4,5-trisphosphate, both cyclic ADP-ribose (cADPR)3 and nicotinic acid adenine dinucleotide phosphate (NAADP) play critical roles in the generation of agonist-evoked calcium signals (2, 3). cADPR releases calcium through activation of the ryanodine receptors located on endoplasmic reticulum stores (2, 4). The mechanism of action of NAADP is less certain (5), although in sea urchin eggs, where its effects were discovered (6), it is clear that NAADP acts on novel calcium-permeable channels located not on the endoplasmic reticulum but instead on acidic lysosome-like stores (7–9). These channels have recently been identified as the two-pore channels (10–12). Remarkably, both cADPR and NAADP are synthesized by the same family of enzymes, the ADP-ribosyl cyclases (13, 14), which therefore play a central role in calcium signaling and have been implicated in a variety of processes ranging from bacterial clearance (15) to social behavior (16).
ADP-ribosyl cyclase activity was originally identified in sea urchin egg homogenates (17), but an enzyme with this activity was later purified (18) and cloned (19) from Aplysia californica ovotestes. This enzyme was shown to catalyze the cyclization of NAD to form cADPR (18, 20) and the replacement of the nicotinamide moiety in NADP with nicotinic acid to form NAADP (21). Based on sequence similarity, it became evident that the mammalian proteins, CD38 (22) and CD157 (23), were also ADP-ribosyl cyclases. More recently, SmNACE (24), a homologue from the blood fluke Schistosoma mansoni, and an expanded family of three ADP-ribosyl cyclases (SpARC1–3) from the sea urchin (25) have been identified. Somewhat paradoxically, many members of the ADP-ribosyl cyclase family, including CD38, the most extensively studied, are located predominantly on the cell surface and therefore away from their cytosolic substrates (13, 14). This topological barrier, however, is circumvented through identified transporter molecules that mediate both substrate efflux and product influx across the plasma membrane (26). Recent studies have also shown that members of the sea urchin ADP-ribosyl cyclase family are present within intracellular organelles where again they are able to access cytosolic substrates through presumed transporters (25, 27). Aplysia ADP-ribosyl cyclase also appears to localize to cytoplasmic organelles in eggs (18) and has been shown more recently to undergo redistribution from the cytoplasm to the nucleus upon depolarization in neurons (28). Targeting of ADP-ribosyl cyclase to different subcellular locales may therefore result in localized production of calcium-mobilizing messengers that may contribute to the production of spatially restricted calcium signals.
Reduced cellular levels of cADPR (15, 29, 30) and NAADP (31) in certain tissues from CD38 knock-out mice underscore the physiological importance of this ADP-ribosyl cyclase in generating calcium-mobilizing messengers in vivo. Such analyses have also highlighted the likely existence of novel ADP-ribosyl cyclases in tissues such as the brain (32) and uterus (33) where messenger levels appear to be unperturbed in these animals. Indeed, several novel activities (34–43), including a soluble enzyme in sea urchin eggs (44), have been described for which molecular correlates are currently lacking. It therefore appears that not all ADP-ribosyl cyclases have been described yet at the molecular level.
ADP-ribosyl cyclases show only modest sequence similarity (30–40%). However, their overall three-dimensional structures, where known, are almost identical (45–47). The active site has been defined for both Aplysia ADP-ribosyl cyclase (48) and CD38 (49) in which a highly conserved glutamate is likely the catalytic residue. Despite this conservation in three-dimensional structures, ADP-ribosyl cyclases show remarkable differences in their catalytic properties. For example, Aplysia ADP-ribosyl cyclase produces primarily cADPR from NAD (18, 20), and in contrast, CD38 possesses robust NAD glycohydrolase activity (22) thereby producing predominantly ADP-ribose and only modest amounts of cADPR (2–3% of reaction products). Additionally, CD38 is capable of hydrolyzing cADPR directly to ADP-ribose, whereas Aplysia ADP-ribosyl cyclase is comparatively a very poor cADPR hydrolase (13, 14, 50). Notably, ADP-ribose also modulates calcium fluxes through regulation of the transient receptor potential channel and TRPM2 present in the plasma membrane (51) and possibly also on lysosomal calcium stores (52, 53). Further differences in catalytic activity of ADP-ribosyl cyclases are evident with the surrogate substrate, nicotinamide guanine dinucleotide (54). Both SmNACE (24) and SpARC1 (25), although able to cyclize nicotinamide guanine dinucleotide to cyclic GDP ribose, produce much less cADPR from NAD (24) than CD38, whereas SpARC2 (55) and several unidentified mammalian ADP-ribosyl cyclases (32, 36, 39) are unable to cyclize nicotinamide guanine dinucleotide.
Understanding the molecular basis for such unprecedented differences in catalysis represents a key challenge that will ultimately aid in understanding the functions of these enzymes in vivo. Intriguingly, although glycohydrolase activity is very low for Aplysia ADP-ribosyl cyclase using the natural substrate, NAD, this activity is unmasked by the introduction into its structure of an additional cystine pair, as found in CD38 (56), and also by using a fluorinated analogue of NAD that cannot be cyclized (57). NAD glycohydrolase activity can also be readily detected in Aplysia ADP-ribosyl cyclase upon mutation of phenylalanine 174, which together with the associated reduction in cyclase activity results in a mutant enzyme displaying properties reminiscent of CD38 (58). Conversely, mutation of the corresponding residue (threonine 221) in CD38 increases cyclase activity at the expense of NAD glycohydrolase activity. Indeed, the concomitant mutation of this residue and of glutamate 146, a residue previously implicated in determining the cyclase/NAD glycohydrolase ratio (59), results in a catalytic profile very similar to that of Aplysia ADP-ribosyl cyclase, including poor cADPR hydrolase activity (58). Mutagenesis of the SmNACE has also resulted in the identification of a single residue that increases ADP-ribosyl cyclase activity (60).
Although ADP-ribosyl cyclases are capable of synthesis of both cADPR and NAADP, how messenger production is controlled in vivo is not clear. It is possible that the activities of the same enzyme may be differentially regulated. Alternatively, different homologues may be endowed with different relative activities. Here, we report the molecular cloning of SpARC4, a novel member of the ADP-ribosyl cyclase family from the sea urchin, and we show that this protein is a GPI-anchored plasma membrane glycoprotein capable of synthesizing both cADPR and NAADP. In contrast to other ADP-ribosyl cyclases, and in analogy with the Aplysia enzyme, SpARC4 preferentially produces cADPR. We identify a unique acidic-basic cluster of amino acids within the N terminus and a nonconserved tyrosine residue within the active site. Whereas manipulation of the former had little impact on catalysis, tyrosine 142 was found to control the relative production of cADPR and NAADP such that its mutation was sufficient to convert SpARC4 into an enzyme that preferentially produces NAADP.
