

Abstract
Uncompetitive N-methyl-d-aspartate (NMDA) receptor antagonists with fast off-rate (UFO) may represent promising drug candidates for various neurodegenerative disorders. In this study, we report that bis(propyl)-cognitin, a novel dimeric acetylcholinesterase inhibitor and γ-aminobutyric acid subtype A receptor antagonist, is such an antagonist of NMDA receptors. In cultured rat hippocampal neurons, we demonstrated that bis(propyl)-cognitin voltage-dependently, selectively, and moderately inhibited NMDA-activated currents. The inhibitory effects of bis(propyl)-cognitin increased with the rise in NMDA and glycine concentrations. Kinetics analysis showed that the inhibition was of fast onset and offset with an off-rate time constant of 1.9 s. Molecular docking simulations showed moderate hydrophobic interaction between bis(propyl)-cognitin and the MK-801 binding region in the ion channel pore of the NMDA receptor. Bis(propyl)-cognitin was further found to compete with [3H]MK-801 with a Ki value of 0.27 μm, and the mutation of NR1(N616R) significantly reduced its inhibitory potency. Under glutamate-mediated pathological conditions, bis(propyl)-cognitin, in contrast to bis(heptyl)-cognitin, prevented excitotoxicity with increasing effectiveness against escalating levels of glutamate and much more effectively protected against middle cerebral artery occlusion-induced brain damage than did memantine. More interestingly, under NMDA receptor-mediated physiological conditions, bis(propyl)-cognitin enhanced long-term potentiation in hippocampal slices, whereas MK-801 reduced and memantine did not alter this process. These results suggest that bis(propyl)-cognitin is a UFO antagonist of NMDA receptors with moderate affinity, which may provide a pathologically activated therapy for various neurodegenerative disorders associated with NMDA receptor dysregulation.
Keywords: Alzheimer Disease; Glutamate Receptors Ionotropic (AMPA, NMDA); Ion Channels; Ischemia; Neurodegeneration; Neurological Diseases; LTP; MCAO; Bis(propyl)-cognitin; Uncompetitive
Introduction
Neurodegenerative disorders are among the leading cause of death in the elderly, and this medical problem is expected to become more serious as the population continues to age. N-Methyl-d-aspartate (NMDA)3 receptor overactivation-induced excitotoxicity is clearly implicated in a variety of acute and chronic neurodegenerative disorders (1–4). Thus, the NMDA receptor has been considered an attractive therapeutic target for the development of neuroprotectants. However, the NMDA receptor, as the major excitatory neurotransmitter receptor in the central nervous system, mediates many important physiological processes, such as excitatory neurotransmission, synaptic plasticity, learning and memory, and cell survival (5–8). The therapeutic potential of many NMDA receptor antagonists, therefore, was limited, and they failed in clinical trials because of their psychotropic side effects and memory-impairing effects resulting from the blockade of NMDA receptor-mediated physiological activities (9, 10). Clinically well tolerated and effective NMDA receptor antagonists for the therapy of neurodegenerative disorders have been pursued over the past several decades.
The strategies applied to develop effective and well tolerated neuroprotective drugs are based on the principle that drugs should interact with their targets only during states of pathological and not physiological activation. Such drugs have been coined pathologically activated therapeutic (PAT) drugs by Lipton (11). For the treatment of neurodegenerative disorders accompanied by NMDA receptor-mediated excitotoxicity, uncompetitive NMDA receptor antagonists with fast off-rate (UFO), but not competitive and noncompetitive ones, fit in with the PAT strategy. An uncompetitive antagonist is defined as a drug whose inhibitory effect is contingent upon prior activation of the channel by agonist, and a fixed concentration of antagonist blocks the response to high concentrations of agonist to a greater extent than the response to low concentrations of agonist (11). A clear advantage of compounds of this type is that they preferentially blocked overactivated (in a tonic rather than phasic manner) channels while sparing physiological neurotransmission mediated by NMDA receptors (12–14). However, for uncompetitive antagonists, the off-rate and affinity further determine the clinical tolerability. Taking dizocilpine (MK-801) as an example, because it binds very tightly with and dissociates very slowly from the receptor, it blocks excessive as well as normal activation, resulting in deleterious side effects. Thus, uncompetitive NMDA receptor antagonists with moderate to low receptor affinity and with fast on/off-rate kinetics have been the center of interest in the search for neuroprotective drugs with beneficial therapeutic potentials for the treatment of various acute and chronic neurodegenerative disorders (10, 15–17).
Over the past decade, our group has developed a series of dimeric acetylcholinesterase (AChE) inhibitors derived from tacrine linked with different numbers of methylene (-CH2-) groups (18). These dimers have been demonstrated to inhibit AChE and γ-aminobutyric acid subtype A (GABAA) receptors in a tether-dependent manner (19, 20). The reduction of acetylcholine breakdown by AChE inhibitors may increase acetylcholine levels in the brain and contribute to learning and memory. Furthermore, blockade of GABAA receptors can increase the release of acetylcholine (21) and facilitates long term potentiation (LTP) induction (22), whereas activation of GABAA receptors may deteriorate aging-associated memory decline (23). In our previous study, bis(heptyl)-cognitin (also known as bis(7)-tacrine), heptylene-linked tacrine dimer, has been found to prevent glutamate-induced excitotoxicity by blocking NMDA receptors. However, the effectiveness of bis(heptyl)-cognitin neuroprotection decreases with the increase of glutamate concentration (24). Therefore, besides their AChE and GABAA receptor inhibitions in a tether-dependent manner, at the beginning of this study, we speculated that this series of multifunctional dimers might possess tether-dependent effects on NMDA receptors and different manners of neuroprotection. By comparison of their actions on NMDA receptors, bis(propyl)-cognitin emerges as a promising neuroprotective drug candidate among these novel dimers. We demonstrate that this dimer is an uncompetitive NMDA receptor antagonist with moderate affinity, fast off-rate, effective neuroprotective activities against glutamate-induced neuronal cell death without obvious cytotoxicity in vitro and middle cerebral artery occlusion (MCAO)-induced brain damage in vivo, and enhancement of LTP. These findings strongly suggest that this dimer may be developed to a PAT drug possessing considerable therapeutic potential for various associated neurodegenerative disorders.
EXPERIMENTAL PROCEDURES
Primary Neuron Cultures
Primary cultured hippocampal neurons were obtained from 18-day-old Sprague-Dawley rat embryos as previously described with modifications (25). Briefly, The hippocampi were dissected and incubated with 0.25% trypsin at 37 °C for 12 min. Cells were then mechanically dissociated by using a Pasteur pipette with a fire-narrowed tip in culture medium and plated at a density of 2 × 105 cells/ml on 35-mm culture dishes precoated with poly-l-lysine (10 μg/ml). Cells were maintained in neurobasal/B27 medium containing 0.5 mm glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin under a humidified atmosphere of 5% CO2, 95% air at 37 °C. Half-changes of medium were done twice weekly. Cells were used for electrophysiological recording 7–15 days after plating.