MATERIALS AND METHODS
Molecular Cloning of SpARC4
The sequences of the primers used are listed in supplemental Table S1. Total RNA was isolated from the prism stage (46 h) embryos of Strongylocentrotus purpuratus using the RNeasy kit (Qiagen), according to manufacturer's instructions. The samples were treated with DNase to eliminate any genomic DNA contamination. First strand cDNA synthesis was performed using the Improm-II kit (Promega) and either gene-specific primer 1R (for 5′-RACE) or oligo(dT)17 adaptor primers (for 3′-RACE). Typical reactions (40 μl) contained 2 μg of RNA. For 5′-RACE, the poly(A) tailing reaction was performed using terminal transferase (Promega) as per the manufacturer's instructions. Nested PCR was performed using the oligo(dT)17 adaptor or adaptor primer as the forward primers with the gene-specific reverse primers 3R or 2R, respectively. A 750-bp product was amplified and sequenced. This extended the 5′-sequence to an in-frame stop codon. For 3′-RACE, PCR was performed using the nested gene-specific forward primers 1F and 2F together with the adaptor primer as the reverse primer. An 850-bp product was amplified, which extended the 3′-sequence to an in-frame stop codon. All the above gene-specific primers were based on genomic contig 640882 identified by an initial BLAST search of the S. purpuratus genome sequencing project at the Baylor College of Medicine with the A. californica ADP-ribosyl cyclase sequence (accession number P29241). An additional pair of gene-specific primers 6F and 3′-UTR 1R were designed on the sequences obtained by RACE and used to amplify the full-length coding sequence +54 bp of 3′-untranslated region of SpARC4. The 1089-bp product was cloned into pCR®II-TOPO® (Invitrogen) and sequenced in both directions. The sequence was deposited at EMBL under the accession number FN645665. All PCRs were performed using Platinum Hi-fidelity TaqDNA polymerase enzyme (Invitrogen) and 35 cycles of denaturation (30 s at 94 °C), annealing (30 s at 50 °C), and extension (1 min at 68 °C) following an initial denaturation step (2 min at 94 °C).
Plasmids
A plasmid encoding untagged SpARC4 was generated by amplification of the full-length coding sequence using primers 7F and 6R followed by cloning of the product into pCS2+ at the EcoRI and XbaI sites. Plasmids encoding for SpARC4, tagged at their N termini with six Myc tags, were generated by cloning products into the EcoRI and XbaI sites of pCS2+ MT into which the sequence corresponding to the signal peptide of SpARC2 (residues 1–23) had been inserted upstream of the Myc tags at the BamHI and ClaI sites (25). Full-length (mature) SpARC4 (SpARC4 Myc) was generated by inserting the product amplified using primer N-Myc 1F and 6R corresponding to amino acids 39–343. SpARC4 lacking the C-terminal 17 amino acids (SpARC4 ΔC Myc) was generated by inserting the product amplified using primers N-Myc 1F and CT-Trun 1R corresponding to amino acids 39–326. SpARC4 lacking the N-terminal 17 amino acids from the mature SpARC4 (SpARC4 ΔN Myc) was generated by inserting the product amplified using primer N-Myc 2F and 6R corresponding to amino acids 56–343. Plasmids encoding for SpARC4R52A/R53A Myc, SpARC4Y142W Myc, and SpARC4Y142H Myc were constructed using the QuikChange site-directed mutagenesis kit (Stratagene) with pCS2+ SpARC4 Myc as the template and the following mutagenic primers: R52,53A_sense/R52,53A_antisense, Y142W_sense/Y142W_antisense, and Y142H_sense/Y142H_antisense. All constructs were verified by DNA sequencing. The sequences of the primers used are listed in supplemental Table S1 and a schematic of the constructs is presented in Fig. 2B.
FIGURE 2.

SpARC4 is a glycoprotein targeted to the plasma membrane. A, predicted structure of SpARC4. Putative N-linked glycosylation sites are marked with a Y. B, schematic of the constructs used in this study. C, Western blot analysis, using an anti-Myc antibody, of Xenopus embryos (left) or HEK cells (right) expressing Myc-tagged SpARC4 (SpARC4 Myc). Samples were untreated (−), mock-treated, or digested with PNGase-F prior to analysis. Migration of molecular mass markers (in kDa) is shown on the left side of the panel. D, confocal fluorescence images of X. laevis embryos (top) or HEK cells (bottom) expressing SpARC4 Myc. Expression was detected by immunocytochemistry using an anti-Myc primary antibody. For embryos, the images are typical of the two distribution patterns observed. The asterisks highlight cells not expressing the protein. For HEK cells, both permeabilized and nonpermeabilized cells were processed. Scale bar, 5 μm.
Sequence Analysis
Sequences were aligned using ClustalW. Signal peptides/anchors were predicted by SignalP 3.0. GPI modification site was identified by big-PI predictor and GPI-SOM. N-Glycosylation sites were predicted by NetNGlyc 1.0. The pro-protease cleavage site was predicted by ProP 1.0.
Microinjection of Xenopus laevis Embryos
Xenopus embryos at the 2–4-cell stage were injected with 0.8 ng of mRNA per blastomere and cultured in 5% normal amphibian medium (61) at 14 °C until the late blastula/early gastrula stage. For immunocytochemistry only a single blastomere was injected to induce mosaic expression.
Transfection of HEK Cells
Human embryonic kidney (HEK) cells were grown and maintained in Dulbecco's modified Eagle's medium supplemented with 10% serum, penicillin (100 units/ml), and streptomycin (100 μg/ml) at 37 °C in a humidified 5% CO2 chamber. The cells were seeded in an antibiotic-free medium 24 h prior to transfection on either 6-well plates (for Western blotting/secretion assays) or 24-well plates containing poly-l-lysine (20 μg/ml)-coated 1-cm diameter coverslips (for immunocytochemistry). Upon reaching 90% confluency, the cells were transiently transfected with plasmid DNA using Lipofectamine 2000 transfection reagent (Invitrogen) according to manufacturer's instructions. Cells were used 17 h post-transfection.
Preparation of Homogenates
For Western blot analysis, homogenates were prepared at 4 °C in a buffer composed of 20 mm Tris-HCl, pH 7.2, 50 mm NaCl, 10 mm magnesium acetate, and 1% Triton X-100. For Xenopus embryos, the embryos were resuspended at a ratio of 20 μl of buffer/embryo and incubated for 30 min, and the supernatant was collected following centrifugation at 21,000 × g for 1 min. For HEK cells, cells were resuspended at a ratio of 100 μl of buffer/confluent well of a 6-well plate and incubated for 60 min, and the supernatant was collected following centrifugation at 100,000 × g for 60 min. For enzyme activity measurements, Xenopus embryos were homogenized in a hypotonic buffer (20 mm HEPES, pH 7.2, with CompleteTM EDTA-free protease inhibitors), followed by three 5-s bursts of sonication at 4 °C. The homogenates were stored at −20 °C in single use aliquots until required.
Enzyme Treatments
For deglycosylation experiments, embryo homogenates (half-embryo equivalents) were denatured and treated with or without PNGase-F (New England Biolabs) according to manufacturer's instructions. For PI-PLC treatment, adherent HEK cells were incubated either in the absence or presence of 1 unit/ml PI-PLC (Sigma) at 37 °C for 1 h in HEPES-buffered saline composed of 156 mm NaCl, 3 mm KCl, 2 mm MgSO4, 1.25 mm KH2PO4, 10 mm d-glucose, 2 mm CaCl2, and 10 mm HEPES, pH 7.4. The medium was then collected and concentrated 10-fold using an Amicon Ultra 4 centrifugal filter (10-kDa cut-off).
Secretion Assays
HEK cells were transfected with plasmids encoding SpARC4 Myc or SpARC4 ΔC Myc and the media replaced with serum and antibiotic-free media, 5 h post-transfection. Cells were cultured for 12–15 h before preparation of cell homogenates and collection of media. The latter were concentrated 10-fold using Amicon Ultra 4 centrifugal filter (10-kDa cut-off).
Western Blot Analysis
Xenopus embryo homogenates (0.33 eq) or HEK cell homogenates (25–50 μg of protein) or media were reduced and denatured with 4 mm dithiothreitol at 90 °C for 10 min and loaded onto NuPAGE 4–12% BisTris gels (Invitrogen). The gel was run with either MOPS or MES running buffer (Invitrogen), and the resolved proteins were transferred to a nitrocellulose membrane (Applied Biosystems). The blots were blocked with 5% dried skimmed nonfat milk powder dissolved in Tris-buffered saline (25 mm Tris-HCl, 137 mm NaCl, 2.7 mm KCl, pH 7.4) supplemented with 0.1% Tween 20 (TBS-T) overnight at 4 °C. The blot was washed three times with TBS-T and incubated with the primary antibody (mouse anti-Myc monoclonal antibody, clone 9e10; Insight Biotechnology; 1:1000 dilution) for 1 h at 25 °C. Following a further three washes with TBS-T, the blot was incubated with the secondary antibody (anti-mouse IgG conjugated to horseradish peroxidase; Invitrogen; 1:1000 dilution) for 1 h at 25 °C. The blots were developed either with Pierce ECL substrate (Thermo Scientific) or ECLTM Advanced System (GE Healthcare) according to the manufacturer's guidelines.
Immunocytochemistry
Xenopus embryos were fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) and stored at 4 °C. The embryos were then permeabilized by two washes with Tris-buffered saline containing 0.1% Triton X-100 followed by three washes with TBS-T (30 min each). The embryos were then blocked with 20% goat serum in TBS-T for 1 h at room temperature. Embryos were incubated with the primary antibody (mouse anti-Myc monoclonal antibody, clone 9e10; 1;100 dilution) overnight at 4 °C in blocking solution. The embryos were then washed four times with TBS-T containing 2 mg/ml bovine serum albumin and blocked again in a solution containing TBS-T supplemented with 2 mg/ml bovine serum albumin and 20% goat serum for 1 h. Embryos were incubated with the secondary antibody (goat polyclonal anti-mouse IgG conjugated with Cy-3; Zymed Laboratories Inc.; 1:100 dilution) overnight at 4 °C in the same blocking solution. The embryos were then washed four times with TBS-T containing 2 mg/ml bovine serum albumin (30 min each).
For transfected HEK cells, cells were fixed in 4% paraformaldehyde in PBS for 10 min at 25 °C followed by three washes with PBS at 25 °C. The cells were permeabilized with PBS + 0.1% Triton X-100 for 10 min at 25 °C with gentle agitation. The cells were then washed again (three times with PBS) and incubated with blocking solution (5% fetal bovine serum and 1% bovine serum albumin in PBS) for 60 min at 25 °C with gentle agitation. The coverslips were incubated with primary antibody (mouse monoclonal anti-Myc 9e10; 1:100 dilution) in blocking solution at 37 °C for 60 min followed by three washes with PBS + 0.1% Tween 20). The coverslips were then incubated with the secondary antibody (goat polyclonal anti-mouse IgG conjugated with Cy-3; 1:100 dilution) in blocking solution at 37 °C for 60 min followed by three washes with PBS + 0.1% Tween 20. The coverslips were mounted on glass slides with 1,4-diazabicyclo[2.2.2]octane, air-dried, and sealed with nail varnish. In some experiments, immunocytochemistry was performed using nonpermeabilized cells. In these cases, live cells were incubated with the primary antibody in HEPES-buffered saline for 60 min at 4 °C. The cells were then fixed, washed with PBS, and incubated with the secondary antibody as described above.
Confocal Microscopy
Fluorescence images of Xenopus embryos and HEK cells were captured using a Zeiss LSM 510 confocal microscope equipped with a 20× or 63× objective. The wavelengths for excitation and emission for Cy3 were 543 and 560–615 nm, respectively.
Enzyme Activity Measurements
Xenopus embryo homogenates (typically 20 μl) were diluted 5-fold into reaction mixtures composed of either 1 mm NAD and 20 mm HEPES, pH 7.2, for the measurement of ADP-ribosyl cyclase activity or 1 mm NADP and 50 mm nicotinic acid, pH 4.8, for the measurement of base-exchange activity. The reactions were allowed to proceed for up to 4 h at room temperature, diluted 10-fold with water, and heated at 60 °C for 5 min. The mixture was then centrifuged for 1 min at 21,000 × g to remove any particulate matter. The supernatants were injected onto an AGMPI (Bio-Rad) anion exchange HPLC column (3 × 150 mm) at a flow rate of 1 ml/min, and bound nucleotides were separated with a concave-up gradient of trifluoroacetic acid as described previously (25). Absorbance was recorded at 254 nm using a UV spectrophotometer, and the areas of the resolved peaks were calculated using Breeze Software (Waters). A mixture of standards (1 mm NAD, NADP, cADPR, and NAADP) was separated in parallel for calibration.
RESULTS
Our original BLAST queries of the S. purpuratus genome with known ADP-ribosyl cyclase sequences led to the isolation of SpARC1–3 (25). Here, we report the molecular cloning of a new ADP-ribosyl cyclase herein named SpARC4. The full-length coding sequence was determined using a combination of 5′- and 3′-RACE (see “Materials and Methods”). SpARC4 is predicted to encode a polypeptide comprising 343 amino acids. An alignment of the deduced amino acid sequence of SpARC4 with those of SpARC1–3 is shown in Fig. 1A. Sequence identity between SpARC4 and other family members was ∼25% and sequence similarity was ∼40% (Fig. 1B). We also identified three expressed sequence tags from the sea urchin Paracentrotus lividus that were 65–66% identical and 74% similar to SpARC4 (supplemental Fig. S1). The 10 “core” cysteine residues found in Aplysia ADP-ribosyl cyclase are conserved in SpARC4 (Fig. 1A, asterisks). These data further highlight expansion of the ADP-ribosyl cyclase family.
FIGURE 1.

Molecular cloning of a novel member of the ADP-ribosyl cyclase family. A, multiple sequence alignment of the deduced amino acid sequences of SpARC4 and that of previously identified ADP-ribosyl cyclases from S. purpuratus (SpARC1–3). Putative cleavage sites of the predicted signal peptide and GPI anchor are marked with the filled and open arrowheads, respectively. The presence of an acidic-basic cluster of amino acids in SpARC4 is marked with a box. Conserved cysteine residues between isoforms are marked with an asterisk. B, table showing percentage sequence identities (shaded) and percentage sequence similarities (above diagonal) derived from pairwise alignments between SpARC family members.
SpARC4 is predicted to have an N-terminal signal peptide of 34 residues, three N-linked glycosylation sites (one within the putative signal peptide), and a C-terminal GPI anchor (Fig. 1A). The predicted structure of SpARC4 is schematically depicted in Fig. 2A. To define the properties of SpARC4, the series of expression constructs represented in Fig. 2B were generated. We first expressed the full-length Myc-tagged SpARC4 (SpARC4 Myc) in Xenopus embryos and HEK cells (Fig. 2C). Western blot analysis using an anti-Myc antibody revealed the presence of a major polypeptide of ∼55 kDa in embryos expressing SpARC4 Myc (Fig. 2C). The apparent molecular mass of SpARC4 Myc was modestly reduced following treatment of homogenates with PNGase-F, which releases N-linked glycans (Fig. 2C). The size of the PNGase-F-treated protein was in reasonable agreement with the predicted molecular mass of mature SpARC4 Myc (44 kDa). In HEK cells expressing SpARC4 Myc, two major bands of ∼55 and 45 kDa were observed (Fig. 2C). Treatment with PNGase-F reduced the apparent molecular mass of the larger band, but the migration of the lower band was not affected. The identity of the latter remains to be established but may correspond to unprocessed SpARC4. These data indicate that SpARC4 is N-glycosylated, consistent with the presence of consensus N-glycosylation sites.