Cerebellar granule neurons (CGNs) were prepared from 8-day-old Sprague-Dawley rats (the animal care facility, Hong Kong University of Science and Technology) as described previously (24). Briefly, neurons were seeded on 96-well plates precoated with poly-l-lysine (10 μg/ml) at a density of 2 × 106 cells/ml in basal modified Eagle's medium containing 10% fetal bovine serum, 25 mm KCl, 2 mm glutamine, and penicillin (100 units/ml)/streptomycin (100 μg/ml). Cytosine arabinoside (10 μm) was added to the culture medium 24 h after plating to limit the growth of non-neuronal cells. With the use of this protocol, 95–99% of the cultured cells were granule neurons. All experiments were performed in CGNs at 7 or 8 days in vitro.
HEK293T Cell Culture and Transfection
HEK293T cells (ATCC, Manassas, VA) were grown as a monolayer using passage numbers less than 25 and maintained in Dulbecco's modified Eagle's medium (Invitrogen), supplemented with 10% fetal bovine serum (Invitrogen) and 2 mm l-glutamine in a humidified incubator at 37 °C with 5% CO2. At 1 day prior to transfection, exponentially growing cells were plated at a density of 1 × 105 cells/ml on 35-mm culture dishes precoated with poly-l-lysine (10 μg/ml). The cells were transiently transfected using Lipofectamine/PLUS reagents (Invitrogen) with a ratio of cDNA for rat NMDA receptor subunits for NR1-1a/NR2A (gift of Dr. Anne Stephenson from the Department of Pharmaceutical and Biological Chemistry, School of Pharmacy, University of London) of 1:3. An NR1-1a (N616R) mutation was introduced by site-directed mutagenesis using the QuikChange kit (Stratagene, La Jolla, CA) according to the manufacturer's directions and confirmed by DNA sequencing. Following transfection, the cells were maintained for 24 h in a glutamine-free medium containing 100 μm d(-)-2-amino-5-phosphonopentanoic acid and 200 μm ketamine to prevent NMDA receptor-mediated excitotoxicity.
Whole-cell Electrophysiological Recordings
Whole-cell patch clamp recordings were carried out at room temperature (22–24 °C) on the stage of an inverted phase-contrast microscope using an Axopatch 200B patch amplifier (Axon Instruments, Burlingame, CA). Before each experiment, the culture medium was removed, and the cells were rinsed completely and continuously superfused with a solution containing 150 mm NaCl, 5 mm KCl, 0.25 mm CaCl2, 10 mm glucose, 0.001 mm glycine, 0.001 mm tetrodotoxin, 0.01 mm (−)-bicuculline methiodide, and 10 mm HEPES (the pH was adjusted to 7.4 with NaOH, and the osmolarity was adjusted to ∼340 mosm with sucrose). The low concentration of Ca2+ was used to minimize the calcium-dependent desensitization of NMDA-activated currents. Pipettes pulled from borosilicate glass (TW-150F, World Precision Instruments, Sarasota, FL) had resistances of 2–4 megaohms when filled with pipette solution containing 140 mm CsCl, 10 mm EGTA, 10 mm HEPES, and 5 mm MgATP with pH 7.3 and 315 mosm in osmolarity. A small patch of membrane underneath the tip of the pipette was aspirated to form a gigaseal, and then a more negative pressure was applied to rupture it, thus establishing a whole-cell configuration. The adjustment of capacitance compensation and series resistance compensation was done before recording the membrane currents. The holding potential was set at −50 mV except when otherwise indicated. Data were acquired using pClamp 9.0 software (Axon Instruments). Currents were filtered at 2 kHz and digitized at 5 kHz.
Drug Application
Drugs used in the experiments included NMDA, AMPA, kainate, tacrine, memantine, MK-801, tetrodotoxin, and (−)-bicuculline methiodide. All of these drugs were purchased from Sigma. Bis(n)-cognitins were prepared according to the method described previously (19). Each drug was dissolved daily in the Mg2+-free external solution just before use and applied with a fast step perfusion system (SF-77B; Warner Instruments, Hamden, CT) through 3-barrel square glass tubing. The distance from the tube mouth to the cell examined was about 100 μm. The application of each drug could be driven by gravity and controlled by the corresponding valve. The solution exchange rate was estimated according to Chen et al. (26) with a time constant of 2.38 ± 0.82 ms (n = 21, mean ± S.E.). Cells or patches were constantly bathed in the normal external solution flowing from one tube connected to a larger reservoir between drug applications (0.5–0.8 ml/min).
Molecular Docking Analysis
The homology model of the ion channel of NMDA receptors was constructed based on the x-ray crystal structure of the potassium channel KcsA (Protein Data Bank code 1BL8), and performed using ICM sequence-structure alignment and the BuildModel algorithm. ICM sequence-structure alignment is based on the primary sequences of subunits of NR1, NR2B, and the primary sequence of potassium channel KcsA (Escherichia coli). The homology model was optimized by Monte Carlo minimization. Three independent runs of the refinement procedure were performed, each run including 100,000 Monte Carlo minimization steps. The ICM Protein Health tool was used to check the quality of the refinement. Molecular docking was performed using the ICM-Pro 3.4-8a program (Molsoft). According to the ICM method, the molecular system was described using internal coordinates as variables. Energy calculations were based on the ECEPP/3 force field with a distance-dependent dielectric constant. The biased probability Monte Carlo minimization procedure was used for global energy optimization. The binding between the compounds and the NMDA receptor was evaluated with a full-atom ICM ligand binding score calculated as follows, Sbind = Eint + TΔSTor + Evw + α1Eel + α2Ehb + α3Ehp + α4Esf, where Evw, Eel, Ehb, Ehp, and Esf are van der Waals, electrostatic, hydrogen bonding, and nonpolar and polar atom solvation energy differences between bound and unbound states, respectively. Eint is the ligand internal strain, ΔSTor is its conformational entropy loss upon binding, T = 300 K, and αi are ligand- and receptor-independent constants. A more negative binding score represents a more favorable binding interaction.
Receptor-ligand Binding Assay
The binding assay was performed as described (24) with modifications. Synaptic plasma membrane was prepared from the cerebella of 15-day-old Sprague-Dawley rats by using discontinuous sucrose density gradients. The receptor ligand binding was performed in triplicate using 150–200 μg of synaptic plasma membrane protein and 4 nm [3H]MK-801 (American Radiolabeled Chemicals Inc., St. Louis, MO) incubated with different concentrations of testing compounds; nonspecific binding was determined by an excess of the unlabeled MK-801. After collecting the samples on Whatman GF/B filters by rapid filtration with an MD-24 sample harvester, filtrated tissue on filters was soaked into scintillation mixtures overnight and measured in a scintillation counter (Wallac 1209, Rackbeta (Turku, Finland)). Specific 3H-ligand binding to receptors was determined by subtracting the nonspecific count from the total, which is defined by 0.1 mm unlabeled MK-801.
Measurement of Neurotoxicity
The percentage of surviving neurons in the presence of bis(propyl)-cognitin and/or glutamate was estimated by determining the activity of mitochondrial dehydrogenases with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (24). The assay was performed according to the specifications of the manufacturer (MTT kit I, Roche Applied Science). Briefly, the neurons were cultured in 96-well plates, 10 μl of 5 mg/ml MTT labeling reagent was added to each well containing cells in 100 μl of medium, and the plate was incubated for 4 h in a humidified incubator at 37 °C. After the incubation, 100 μl of the solvating solution (0.01 m HCl in 10% SDS solution) was added to each well for 17–18 h. The absorbance of the samples was measured at a wavelength of 570 nm with 630 nm as a reference wavelength. Unless otherwise indicated, the extent of MTT conversion in cells exposed to glutamate is expressed as a percentage of the control.
Transient Focal Cerebral Ischemia
The MCAO model was made on Male Sprague-Dawley rats (280–320 g) after anesthesia by injection of chloral hydrate (350 mg/kg intraperitoneally) as described previously with minor modifications (27). Briefly, a nylon filament with a rounded tip was advanced from the external carotid artery into the lumen of the internal carotid artery until laser Doppler flowmetry (PF5000, Perimed (Stockholm, Sweden)) showed a sharp decrease of the regional blood flow of middle cerebral artery to ∼20% of the base-line value. Two hours after onset of ischemia, reperfusion was allowed by withdrawal of the suture thread. The animals from the sham group received the same operation procedure except without the insertion of the nylon filament. 24 h after the reperfusion, neurological deficits were evaluated by a 3-point grading scale (28). Immediately after scoring of neurological deficits, rats were killed with an overdose of 10% chloral hydrate. Brains were sectioned into seven 2-mm coronal slices by use of a rat brain matrix (RBM 4000C; ASI Inc., Warren, MI) and stained with 2% 2,3,5-triphenyltetrazolium chloride. The infarct volume % = (contralateral volume − ipsilateral non-ischemic volume)/contralateral volume × 100%, and the edema extent = (ipsilateral volume − contralateral volume)/contralateral volume × 100%.
Preparation of Slices
All experiments were carried out on transverse slices of the Wistar rat hippocampus (males, age 3–4 weeks, weight 40–80 g). The brains were rapidly removed after decapitation and placed in cold oxygenated (95% O2, 5% CO2) media. Slices were cut at a thickness of 350 μm using a Campden Vibroslice and placed in a storage container containing oxygenated media at room temperature (20–22 °C) for 1 h. The slices were then transferred to a recording chamber for submerged slices and continuously superfused at a rate of 5–6 ml/min at 30–32 °C. The control media contained 120 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 26 mm NaHCO3, 2.0 mm MgSO4, 2.0 mm CaCl2, 10 mm d-glucose. All solutions contained 100 μm picrotoxin (Sigma) to block GABAA receptor-mediated response.
LTP Recording
The slices were transferred to the recording chamber, in which it was held submerged between two nylon nets and maintained at 30–32 °C. The chamber consisted of a circular well of a low volume (1–2 ml) and was perfused constantly at a rate of 4–5 ml/min. Standard electrophysiological techniques were used to record field potentials. All experiments were carried out in the dentate gyrus, with presynaptic stimulation applied to the medial perforant pathway of the dentate gyrus using a bipolar insulated tungsten wire electrode, and field excitatory postsynaptic potentials (EPSPs) recorded at a control test frequency of 0.033 Hz from the middle one-third of the molecular layer of the dentate gyrus with a glass microelectrode. In each experiment, an input-output curve (afferent stimulus intensity versus EPSP amplitude) was plotted at the test frequency. For all experiments, the amplitude of the test EPSP was adjusted to one-third of maximum (1–1.2 mV), and base-line synaptic transmission was monitored for 30 min before drug administration or delivering either high or low frequency stimulation. LTP was evoked by high frequency stimulation (HFS) consisting of eight trains, each of eight stimuli at 200 Hz, intertrain interval of 2 s, with the stimulation voltage increased during the HFS so as to elicit an initial EPSP of the train of double the normal test EPSP amplitude. Agents tested in this study were perfused over the slices for 60 min prior to HFS. Control (vehicle alone) and experimental levels of LTP were measured on slices prepared from the same hippocampus. Electrical signals were collected with an Axoclamp-700B and were analyzed using pClamp 10.2 (Axon Instruments). All values were normalized to this base line, and all values of LTP reported here were calculated as the changes in field EPSP amplitude measured 60 min after tetanic stimulation.
Data Analysis
All data are the means ± S.E. for n independent observations, and statistical analyses were performed using Student's t test. Concentration-response curves were analyzed using a four-parameter logistic equation of the form, Y = Ymin + (Ymax − Ymin)/(1 + 10∧((logEC50 − X)*Hill slope)), where Y is the response at a given concentration, Ymax and Ymin are the maximum and minimum response, X is the logarithmic value of the concentration, and Hill slope is the slope factor of the curve. EC50 (IC50 for inhibition) is the concentration that gives a response halfway between Ymax and Ymin.
RESULTS
Inhibition of NMDA-activated Currents by Bis(n)-cognitins
To look into tether-dependent effects of the series of tacrine dimers (i.e. bis(n)-cognitins) on NMDA receptors, we examined IC50 values of bis(n)-cognitins to inhibit NMDA-activated currents and voltage dependence of inhibition by using patch clamp techniques. In primary cultured rat hippocampal neurons, 30 μm NMDA-activated currents exhibited slight or no desensitization. As shown in Fig. 1A, bis(n)-cognitins, when co-applied with NMDA, inhibited NMDA-activated currents (INMDA) at a holding potential of −50 mV in a concentration-dependent manner. The IC50 value and slope factor for bis(propyl)-cognitin were 0.52 ± 0.05 μm and 0.74 ± 0.04, respectively. By comparison of the inhibitory effects of bis(n)-cognitins at holding potentials from −70 to +50 mV, bis(propyl)-cognitin showed the strongest voltage-dependent decrease of inhibition to INMDA (Fig. 1B). The reversal potential value was not changed by bis(propyl)-cognitin. Over the concentrations examined, none of bis(n)-cognitins applied alone activated detectable currents.
FIGURE 1.

Tether length differential potency and voltage dependence of bis(n)-cognitins. A, the graph shows the inhibitory effects of memantine, tacrine, and bis(n)-cognitins (B2C, B3C, B5C, B7C, and B10C) on 30 μm NMDA-activated currents at a holding potential of −50 mV in cultured rat hippocampal neurons. Each point represents the mean of percentage inhibition from 5–7 neurons. The IC50 values are indicated in the graph. Inset, molecular structure of bis(propyl)-cognitin. B, voltage dependence of tacrine and bis(n)-cognitin blockade of NMDA-activated currents is analyzed by plotting the mean of percentage inhibition against holding potentials.
Bis(propyl)-cognitin Selectively Inhibits INMDA in an Agonist-dependent Manner
We further examined the selectivity of bis(propyl)-cognitin inhibition among ionotropic glutamate receptors. As shown in Fig. 2A, bis(propyl)-cognitin had no detectable effect on steady-state currents activated by 30 μm AMPA or kainic acid even at a concentration of 10 μm, although it inhibited INMDA by about 65% at 1 μm. We then tested whether preapplication of bis(propyl)-cognitin had any effect on the subsequent NMDA-activated currents. It was evident from Fig. 2B that preapplied bis(propyl)-cognitin had no detectable effect on NMDA-activated currents, and bis(propyl)-cognitin could only exert an inhibitory effect when NMDA receptors were activated by co- or preapplication of NMDA, suggesting an agonist-dependent mechanism of bis(propyl)-cognitin inhibition. Fig. 2C further demonstrated that current amplitude of NMDA co-applied with bis(propyl)-cognitin decreased gradually and reached a steady-state level within several applications of NMDA in the presence of bis(propyl)-cognitin, and the NMDA-activated current amplitude gradually recovered from the blockade upon the repeated application of NMDA in the absence of bis(propyl)-cognitin.