To determine the subcellular localization of SpARC4, we performed immunocytochemical analysis in conjunction with confocal microscopy. Fig. 2D shows the distribution of SpARC4 Myc expressed in Xenopus embryos. Two patterns were observed as follows: clear cell surface staining (Fig. 2D, top left) or a punctate intracellular distribution often concentrated at the periphery (Fig. 2F, top right). In these experiments, only a single blastomere of a 2–4-cell embryo was injected to effect mosaic expression. Indeed, cells that did not express the protein were clearly evident in embryos (Fig. 2D, asterisks), thereby defining background staining and thus attesting to the specificity of the labeling. In HEK cells, SpARC4 localized mainly to the plasma membrane, although diffuse intracellular staining was also evident (Fig. 2D, bottom left). Localizations were also performed using intact (nonpermeabilized) cells (Fig. 2D, bottom right). Surface staining was readily detectable indicating that the epitope is extracellular, further confirming plasma membrane residence of this protein.
Given the consensus sequence for GPI anchoring, we next examined the effect of bacterial PI-PLC, which releases GPI-anchored proteins from the plasma membrane. We first treated cells expressing SpARC4 Myc with PI-PLC and analyzed the media for SpARC immunoreactivity by Western blotting. As shown in Fig. 3A, this treatment resulted in the appearance of released SpARC4 Myc in the medium. These data suggest that SpARC4 is attached to the plasma membrane by a GPI anchor. In parallel experiments, we examined cell surface expression of SpARC4 Myc by immunocytochemistry. As shown in Fig. 3B, PI-PLC treatment caused a reduction in expression of SpARC4, again consistent with the presence of a GPI anchor. Finally, we deleted the C terminus harboring the putative GPI-anchoring site and compared secretion of the full-length and truncated SpARC4 Myc. As shown in Fig. 3C, SpARC4 ΔC Myc was present in the medium at higher levels than the full-length construct. Both constructs were expressed at comparable levels within cells (data not shown). These data suggest that SpARC4 is attached to the plasma membrane at its C terminus via a GPI anchor.
FIGURE 3.
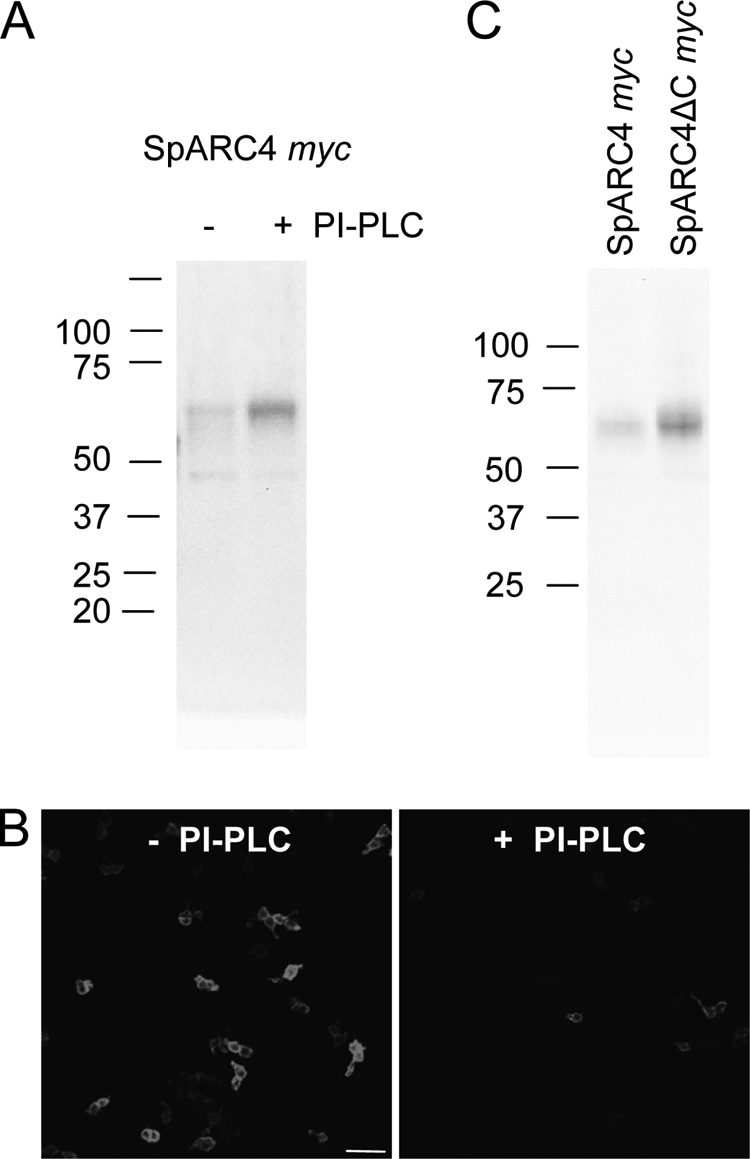
SpARC4 is a GPI-anchored protein. HEK cells expressing SpARC4 Myc were treated with (+) or without (−) PI-PLC, and the presence of SpARC4 was determined in media samples by Western blot analysis (A) and on the cell surface by immunocytochemistry (B). Scale bar, 50 μm. C, Western blot analysis of media samples from HEK cells expressing SpARC4 Myc or Myc-tagged SpARC4 lacking the C-terminal 17 amino acids (SpARC4 ΔC Myc).
We have previously used an HPLC assay and Xenopus embryos overexpressing SpARC1 and SpARC2 to characterize the enzymatic properties of the heterologously translated proteins (25). Here, we use the same approach to characterize SpARC4. In the first set of experiments, homogenates expressing SpARC4 Myc were incubated with NAD. As shown in the HPLC in Fig. 4A, NAD, after a 3-h incubation, was converted to a product with retention time of 17.7 ± 0.14 min corresponding to that of cADPR (17.3 ± 0.1 min). ADP-ribose production was not routinely detected (retention time = 26 min) under these experimental conditions. We next tested whether SpARC4 could generate NAADP. For these experiments, homogenates were incubated with NADP and nicotinic acid under acidic conditions to favor the base-exchange reaction. As shown in Fig. 4B, NADP, after a 3-h incubation, was converted to a product (retention time = 31.4 ± 0.04 min) that co-eluted with NAADP (retention time = 31.3 ± 0.03 min). Thus, SpARC4 was capable of both the cyclization and base-exchange reactions indicating that it is a bona fide multifunctional ADP-ribosyl cyclase.
FIGURE 4.

SpARC4 is a multifunctional ADP-ribosyl cyclase. A and B, homogenates prepared from Xenopus embryos expressing SpARC4 Myc were incubated with 1 mm NAD (A) or 1 mm NADP + 50 mm nicotinic acid (NA) (B) and the reaction mixtures separated by HPLC. Typical chromatograms after a 0.1-h (black traces) or 3-h (red traces) incubation are shown. C and D, time courses for production of cADPR (circles) and NAADP (squares) by Xenopus embryo homogenates expressing SpARC4 Myc (C) and SpARC2 Myc (D).
A time course for cADPR and NAADP production by SpARC4 is shown in Fig. 4C. Strikingly, SpARC4 Myc produced markedly more cADPR than NAADP under these experimental conditions of saturating substrate concentrations. This is in direct contrast to SpARC1 and SpARC2 which, as reported previously (25), produce more NAADP than cADPR when assayed identically. The results with SpARC2 are confirmed here (Fig. 4D). The initial rates of cADPR and NAADP production are listed in Table 1. SpARC4 untagged also displayed the same preference for producing cADPR as SpARC4 Myc (Table 1), indicating that the unusual catalytic activity associated with SpARC4 was not a consequence of tagging the protein.