FIGURE 2.
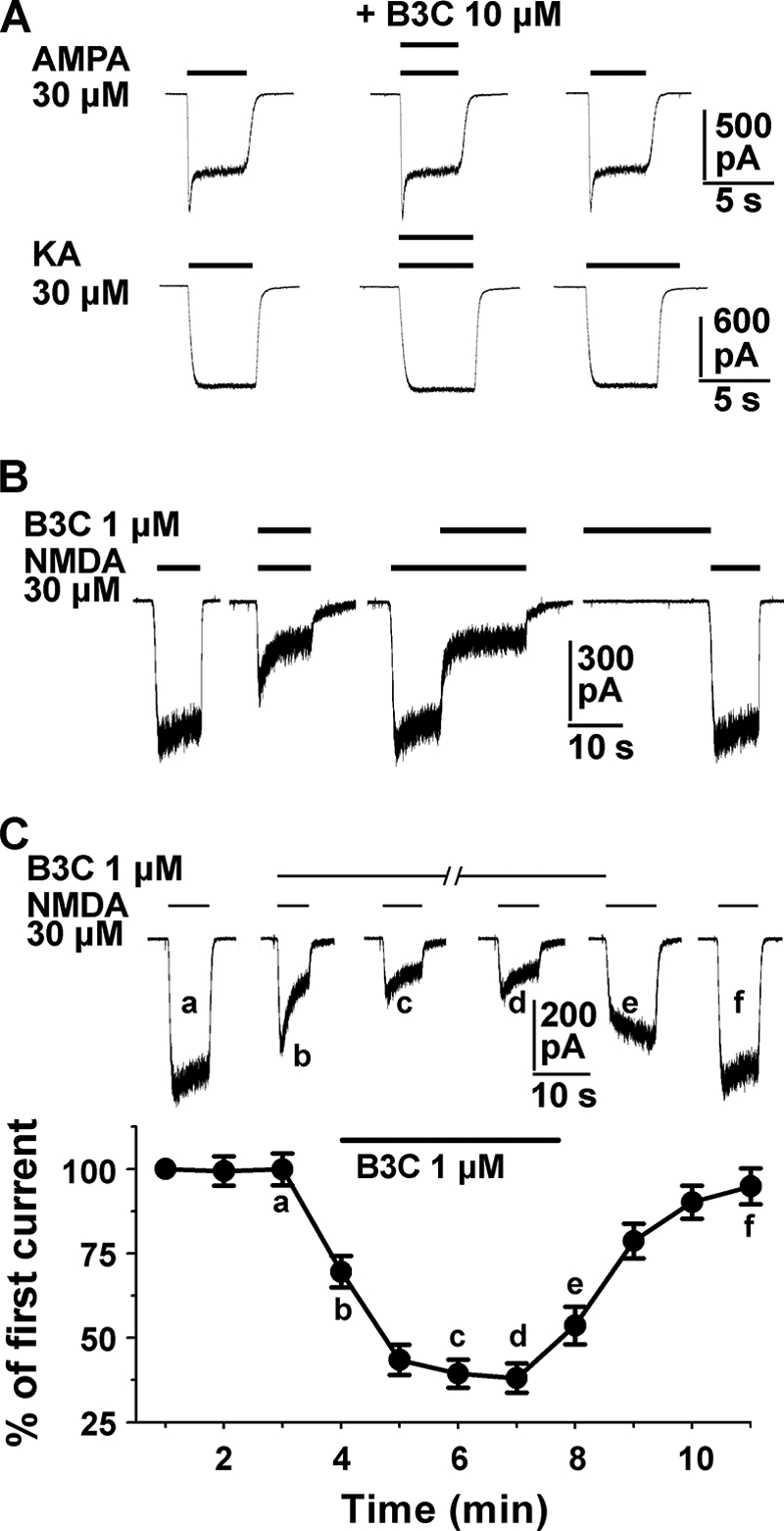
Selective, agonist- and use-dependent effects of bis(propyl)-cognitin inhibition. A, current traces show the effect of 10 μm bis(propyl)-cognitin (B3C) on 30 μm AMPA- or kainic acid (KA)-activated currents. Each row of traces is recorded from the same neuron and repeated in five different neurons. Bis(propyl)-cognitin is co-applied with AMPA or kainic acid for the time course indicated above each trace. B, representative current traces recorded from the same neuron show three different modes of drug application: co-application of bis(propyl)-cognitin (0.3 μm) and NMDA (30 μm), co-application made after the onset of the response to NMDA, and sequential application of NMDA following bis(propyl)-cognitin preapplication. Note the different effects of bis(propyl)-cognitin under these different conditions. A similar result was obtained in four different neurons. C, representative current traces (top) and graph (bottom, n = 8) show the control NMDA response and the cumulative inhibition incurred by consecutive co-application of NMDA and 1 μm bis(propyl)-cognitin, followed by the response to NMDA alone for recovery. Each response, normalized to the first NMDA-activated current, is activated at intervals of 1 min. The solid bars indicate the application of NMDA and/or bis(propyl)-cognitin.
Bis(propyl)-cognitin Inhibits INMDA in an Uncompetitive Manner
To distinguish possible patterns (competitive, noncompetitive, or uncompetitive mechanism) of interaction between bis(propyl)-cognitin and NMDA receptors, we examined the effect of a fixed concentration of bis(propyl)-cognitin (0.3 μm) on currents activated by NMDA at concentrations from 1 to 1000 μm. As shown in Fig. 3A, the concentration-response curve for NMDA-activated currents was shifted downward, with the maximum response reduced by about 50%. The EC50 value (29.89 ± 3.37 μm versus a control value of 42.24 ± 3.84 μm; p < 0.05) and the slope factor of the NMDA concentration-response curve (0.99 ± 0.02 versus a control value of 1.11 ± 0.03; p < 0.05) were decreased in the presence of bis(propyl)-cognitin. A further plot of percentage inhibition against NMDA concentrations revealed that the inhibitory effect increased with the increase of NMDA concentration (Fig. 3A, inset). Furthermore, the concentration-response curve for bis(propyl)-cognitin inhibition was shifted leftward, with the IC50 value reduced from 0.52 μm (nH = 0.74) to 0.21 μm (nH = 0.76) (p < 0.05).
FIGURE 3.

Bis(propyl)-cognitin is an uncompetitive inhibitor of NMDA receptors. A, NMDA dose-response curves in the absence (○) and presence (●) of 0.3 μm bis(propyl)-cognitin (B3C). The steady state response for each concentration of NMDA in the absence or presence of bis(propyl)-cognitin is normalized to the response produced by 30 μm NMDA in the absence of bis(propyl)-cognitin (marked with an asterisk). The responses are the means ± S.E. (error bars) from 9–15 neurons at each concentration of NMDA. Inset, plot showing the percentage inhibition of B3C and d(-)-2-amino-5-phosphonopentanoic acid (AP5) against NMDA concentrations ranging from 3 to 300 μm. B, dose-response relationships for bis(propyl)-cognitin inhibition of INMDA obtained under the conditions of 0.1 μm (●) and 10 μm (■) glycine. The curves were fitted using the logistic equation under “Experimental Procedures.”
Similarly, we investigated the effect of glycine on the bis(propyl)-cognitin inhibition of INMDA by changing the glycine concentrations from 0.1 to 10 μm in the external solution. As reported by others, the decrease of glycine concentrations resulted in a decrement of INMDA amplitude (data not shown). It could be seen from Fig. 3B that the inhibitory effect of bis(propyl)-cognitin on INMDA increased with the increase of glycine concentration in the external solution. The concentration-response curve for bis(propyl)-cognitin inhibition in the presence of 10 μm glycine was shifted leftward, with the IC50 value decreased to 0.28 ± 0.02 μm from 1.23 ± 0.08 μm in the presence of 0.1 μm glycine and with the slope factor basically unchanged (0.79 for 10 μm glycine versus 0.74 for 0.1 μm glycine, p > 0.05).
Bis(propyl)-cognitin Inhibition of INMDA Is of Fast Onset and Offset Kinetics
The kinetics of onset and offset (τon and τoff) of the inhibition were further investigated by fitting the changes of current amplitudes upon the application and removal of bis(propyl)-cognitin with single exponential functions. An example of single exponential fits to the onset and offset of inhibition is shown in Fig. 4A. The inverse of the onset and offset time constants derived from such fits was plotted as a function of bis(propyl)-cognitin concentrations and fitted with linear functions in Fig. 4B. The inverse of τon was significantly correlated with bis(propyl)-cognitin concentrations (p < 0.001), indicating that bis(propyl)-cognitin inhibition proceeded more quickly at higher concentrations. The inverse of τoff was not correlated with bis(propyl)-cognitin concentrations (p > 0.05). The mean value of τoff was 1911.5 ± 207.2 ms. Based on the on- and off-rate time constants, a Kd value of 0.68 μm was calculated for bis(propyl)-cognitin by the equation, Kd = koff/kon (kon = (1/τon − 1/τoff)/[B3C]; koff = 1/τoff), which was close to the value (0.52 μm) obtained from the concentration-response relationship.
FIGURE 4.

Onset and offset time constants of bis(propyl)-cognitin inhibition. A, traces show currents activated by 30 μm NMDA and its inhibition by 1 μm bis(propyl)-cognitin applied after the activation of NMDA receptors. The periods of drug application are indicated by the solid bar above the trace. White dotted lines drawn through the current trace (black part) are the theoretical fit of onset (τon) and offset (τoff) of bis(propyl)-cognitin inhibition using single exponential functions. B, graph plotting average τon (■) and τoff (●) values against bis(propyl)-cognitin concentration shows the concentration dependence of onset and offset kinetics. Error bars, S.E.
Acting Site(s) Mapping for Bis(propyl)-cognitin on the NMDA Receptor
To gain insight into the acting site(s) of bis(propyl)-cognitin on the NMDA receptor, molecular docking simulation was performed. Bis(propyl)-cognitin showed favorable interaction with the active site of the ion channel of NMDA receptors, with a binding score of −29.76. As a reference, a binding score of −33 was obtained for the known NMDA receptor uncompetitive antagonist memantine. The NH group of bis(propyl)-cognitin formed an additional hydrogen bond with the CO group of Val640 of the NR2B (2.79 Å). The cyclohexane in the bis(propyl)-cognitin molecule was located in the depth of the hydrophobic pocket formed by Asn616 of the NR1 subunit and by Phe614 and Asn615 of the NR2B subunit (Fig. 5A). Furthermore, as illustrated in Fig. 5B, bis(propyl)-cognitin apparently inhibited [3H]MK-801 binding to NMDA receptors, with an IC50 value of 0.36 ± 0.07 μm (Ki = 0.27 ± 0.05 μm). It has been reported that the Asn616 residue in the pore region is closely involved in the acting pocket of many open channel blockers (29). We next examined the effect of NR1(N616R) mutation on bis(propyl)-cognitin inhibition. As indicated in Fig. 5C, single residue mutation in NR1 significantly reduced the inhibitory effect of bis(propyl)-cognitin, with the IC50 value increased from 0.97 ± 0.13 μm to 45.51 ± 4.32 μm (n = 7, p < 0.001).
FIGURE 5.

Acting site of bis(propyl)-cognitin on the NMDA receptor. A, a close-up view of the low energy pose of bis(propyl)-cognitin in the NMDA receptor channel pore generated by molecular docking. The NMDA receptor is depicted in ribbon form, bis(propyl)-cognitin (tan) is depicted as a stick model, and the surface of the ligand pocket is shown as transparent gray skin. The homology model of the ion channel of NMDA receptor is constructed based on the x-ray crystal structure of the potassium channel KcsA (Protein Data Bank code 1BL8). B, graph shows the inhibition of [3H]MK-801 binding to rat in rat cerebellar cortex membrane preparations by bis(propyl)-cognitin (B3C). Membranes from rat cerebellar cortex were incubated with [3H]MK-801 (4 nm) and bis(propyl)-cognitin at gradually increasing concentrations. The data, expressed as percentage of control, are means according to three independent experiments. The Ki value was calculated from the corresponding IC50 Value according to the equation, Ki = IC50/(1 + C/Kd), where C is the concentration of radioligand and Kd is the dissociation constant obtained from the saturation experiment (12 nm). C, representative current traces and plots show the concentration-response relationships for bis(propyl)-cognitin inhibition of currents mediated by recombinant NR1/NR2A (wild type (WT)) and NR1(N616R)/NR2A receptors expressed in HEK293T cells. Error bars, S.E.
Bis(propyl)-cognitin Is Increasingly Effective in Protecting against Escalating Levels of Glutamate-induced Excitotoxicity
We have previously reported that bis(heptyl)-cognitin prevents glutamate-induced neuronal apoptosis in CGNs (24). In the current study, the model of glutamate-induced excitotoxicity in CGNs was used to investigate further the neuroprotective activities of bis(propyl)-cognitin in vitro. Up to 30 μm, bis(propyl)-cognitin showed no noticeable toxic effect on CGNs when applied alone (data not shown). When CGNs were pretreated with bis(propyl)-cognitin at concentrations from 0.1 to 30 μm for 2 h and exposed to 75 μm glutamate for 24 h, it was found that bis(propyl)-cognitin prevented glutamate-induced neuronal cell death in a concentration-dependent manner with an EC50 value of 1.33 ± 0.03 μm. The neuroprotection of bis(propyl)-cognitin was more substantial than that of the same concentration of memantine (Fig. 6A). Furthermore, it has been shown in our previous study that the effectiveness of the neuroprotection of bis(heptyl)-cognitin decreases with the increase of glutamate concentrations, and it can hardly protect against 300–500 μm glutamate-induced excitotoxicity (24). Very interestingly, however, the neuroprotective activity of bis(propyl)-cognitin, in contrast to bis(heptyl)-cognitin, increased with the escalation of glutamate concentrations when CGNs were pretreated with 3 μm bis(propyl)-cognitin and then exposed to glutamate (from 25 up to 500 μm) (Fig. 6B).