TABLE 1.
Initial rates of cADPR and NAADP production
Data are presented as means ± S.E. from the indicated number of independent Xenopus embryo preparations.
| Construct | cADPR production | NAADP production | n |
|---|---|---|---|
| nmol/h/embryo | nmol/h/embryo | ||
| SpARC4 Myc | 2.61 ± 0.49 | 0.77 ± 0.16 | 9 |
| SpARC4 untagged | 2.96 ± 1.19 | 1.02 ± 0.51 | 3 |
| SpARC4 ΔN Myc | 3.9 ± 0.85 | 1.36 ± 0.32 | 3 |
| SpARC4R52A/R53A Myc | 2.19 ± 0.42 | 0.97 ± 0.09 | 3 |
| SpARC4Y142W Myc | 0.13 ± 0.02 | 0.15 ± 0.03 | 3 |
| SpARC4Y142H Myc | 0.1 ± 0.04 | 1.06 ± 0.24 | 3 |
| SpARC2 Myc | 0.05 ± 0.01 | 0.39 ± 0.07 | 6 |
Inspection of the sequence downstream of the predicted signal peptide of SpARC4 revealed the presence of a cluster of acidic amino acids followed by basic residues (Fig. 1A, box). This acidic-basic cluster is unique to SpARC4 among ADP-ribosyl cyclases. To investigate the potential functional role of this cluster, we generated a construct in which the sequence was deleted (SpARC4 ΔN Myc). This construct like the full-length construct was readily expressed in both Xenopus embryos and HEK cells (Fig. 5A). Immunocytochemical analysis indicated that it localized to the plasma membrane (Fig. 5B). Measurement of cADPR and NAADP production did not reveal any differences between SpARC4 ΔN Myc and SpARC4 Myc (Fig. 5C). Thus, SpARC4 ΔN Myc also preferentially produced cADPR (Fig. 5C and Table 1). We noted that the basic amino acids within the unique region of SpARC4 conform to a consensus sequence (RXXR) for cleavage by pro-convertase, raising the possibility that SpARC4 may be proteolytically processed. However, we could not detect the putative N-terminal cleaved product containing the Myc tag (predicted size ∼15 kDa) by Western blotting when gels were run under conditions to optimally resolve small proteins and when the blots were overdeveloped (Fig. 5D). Additionally, a mutant construct (SpARC4R52A/R53A Myc), in which arginines 52 and 53 were replaced with alanine (to disrupt the consensus cleavage site), was readily expressed (Fig. 5A) and localized to the plasma membrane (Fig. 5B) and was catalytically similar to SpARC4 Myc (Fig. 5C, Table 1). Taken together, the acidic-basic cluster appears not to affect the catalytic properties of SpARC4.
FIGURE 5.

Lack of effect of the acidic-basic cluster on SpARC4 activity. A, Western blot analysis of Xenopus embryos (left) or HEK cells (right) expressing Myc-tagged SpARC4 lacking either the N-terminal 17 amino acids (SpARC4 ΔN Myc) or in which arginines 52 and 53 were mutated to alanine (SpARC4R52A/R53A Myc). SpARC4 Myc was analyzed in parallel. B, immunocytochemical localization of SpARC4 ΔN Myc and SpARC4R52A/R53A Myc in HEK cells. Scale bar, 5 μm. C, time courses for production of cADPR and NAADP by SpARC4 Myc, SpARC4 ΔN Myc, and SpARC4R52A/R53A Myc. D, Western blot analysis of SpARC4 Myc and SpARC4R52A/R53A. Arrowhead marks the expected migration of the putative cleavage product. Blots were deliberately overexposed.
Several residues that form the active site of Aplysia ADP-ribosyl cyclase, CD38 and CD157, have been identified by site-directed mutagenesis and structural studies. These include a tryptophan residue (at positions 77 and 125 in Aplysia and human CD38, respectively), which is strictly conserved in these ADP-ribosyl cyclases (48, 49). Intriguingly, multiple sequence alignment identifies a tyrosine residue at the corresponding position 142 of SpARC4, suggesting that this enzyme, in analogy with S. mansoni NACE (24), possesses a noncanonical active site (Fig. 6A). To assess the potential functional importance of tyrosine 142, we generated a mutant construct in which this residue was replaced with tryptophan (SpARC4Y142W Myc). We also generated a second construct in which tyrosine was replaced with histidine (SpARC4Y142H Myc). The latter mutation corresponds to the residue found in SmNACE (Fig. 6A). Both mutant constructs were readily expressed in Xenopus embryos and HEK cells to comparable levels as the wild type construct (Fig. 6B). Additionally, both mutant constructs localized to the plasma membrane (Fig. 6C). Production of cADPR and NAADP for the two constructs is shown in Fig. 6D. Remarkably, whereas the wild type construct preferentially produced cADPR, production of cADPR and NAADP was comparable for SpARC4Y142W Myc (Fig. 6D). Overall activity, however, was reduced relative to the wild type enzyme (Table 1). These data highlight a critical role for tyrosine 142 in determining both the catalytic efficiency of SpARC4 and the relative production of cADPR and NAADP. The results with SpARC4Y142H Myc were even more striking. This single mutation greatly affected the ADP-ribosyl cyclase activity of SpARC4 without perturbing the transglycosidation process (Fig. 6D). Thus, there was a complete reversal in the preference for the different reactions such that the mutant cyclase preferentially produced NAADP (Fig. 6D and Table 1), very much like SpARC2 (Fig. 4D). The ratio of the initial rates of cADPR and NAADP production for the different constructs from several independent preparations is summarized in Fig. 7. This analysis has the advantage in that it is independent of the absolute expression levels of the constructs, which vary between preparations. These data show that production of cADPR and NAADP by SpARC4 is controlled by a single active site residue, which, when mutated, can convert SpARC4 into a SpARC2-like enzyme.
FIGURE 6.

Regulation of SpARC4 activity by noncanonical active site residue. A, multiple sequence alignment of ADP-ribosyl cyclases showing lack of conservation of an active site tyrosine (asterisk) in SpARC4 and Schistosoma ADP-ribosyl cyclase. Abbreviations used are as follows: Ac, Aplysia californica; Hs, Homo sapiens; Sm, Schistosoma mansoni; Sj, Schistosoma japonica. B, Western blot analysis of Xenopus embryos (left) or HEK cells (right) expressing SpARC4 Myc or Myc-tagged SpARC4 in which tyrosine 142 was mutated to either tryptophan (SpARC4Y142W Myc) or histidine (SpARC4Y142H Myc). C, immunocytochemical localization of SpARC4Y142W Myc and SpARC4Y142H Myc in HEK cells. Scale bar, 5 μm. D, time courses for production of cADPR and NAADP by SpARC4 Myc, SpARC4Y142W Myc, and SpARC4Y142H Myc.
FIGURE 7.