FIGURE 6.

Neuroprotection of bis(propyl)-cognitin in vitro. A, CGNs, at 8 days in vitro, were preincubated with bis(propyl)-cognitin (B3C) at different concentrations as indicated for 2 h and then exposed to 75 μm glutamate for another 24 h. Cell viability is measured using an MTT assay. All of the data, expressed as percentage of untreated control, are the means ± S.E. (error bars) of three separate experiments. Inset, plots show dose-response relationships for bis(propyl)-cognitin and memantine protection. B, CGNs, at 8 days in vitro, were preincubated with 3 μm bis(propyl)-cognitin (B3C) or bis(heptyl)-cognitin (B7C) for 2 h and then exposed to different concentrations of glutamate as indicated for another 24 h. The cell survival after the treatment with glutamate or glutamate following bis(propyl)-cognitin or bis(heptyl)-cognitin preincubation is expressed as percentage of untreated control.
Bis(propyl)-cognitin Rescues MCAO-induced Impairments Much More Potently than Memantine
The excitotoxicity associated with the NMDA receptor overactivation is closely involved in the pathogenesis of stroke (15, 30). A transient focal ischemic stroke model of MCAO-induced impairments in rats was employed to determine the neuroprotective effect of bis(propyl)-cognitin in vivo. Adult male Sprague-Dawley rats were subjected to transient MCAO for 2 h by the intraluminal suture method, and intraperitoneal injection of saline or bis(propyl)-cognitin (0.19 and 0.65 μmol/kg) was performed 15 min after MCAO onset. As depicted in Fig. 7, the treatment with bis(propyl)-cognitin produced a dose-dependent improvement in 24-h neurological deficits with neurological scores reduced from 2.2 ± 0.60 (saline group) to 1.6 ± 0.24 and 0.5 ± 0.29 for 0.19 and 0.65 μmol/kg bis(propyl)-cognitin-treated groups, respectively. Moreover, at 0.19 and 0.65 μmol/kg, bis(propyl)-cognitin reduced the total infarct volume significantly from a vehicle control of 34.92 ± 3.79% to 29.43 ± 5.33 and 11.24 ± 1.95% (n = 8–11 in each group, p < 0.01), respectively. Consistent with the previous study (31), memantine at a dose of 93 μmol/kg reduced the total infarct volume to 23.97%.
FIGURE 7.

Neuroprotective activity of bis(propyl)-cognitin in vivo. A, representative 2,3,5-triphenyltetrazolium chloride-stained coronal brain sections from animals of the sham group (left column), vehicle-treated control group (middle column), and bis(propyl)-cognitin (B3C) (0.65 μmol/kg)-treated group (right column), respectively. The white area represents the area of infarction in the brains of stroke rats. Drug or saline was intraperitoneally administered 15 min after the onset of MCAO, and rats were decapitated 24 h later. B, infarct volume for seven sequential coronal sections from anterior to posterior in rats receiving saline (Control) or bis(propyl)-cognitin (0.65 μmol/kg) 15 min after the induction of ischemia. C, total infarct volume (left) and neurological deficit scores (right) at 24 h after reperfusion for saline-, bis(propyl)-cognitin (0.19 and 0.65 μmol/kg)-, and memantine (93 μmol/kg)-treated rats, respectively. *, p < 0.05; **, p < 0.01 compared with control. Error bars, S.E.
Bis(propyl)-cognitin Enhances LTP Induction
Based on the UFO characteristics of bis(propyl)-cognitin in antagonizing NMDA receptors, we wanted to investigate the effect of bis(propyl)-cognitin on NMDA receptor-mediated physiological function in terms of LTP. In rat hippocampal slices, HFS induced NMDA receptor-dependent LTP under control conditions that reached a peak immediately following HFS and then slowly declined over the next 1 h to 45–80% above base line, and the averaged LTP was measured to be 149 ± 3% at 60 min post-HFS (n = 6) (Fig. 8A). We next tested the effect of bis(propyl)-cognitin on LTP induction. Bis(propyl)-cognitin at a concentration of 1 μm did not change the base line but enhanced LTP significantly compared with the LTP control (n = 5, p < 0.01) (Fig. 8), and the LTP was 172 ± 4% at 60 min post-HFS. Interestingly, bis(propyl)-cognitin significantly increased LTP even at 10 μm, although the effect was lower than that at 1 μm. To compare the effect of different NMDA receptor antagonists, memantine and MK-801 were also investigated. It was found that memantine at 3 μm did not alter LTP induction. However, MK-801 at 1 μm was able to significantly inhibit LTP induction (Fig. 8B).
FIGURE 8.

Bis(propyl)-cognitin enhances LTP. A, the graph shows induction of LTP in control (●) and in the presence of bis(propyl)-cognitin (B3C), perfused for 60 min prior to HFS (○). B, statistical analyses of the effect of bis(propyl)-cognitin, memantine, and MK-801 on LTP. The agents were perfused over the slices for 60 min prior to HFS, and LTP induction was recorded. All data are the means ± S.E. of EPSPs at 60 min after HFS (n = 5). *, p < 0.05; **, p < 0.01 versus LTP control.
DISCUSSION
Excitotoxicity mediated by NMDA receptors has been thought to be the final common pathway in a variety of neurological diseases. NMDA receptor antagonists with the UFO properties and moderate affinity are proposed to be effective and well tolerated in treating these diseases (3, 11). In the current study, compared with memantine and other dimeric AChE inhibitors derived from tacrine, the characteristics of bis(propyl)-cognitin to inhibit NMDA receptors and its neuroprotection in vitro and in vivo were investigated. We mainly report three novel points; bis(propyl)-cognitin 1) is an uncompetitive NMDA receptor antagonist with fast off-rate and moderate affinity, 2) possesses superior neuroprotective activities against glutamate-induced excitotoxicity in vitro and MCAO-induced impairments in vivo, indicating that this dimer may prevent glutamate-induced pathological processes, and 3) enhances the LTP induction in hippocampal slices, suggesting that this dimer may not interfere with but even promote the NMDA receptor-mediated physiological functions.