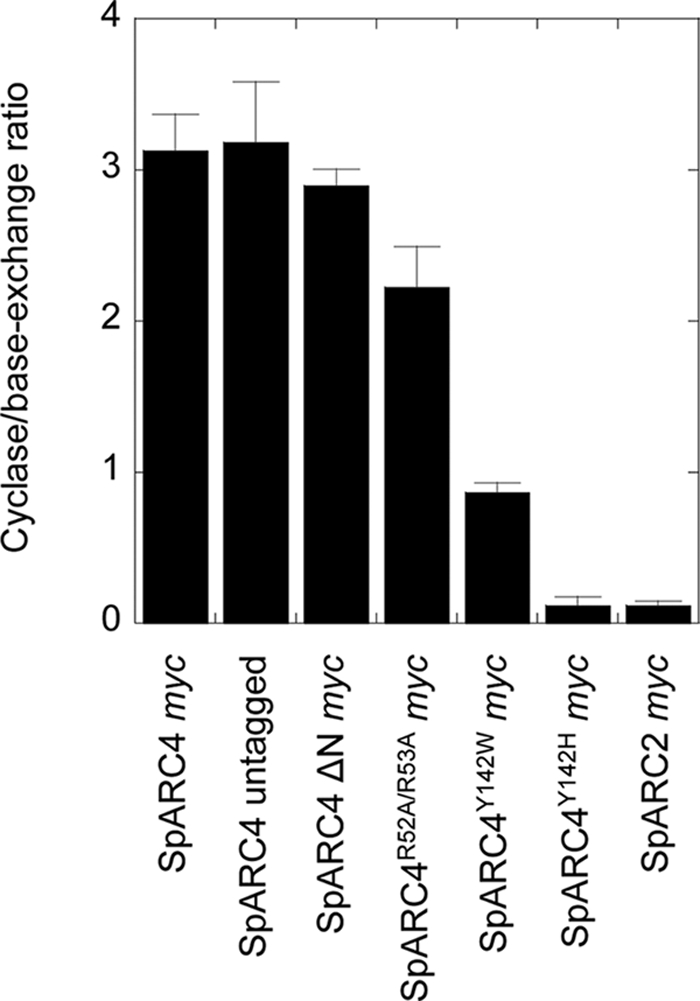
Summary of mutational analysis on SpARC4 activity. Pooled data showing the ratio of ADP-ribosyl cyclase to base-exchange activity for the indicated construct. Data are presented as means ± S.E. of the mean from 3 to 6 independent Xenopus embryo preparations.
DISCUSSION
In this study, we report the molecular cloning of an additional member of the ADP-ribosyl cyclase family and show that it is a glycoprotein targeted to the plasma membrane by a GPI-anchor and that it displays unusual catalytic activity dictated by a single active site residue.
Our understanding of the molecular diversity of ADP-ribosyl cyclases is currently limited. Aplysia and Schistosoma (protostomes) possess a single ADP-ribosyl cyclase gene, and mammals (deuterostomes) possess two. The sea urchin holds a key evolutionary position as a basal deuterostome and as a close relative of the chordates. We have previously characterized an expanded three-member family of ADP-ribosyl cyclases in the sea urchin (25). Here, we highlight further expansion of this family through the cloning of a fourth member, SpARC4. Comparative genomic analysis of ADP-ribosyl cyclases in other deuterostomes should provide insight into the unusual evolution of the ADP-ribosyl cyclase gene family in this lineage.
SpARC4, like SpARC2 (55), mammalian CD157 (23), and SmNACE (24), localizes to the plasma membrane via a GPI anchor (Figs. 2 and 3), which contrasts to the intracellular location of SpARC1 (25). The ecto-cellular location of SpARC4 shared also by mammalian CD38 (22) further suggests that second messenger production may occur at extracellular sites. Whether SpARC4 (and SpARC2) is internalized, e.g. via endocytosis, as reported for CD38 (62) remains to be established, although we did note a punctate intracellular distribution of SpARC4 in some cells upon heterologous expression in Xenopus embryos, which might indicate an endosomal location (Fig. 2).
A clear distinguishing feature of SpARC4 relates to its catalytic activity; under our experimental conditions, SpARC4 appears to produce predominantly cADPR (Fig. 4), whereas SpARC1 and SpARC2 preferentially produce NAADP (25, 55). An unidentified enzyme present on the surface of sea urchin sperm also preferentially produces NAADP (63). Although much attention has focused on determining the relative rates of NAD cyclization and glycohydrolase activity culminating recently in the effective interconversion of CD38 and Aplysia ADP-ribosyl cyclase by site-directed mutagenesis (58), little is known about what controls the relative production of cADPR and NAADP.
Elucidating the molecular basis for the pronounced difference in catalytic activity between SpARC4 and other family members represents a formidable challenge given the very modest sequence identity between them (Fig. 1B). We first focused on a cluster of acidic and basic amino acids in the N terminus of SpARC4. This cluster is absent from other SpARCs. Interestingly, this region of the protein encompasses a consensus pro-convertase cleavage sequence raising the possibility that SpARC4 may be proteolytically processed, which in turn may regulate catalysis. Indeed, cleavage at such sites in many other proteins, including tolloid metalloproteases, is required for activity (64). However, as shown in Fig. 5D, we could not detect cleavage of SpARC4 despite the presence of robust pro-protease activity in Xenopus embryos (65, 66). Moreover, catalytic activity of SpARC4 was unaffected when the putative cleavage site was disrupted by site-directed mutagenesis or when the N terminus encompassing the site was deleted (Fig. 5C). Thus, this region of the enzyme appears to not affect catalysis. Its functional role remains to be defined.
Catalysis is thought to proceed via a partitioning kinetic mechanism (13, 14, 50) raising the possibility that active site residue(s) may control the cyclization step relative to the base-exchange pathway. We therefore next focused on position 142 in SpARC4 where a tyrosine residue replaces tryptophan, which is otherwise strictly conserved in the Aplysia enzyme, CD38 and CD157. This substitution was confirmed by expressed sequence tags from a different urchin species (supplemental Fig. S1) indicating that a fourth ADP-ribosyl cyclase is not unique to the purple sea urchin and that the difference in sequence at this position was not due to a cloning error. The noncanonical active site described here for SpARC4 is also a feature of SmNACE (24). In SmNACE, the active site contains a histidine residue (His-103) in place of this conserved tryptophan, which in the other ADP-ribosyl cyclases has been implicated in substrate positioning (48, 49). In the Aplysia enzyme and CD38, mutation of this residue substantially affects both ADP-ribosyl cyclase and base-exchange activity. Notably, studies on CD38 included mutation of tryptophan 125 to tyrosine thus producing a mutant enzyme similar to SpARC4 (49). Conversely, the ADP-ribosyl cyclase activity of SmNACE, which is undetectable with NAD as substrate, was dramatically increased upon mutation of the noncanonical active site histidine 103 to tryptophan (60). This effect was ascribed to the more polar environment in the wild type enzyme favoring entry of water and thus hydrolysis of NAD, as opposed to the competing intramolecular cyclization of NAD (60). In contrast, SpARC4 displays robust ADP-ribosyl cyclase activity relative to other SpARCs and is more active than its tryptophan mutant (Fig. 6). These data suggest that other residues are likely to contribute to catalysis in SpARC4.
Despite the conservative nature of the substitution at the active site of SpARC4 (tryptophan to tyrosine), we show that this residue plays a critical role in determining the production of cADPR over NAADP. This is most pronounced in SpARC4Y142H, where the ADP-ribosyl cyclase activity was decreased by 1 order of magnitude, but in sharp contrast with SpARC4Y142W, the mutation did not affect the transglycosidation reaction. Consequently, a complete switch in relative ADP-ribosyl cyclase and base-exchange activity is observed resulting in a catalytic profile very similar to SpARC1 and SpARC2 (Fig. 6). Previous mutagenesis analyses of Aplysia ADP-ribosyl cyclase and CD38 have generally focused on cyclase/glycohydrolase ratios, and where measured, base-exchange activity of the mutants tended to mirror changes in cyclase activity. One notable exception is glutamate 146 in CD38, which when mutated did in some instances (e.g. E146D and E146G) result in reciprocal changes in cyclase and base-exchange activity (59). Thus, this residue might also dictate the relative production of cADPR and NAADP.
The noncanonical active sites of SmNACE and SpARC4 are highly unusual. The presence of residues other than tryptophan at this position in ADP-ribosyl cyclases appears unique to these enzymes based on inspection of a variety of ADP-ribosyl cyclase sequences across phyla (data not shown). Thus, selection pressure appears to have been relaxed for these homologues. The resulting changes in activity in the sea urchin might be of particular significance given the multiplicity of isoforms in this organism. We speculate that different ADP-ribosyl cyclase isoforms with different activities might regulate specific cellular functions.
Supplementary Material
Acknowledgments
We thank Dev Churamani for help with confocal microscopy and George Dickinson and Chi Li for useful discussions.
This work was supported in part by Grants BB/D018110/1 and BB/G013721/1 from the Biotechnology and Biological Sciences Research Council (to S. P.).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Table S1 and Fig. S1.
The nucleotide sequence was deposited at EMBL under the accession number FN645665.
- cADPR
- cyclic ADP-ribose
- NAADP
- nicotinic acid adenine dinucleotide phosphate
- HEK
- human embryonic kidney
- PBS
- phosphate-buffered saline
- RACE
- rapid amplification of cDNA ends
- GPI
- glycosylphosphatidylinositol
- HPLC
- high pressure liquid chromatography
- MES
- 4-morpholineethanesulfonic acid
- MOPS
- 4-morpholinepropanesulfonic acid
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- PI-PLC
- phosphatidylinositol-specific phospholipase C
- PNGase-F
- peptide:N-glycosidase-F
- RACE
- rapid amplification of cDNA ends
- NACE
- NAD(P)+-catabolizing enzyme.
REFERENCES
- 1.Berridge M. J., Lipp P., Bootman M. D. (2000) Nat. Rev. Mol. Cell Biol. 1, 11–21 [DOI] [PubMed] [Google Scholar]
- 2.Lee H. C. (2001) Annu. Rev. Pharmacol. Toxicol. 41, 317–345 [DOI] [PubMed] [Google Scholar]
- 3.Guse A. H., Lee H. C. (2008) Sci. Signal. 1, re10. [DOI] [PubMed] [Google Scholar]
- 4.Galione A., Lee H. C., Busa W. B. (1991) Science 253, 1143–1146 [DOI] [PubMed] [Google Scholar]
- 5.Guse A. H. (2009) Curr. Biol. 19, R521–R523 [DOI] [PubMed] [Google Scholar]
- 6.Lee H. C., Aarhus R. (1995) J. Biol. Chem. 270, 2152–2157 [DOI] [PubMed] [Google Scholar]
- 7.Berridge G., Dickinson G., Parrington J., Galione A., Patel S. (2002) J. Biol. Chem. 277, 43717–43723 [DOI] [PubMed] [Google Scholar]
- 8.Churchill G. C., Okada Y., Thomas J. M., Genazzani A. A., Patel S., Galione A. (2002) Cell 111, 703–708 [DOI] [PubMed] [Google Scholar]
- 9.Patel S., Docampo R. (2010) Trends Cell Biol. 20, 277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brailoiu E., Churamani D., Cai X., Schrlau M. G., Brailoiu G. C., Gao X., Hooper R., Boulware M. J., Dun N. J., Marchant J. S., Patel S. (2009) J. Cell Biol. 186, 201–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Calcraft P. J., Ruas M., Pan Z., Cheng X., Arredouani A., Hao X., Tang J., Rietdorf K., Teboul L., Chuang K. T., Lin P., Xiao R., Wang C., Zhu Y., Lin Y., Wyatt C. N., Parrington J., Ma J., Evans A. M., Galione A., Zhu M. X. (2009) Nature 459, 596–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brailoiu E., Hooper R., Cai X., Brailoiu G. C., Keebler M. V., Dun N. J., Marchant J. S., Patel S. (2010) J. Biol. Chem. 285, 2897–2901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee H. C., Munshi C., Graeff R. (1999) Mol. Cell. Biochem. 193, 89–98 [DOI] [PubMed] [Google Scholar]
- 14.Schuber F., Lund F. E. (2004) Curr. Mol. Med. 4, 249–261 [DOI] [PubMed] [Google Scholar]
- 15.Partida-Sánchez S., Cockayne D. A., Monard S., Jacobson E. L., Oppenheimer N., Garvy B., Kusser K., Goodrich S., Howard M., Harmsen A., Randall T. D., Lund F. E. (2001) Nat. Med. 7, 1209–1216 [DOI] [PubMed] [Google Scholar]
- 16.Jin D., Liu H. X., Hirai H., Torashima T., Nagai T., Lopatina O., Shnayder N. A., Yamada K., Noda M., Seike T., Fujita K., Takasawa S., Yokoyama S., Koizumi K., Shiraishi Y., Tanaka S., Hashii M., Yoshihara T., Higashida K., Islam M. S., Yamada N., Hayashi K., Noguchi N., Kato I., Okamoto H., Matsushima A., Salmina A., Munesue T., Shimizu N., Mochida S., Asano M., Higashida H. (2007) Nature 446, 41–45 [DOI] [PubMed] [Google Scholar]
- 17.Clapper D. L., Walseth T. F., Dargie P. J., Lee H. C. (1987) J. Biol. Chem. 262, 9561–9568 [PubMed] [Google Scholar]
- 18.Hellmich M. R., Strumwasser F. (1991) Cell Regul. 2, 193–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Glick D. L., Hellmich M. R., Beushausen S., Tempst P., Bayley H., Strumwasser F. (1991) Cell Regul. 2, 211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lee H. C., Aarhus R. (1991) Cell Regul. 2, 203–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Aarhus R., Graeff R. M., Dickey D. M., Walseth T. F., Lee H. C. (1995) J. Biol. Chem. 270, 30327–30333 [DOI] [PubMed] [Google Scholar]
- 22.Howard M., Grimaldi J. C., Bazan J. F., Lund F. E., Santos-Argumedo L., Parkhouse R. M., Walseth T. F., Lee H. C. (1993) Science 262, 1056–1059 [DOI] [PubMed] [Google Scholar]
- 23.Hirata Y., Kimura N., Sato K., Ohsugi Y., Takasawa S., Okamoto H., Ishikawa J., Kaisho T., Ishihara K., Hirano T. (1994) FEBS Lett. 356, 244–248 [DOI] [PubMed] [Google Scholar]
- 24.Goodrich S. P., Muller-Steffner H., Osman A., Moutin M. J., Kusser K., Roberts A., Woodland D. L., Randall T. D., Kellenberger E., LoVerde P. T., Schuber F., Lund F. E. (2005) Biochemistry 44, 11082–11097 [DOI] [PubMed] [Google Scholar]
- 25.Churamani D., Boulware M. J., Geach T. J., Martin A. C., Moy G. W., Su Y. H., Vacquier V. D., Marchant J. S., Dale L., Patel S. (2007) PLoS. ONE. 2, e797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.De Flora A., Zocchi E., Guida L., Franco L., Bruzzone S. (2004) Ann. N.Y. Acad. Sci. 1028, 176–191 [DOI] [PubMed] [Google Scholar]