Bis(propyl)-cognitin is an uncompetitive NMDA receptor antagonist. We have reported that bis(heptyl)-cognitin, a dimeric AChE inhibitor, is an antagonist of NMDA receptors (24). We speculate that other dimers from this series might also block NMDA receptors. Indeed, bis(n)-cognitins can selectively inhibit NMDA-activated currents with different extents of potency and voltage dependence. We wonder whether some of these dimers are NMDA receptor antagonists with the UFO properties. In comparing actions of the series of dimers on NMDA receptors, we single out bis(propyl)-cognitin as a promising UFO agent with moderate affinity based on moderate potency and strong voltage dependence. Uncompetitivity is the major determinant of safety and tolerability for NMDA receptor antagonists in clinical trials (11, 16). To test whether bis(propyl)-cognitin inhibition is competitive, non-competitive, or uncompetitive as defined by Lipton (11), we have examined the effect of increasing NMDA concentrations on bis(propyl)-cognitin inhibition. The concentration-response curve for NMDA-activated currents in the presence of bis(propyl)-cognitin is shifted downward. This means that an increase of NMDA concentration cannot overcome the inhibition of bis(propyl)-cognitin, suggesting that bis(propyl)-cognitin inhibition is not via a competitive mechanism. A further analysis of the agonist concentration dependence of bis(propyl)-cognitin inhibition by plotting the percentage inhibition against NMDA concentrations reveals that the inhibition increases with the increase of NMDA concentration, which is characteristic of an uncompetitive mechanism (10, 31). Different from memantine, which has been demonstrated to act on the NMDA receptor with a quite slow onset (32), bis(propyl)-cognitin inhibition exhibits fast onset and reaches equilibrium during its application (several seconds). This can exclude the possibility that the observed uncompetitive mechanism is an artifact caused by an inadequate time of drug application.
Bis(propyl)-cognitin inhibits NMDA receptors in an agonist-dependent manner. Because the inhibitory effect of uncompetitive antagonists is contingent upon prior activation of the receptor channel by the agonist (11), we have checked the agonist dependence of bis(propyl)-cognitin. We observe that bis(propyl)-cognitin co-applied or applied after the activation of NMDA receptors but not preapplied shows an inhibitory effect on NMDA-activated currents. This observation indicates that bis(propyl)-cognitin association with its acting site on the receptor requires the channel to be open, which is similar to the mechanism of action of memantine and MK-801, two NMDA receptor uncompetitive antagonists (31, 33). These observations strongly support the uncompetitive mechanism of action, which is further supported by the use dependence of action. Use-dependent compounds can only access their site(s) of action when the receptor/channel is in an activated state, resulting in a delay between receptor activation and the antagonist-induced blockade. Therefore, they are less likely to block short periods of activation, which typify physiological activity of NMDA receptors (34).
Bis(propyl)-cognitin inhibition has the fast off-rate property. Effective and safe NMDA receptor antagonists for the treatment of neurodegenerative disorders are required to leave the channel quickly when the membrane is depolarized under physiological conditions. To meet this criterion, the antagonist is expected to inhibit the receptor channel at negative membrane potentials and can leave the channel quickly and have no effect at depolarized membrane potentials (e.g. >−20 mV) (35). The observed strong voltage dependence of bis(propyl)-cognitin inhibition of NMDA-activated currents may meet this requirement, and more importantly, the inhibition exhibits a fast off-rate (τoff ∼2 s), which is much faster than that of MK-801 (τoff ∼90 min) (33). This mechanism of action enables the quick elimination of bis(propyl)-cognitin upon its removal or membrane depolarization, suggesting that this compound should be safer than MK-801 (16). The fast off-rate of bis(propyl)-cognitin also reflects its low affinity toward NMDA receptors. Like memantine (16, 26), the onset time constants are well correlated with bis(propyl)-cognitin concentrations, whereas the offset time constants are independent of its concentrations. The fast onset (millisecond to second time scale) of inhibition can also exclude second messenger-mediated mechanism because the G-protein-coupled cascade requires several tens of seconds for the process of signal transduction (36), supporting the idea that bis(propyl)-cognitin inhibits NMDA receptors directly.
Bis(propyl)-cognitin moderately blocks NMDA receptors at the MK-801 site. To further characterize acting site(s) of bis(propyl)-cognitin, we have applied molecular docking analysis to examine the interaction between bis(propyl)-cognitin and NMDA receptors. Bis(propyl)-cognitin exhibits favorable interaction with the channel pore of the NMDA receptor at a similar binding score of memantine. The interaction of bis(propyl)-cognitin with the channel pore region is further proved by the fact that it moderately competes with [3H]MK-801 binding to NMDA receptors. These results demonstrate that the acting site of bis(propyl)-cognitin may overlap with that of MK-801 in the channel pore of the NMDA receptor. Previous study has shown that mutation of NR1(N616R) significantly reduced the inhibitory potency of Mg2+, memantine, and MK-801 (29). By using HEK293T cells transfected with NR1(N616R)/NR2A, it is also demonstrated that the inhibitory potency of bis(propyl)-cognitin is reduced. We speculate that the mutation interferes with the binding of bis(propyl)-cognitin to the region of MK-801 binding on NMDA receptors. The interaction between bis(propyl)-cognitin and the NMDA receptors further supports the idea that the inhibition is exerted by directly modulating NMDA receptors rather than via a second messenger-mediated mechanism. Together, these results suggest that bis(propyl)-cognitin is an NMDA receptor antagonist at the MK-801 site with moderate affinity. Increasing evidence has shown that the drugs with fast kinetics in moderately inhibiting the targeted receptors and/or enzymes may have higher therapeutic significance and hardly interfere with the physiological function of the brain (35, 37, 38).
The UFO properties and moderate affinity are expected to endow bis(propyl)-cognitin with increasing protective effectiveness against escalating concentrations of glutamate-induced excitotoxicity and enable its quick relief upon depolarization associated with physiological neurotransmission without observable toxicity. Indeed, we have found that, similar to memantine but in contrast to bis(heptyl)-cognitin (24), bis(propyl)-cognitin is increasingly effective in protecting against escalating levels of glutamate-induced excitotoxicity without obvious cytotoxicity. Interestingly, a low micromolar concentration of bis(propyl)-cognitin shows more pronounced improvement in neurological scores and reduction in infarct volume in rats subjected to MCAO than does memantine (31), implying that bis(propyl)-cognitin may possess other relevant targets in vivo. Our preliminary data show that this dimer does possess selective inhibition on neuronal and inducible nitric-oxide synthase instead of endothelial nitric-oxide synthase and inhibition of voltage-gated calcium channels.4
We examined whether bis(propyl)-cognitin interferes with NMDA receptor-mediated physiological functions by investigating its effect on LTP induction in hippocampal slices. Similar to the previous report (39), MK-801 significantly reduces LTP induction, whereas memantine at a relatively low concentration has no significant effect on LTP induction, and the differential effects of MK-801 and memantine on NMDA receptor-dependent LTP have been attributed to their different affinities and voltage dependences to antagonize NMDA receptors. Under the conditions of a resting membrane potential of around −70 mV, the ion channel of the NMDA receptor is blocked by Mg2+ ions. Even if glutamate and glycine bind to the NMDA receptor, the Mg2+ ion block prevents the flow of ions (35). The Mg2+ block can be removed by depolarization of the cell membrane. In other words, the NMDA receptor only allows Ca2+ ions entering the cell if two conditions are fulfilled; the receptor must be activated by ligands, and the neuron must be depolarized (e.g. by the activation of non-NMDA receptors). An important aspect is that the Mg2+ block is reduced, even after slight depolarization of the cell membrane to around −50 mV. With the disruption of energy metabolism in neurodegenerative disorders, glutamate is not cleared properly and may even be inappropriately released; moreover, energetically compromised neurons become depolarized (more positively charged) because in the absence of energy the neurons cannot maintain their ionic homeostasis. This depolarization (around −50 mV) relieves the normal Mg2+ block of NMDA receptor-coupled channels because the relatively positive charge in the cell repels positively charged Mg2+ from the channel pore, and then excessive stimulation of NMDA receptors occurs. Recent studies suggest that NMDA receptors are overstimulated in a tonic rather than a phasic manner in several neurodegenerative disorders. Memantine is an uncompetitive and moderate antagonist of NMDA receptor with an appropriate voltage-dependent inhibition between that of Mg2+ and MK-801. This property may help memantine to inhibit the activation of NMDA receptors caused by a tonic and low concentration (50–100 μm) glutamate under relatively low depolarized conditions (about −50 mV; i.e. the aforementioned pathological conditions), which is closely involved in neuronal damage and deficits in synaptic plasticity, but not to interfere with the activation of NMDA receptors caused by a phasic and high concentration (10 mm) glutamate under high depolarized conditions (about −20 mV), which is closely involved in physiological functions of glutamate and LTP formation in synaptic plasticity (17, 35). As expected, bis(propyl)-cognitin should not inhibit LTP induction because bis(propyl)-cognitin has a profile similar to that of memantine for inhibition of NMDA receptors. However, to our surprise, bis(propyl)-cognitin is observed to significantly enhance the LTP induction at 1 μm and without inhibition even at 10 μm. This may be attributed to the increase of acetylcholine levels caused by AChE inhibition by bis(propyl)-cognitin because previous studies have reported that the hippocampus synaptic transmission is enhanced by the activation of α7 nACh receptors linked with a rise in intracellular calcium and transmitter release (40, 41). On the other hand, bis(propyl)-cognitin has also been reported to inhibit GABAA receptors (20), which may additionally contribute to the facilitation of LTP (21–23). LTP is widely considered to be one of the major cellular mechanisms underlying learning and memory. The enhancement of LTP by bis(propyl)-cognitin suggests that this dimer might provide substantial therapeutic potentials for cognitive impairments associated with neurodegenerative disorders.