- 27.Davis L. C., Morgan A. J., Ruas M., Wong J. L., Graeff R. M., Poustka A. J., Lee H. C., Wessel G. M., Parrington J., Galione A. (2008) Curr. Biol. 18, 1612–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bezin S., Charpentier G., Lee H. C., Baux G., Fossier P., Cancela J. M. (2008) J. Biol. Chem. 283, 27859–27870 [DOI] [PubMed] [Google Scholar]
- 29.Kato I., Yamamoto Y., Fujimura M., Noguchi N., Takasawa S., Okamoto H. (1999) J. Biol. Chem. 274, 1869–1872 [DOI] [PubMed] [Google Scholar]
- 30.Fukushi Y., Kato I., Takasawa S., Sasaki T., Ong B. H., Sato M., Ohsaga A., Sato K., Shirato K., Okamoto H., Maruyama Y. (2001) J. Biol. Chem. 276, 649–655 [DOI] [PubMed] [Google Scholar]
- 31.Kim S. Y., Cho B. H., Kim U. H. (2010) J. Biol. Chem. 285, 576–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ceni C., Muller-Steffner H., Lund F., Pochon N., Schweitzer A., De Waard M., Schuber F., Villaz M., Moutin M. J. (2003) J. Biol. Chem. 278, 40670–40678 [DOI] [PubMed] [Google Scholar]
- 33.Soares S., Thompson M., White T., Isbell A., Yamasaki M., Prakash Y., Lund F. E., Galione A., Chini E. N. (2007) Am. J. Physiol. Cell Physiol 292, C227–C239 [DOI] [PubMed] [Google Scholar]
- 34.Mészáros L. G., Wrenn R. W., Váradi G. (1997) Biochem. Biophys. Res. Commun. 234, 252–256 [DOI] [PubMed] [Google Scholar]
- 35.Matsumura N., Tanuma S. (1998) Biochem. Biophys. Res. Commun. 253, 246–252 [DOI] [PubMed] [Google Scholar]
- 36.Guse A. H., da Silva C. P., Berg I., Skapenko A. L., Weber K., Heyer P., Hohenegger M., Ashamu G. A., Schulze-Koops H., Potter B. V., Mayr G. W. (1999) Nature 398, 70–73 [DOI] [PubMed] [Google Scholar]
- 37.Wilson H. L., Dipp M., Thomas J. M., Lad C., Galione A., Evans A. M. (2001) J. Biol. Chem. 276, 11180–11188 [DOI] [PubMed] [Google Scholar]
- 38.Hohenegger M., Suko J., Gscheidlinger R., Drobny H., Zidar A. (2002) Biochem. J. 367, 423–431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bruzzone S., De Flora A., Usai C., Graeff R., Lee H. C. (2003) Biochem. J. 375, 395–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sternfeld L., Krause E., Guse A. H., Schulz I. (2003) J. Biol. Chem. 278, 33629–33636 [DOI] [PubMed] [Google Scholar]
- 41.Xie G. H., Rah S. Y., Kim S. J., Nam T. S., Ha K. C., Chae S. W., Im M. J., Kim U. H. (2005) Biochem. Biophys. Res. Commun. 330, 1290–1298 [DOI] [PubMed] [Google Scholar]
- 42.Zhang F., Zhang G., Zhang A. Y., Koeberl M. J., Wallander E., Li P. L. (2006) Am. J. Physiol. Heart Circ. Physiol. 291, H274–H282 [DOI] [PubMed] [Google Scholar]
- 43.Billington R. A., Harper C., Bellomo E. A., Publicover S., Barratt C. L., Genazzani A. A. (2006) Fertil. Steril. 86, 891–898 [DOI] [PubMed] [Google Scholar]
- 44.Graeff R. M., Franco L., De Flora A., Lee H. C. (1998) J. Biol. Chem. 273, 118–125 [DOI] [PubMed] [Google Scholar]
- 45.Prasad G. S., McRee D. E., Stura E. A., Levitt D. G., Lee H. C., Stout C. D. (1996) Nat. Struct. Biol. 3, 957–964 [DOI] [PubMed] [Google Scholar]
- 46.Yamamoto-Katayama S., Ariyoshi M., Ishihara K., Hirano T., Jingami H., Morikawa K. (2002) J. Mol. Biol. 316, 711–723 [DOI] [PubMed] [Google Scholar]
- 47.Liu Q., Kriksunov I. A., Graeff R., Munshi C., Lee H. C., Hao Q. (2006) J. Biol. Chem. 281, 32861–32869 [DOI] [PubMed] [Google Scholar]
- 48.Munshi C., Thiel D. J., Mathews I. I., Aarhus R., Walseth T. F., Lee H. C. (1999) J. Biol. Chem. 274, 30770–30777 [DOI] [PubMed] [Google Scholar]
- 49.Munshi C., Aarhus R., Graeff R., Walseth T. F., Levitt D., Lee H. C. (2000) J. Biol. Chem. 275, 21566–21571 [DOI] [PubMed] [Google Scholar]
- 50.Cakir-Kiefer C., Muller-Steffner H., Schuber F. (2000) Biochem. J. 349, 203–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Perraud A. L., Fleig A., Dunn C. A., Bagley L. A., Launay P., Schmitz C., Stokes A. J., Zhu Q., Bessman M. J., Penner R., Kinet J. P., Scharenberg A. M. (2001) Nature 411, 595–599 [DOI] [PubMed] [Google Scholar]
- 52.Lange I., Yamamoto S., Partida-Sanchez S., Mori Y., Fleig A., Penner R. (2009) Sci. Signal. 2, ra23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Patel S., Docampo R. (2009) Sci. Signal. 2, pe69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Graeff R. M., Walseth T. F., Fryxell K., Branton W. D., Lee H. C. (1994) J. Biol. Chem. 269, 30260–30267 [PubMed] [Google Scholar]
- 55.Churamani D., Boulware M. J., Ramakrishnan L., Geach T. J., Martin A. C., Vacquier V. D., Marchant J. S., Dale L., Patel S. (2008) Cell. Signal. 20, 2347–2355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tohgo A., Takasawa S., Noguchi N., Koguma T., Nata K., Sugimoto T., Furuya Y., Yonekura H., Okamoto H. (1994) J. Biol. Chem. 269, 28555–28557 [PubMed] [Google Scholar]
- 57.Zhang B., Muller-Steffner H., Schuber F., Potter B. V. (2007) Biochemistry 46, 4100–4109 [DOI] [PubMed] [Google Scholar]
- 58.Graeff R., Liu Q., Kriksunov I. A., Kotaka M., Oppenheimer N., Hao Q., Lee H. C. (2009) J. Biol. Chem. 284, 27629–27636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Graeff R., Munshi C., Aarhus R., Johns M., Lee H. C. (2001) J. Biol. Chem. 276, 12169–12173 [DOI] [PubMed] [Google Scholar]
- 60.Kuhn I., Kellenberger E., Rognan D., Lund F. E., Muller-Steffner H., Schuber F. (2006) Biochemistry 45, 11867–11878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sive H. L., Grainger R. M., Harland R. M. (2000) Early Development of Xenopus laevis: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 62.Zocchi E., Franco L., Guida L., Piccini D., Tacchetti C., De Flora A. (1996) FEBS Lett. 396, 327–332 [DOI] [PubMed] [Google Scholar]
- 63.Vasudevan S. R., Galione A., Churchill G. C. (2008) Biochem. J. 411, 63–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Leighton M., Kadler K. E. (2003) J. Biol. Chem. 278, 18478–18484 [DOI] [PubMed] [Google Scholar]
- 65.Birsoy B., Berg L., Williams P. H., Smith J. C., Wylie C. C., Christian J. L., Heasman J. (2005) Development 132, 591–602 [DOI] [PubMed] [Google Scholar]
- 66.Nelsen S., Berg L., Wong C., Christian J. L. (2005) Dev. Dyn. 233, 1038–1044 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.