Increasing evidence has shown that multifactorial etiopathogenesis is clearly indicated in neurodegenerative disorders, and multiple-drug therapy is required to address the varied pathological aspects of these diseases (42). Current approaches with one drug-one target only offer limited and transient benefits to patients and do not significantly delay the course of neurodegeneration (43). Multifunctional compounds might provide greater therapeutic efficacy by targeting different sites in the brain concurrently (43, 44). Therefore, new one drug-multiple target approaches have been developed expressly to target multiple sites in the central nervous system with single molecular entities for the treatment of these diseases (42). Combining these novel findings with our previous results (19, 20), we conjecture that over the same concentration range (low to submicromolar range), bis(propyl)-cognitin concurrently possesses anti-NMDA receptors, anti-GABAA receptors, and anti-AChE activities. The synergism may provide bis(propyl)-cognitin as a promising PAT drug candidate for effective prevention and treatment and even modification of the pathological processes of neurodegenerative disorders as well as the alleviation of the cognitive impairments with fewer side effects.
Acknowledgments
We thank Dr. Anne Stephenson for providing the cNDAs of NR1, NR2A, and NR2B and Wei Zhang for technical support.
This work was supported by grants from the Research Grants Council of Hong Kong (PolyU6608/07M, 5609/09M; N_PolyU618/07; AoE/B15/01-II), Hong Kong Polytechnic University (G-YX96, G-U439), the National Nature Science Foundation of China (30770684), Zhejiang (Y207193), the Ministry of Science and Technology of China (2008CB517411), and the Shenzhen Shuangbai Scheme 2008.
J. Luo, W. Li, and Y. Han, unpublished data.
- NMDA
- N-methyl-d-aspartate
- PAT
- pathologically activated therapeutic
- UFO
- uncompetitive NMDA receptor antagonist(s) with fast off-rate
- AChE
- acetylcholinesterase
- GABAA
- γ-aminobutyric acid subtype A
- LTP
- long-term potentiation
- MCAO
- middle cerebral artery occlusion
- CGN
- cerebellar granule neuron
- AMPA
- α-amino-3-hydroxy-5-methyl-4-isoxazole propionate
- MTT
- 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- EPSP
- excitatory postsynaptic potential
- HFS
- high frequency stimulation.
REFERENCES
- 1.Hynd M. R., Scott H. L., Dodd P. R. (2004) Neurochem. Int. 45, 583–595 [DOI] [PubMed] [Google Scholar]
- 2.Waxman E. A., Lynch D. R. (2005) Neuroscientist 11, 37–49 [DOI] [PubMed] [Google Scholar]
- 3.Lipton S. A., Rosenberg P. A. (1994) N. Engl. J. Med. 330, 613–622 [DOI] [PubMed] [Google Scholar]
- 4.Meldrum B., Garthwaite J. (1990) Trends Pharmacol. Sci. 11, 379–387 [DOI] [PubMed] [Google Scholar]
- 5.Petrović M., Horák M., Sedlácek M., Vyklický L., Jr. (2005) Prague Med. Rep. 106, 113–136 [PubMed] [Google Scholar]
- 6.Villmann C., Becker C. M. (2007) Neuroscientist 13, 594–61517911221 [Google Scholar]
- 7.Klimaviciusa L., Safiulina D., Kaasik A., Klusa V., Zharkovsky A. (2008) Neurotoxicology 29, 101–108 [DOI] [PubMed] [Google Scholar]
- 8.Takadera T., Matsuda I., Ohyashiki T. (1999) J. Neurochem. 73, 548–556 [DOI] [PubMed] [Google Scholar]
- 9.Tai K. K., Blondelle S. E., Ostresh J. M., Houghten R. A., Montal M. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 3519–3524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lipton S. A. (2004) NeuroRx 1, 101–110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lipton S. A. (2007) Nat. Rev. Neurosci. 8, 803–808 [DOI] [PubMed] [Google Scholar]
- 12.Stieg P. E., Sathi S., Warach S., Le D. A., Lipton S. A. (1999) Eur. J. Pharmacol. 375, 115–120 [DOI] [PubMed] [Google Scholar]
- 13.Hare W., WoldeMussie E., Lai R., Ton H., Ruiz G., Feldmann B., Wijono M., Chun T., Wheeler L. (2001) Surv. Ophthalmol. 45, Suppl. 3, S284–S289 [DOI] [PubMed] [Google Scholar]
- 14.Lipton S. A. (2005) Curr. Alzheimer Res. 2, 155–165 [DOI] [PubMed] [Google Scholar]
- 15.Planells-Cases R., Montoliu C., Humet M., Fernández A. M., García-Martínez C., Valera E., Merino J. M., Pérez-Payá E., Messeguer A., Felipo V., Ferrer-Montiel A. (2002) J. Pharmacol. Exp. Ther. 302, 163–173 [DOI] [PubMed] [Google Scholar]
- 16.Lipton S. A. (2004) J. Alzheimers Dis. 6, S61–74 [DOI] [PubMed] [Google Scholar]
- 17.Lipton S. A. (2006) Nat. Rev. Drug Discov. 5, 160–170 [DOI] [PubMed] [Google Scholar]
- 18.Li W. M., Kan K. K., Carlier P. R., Pang Y. P., Han Y. F. (2007) Curr. Alzheimer Res. 4, 386–396 [DOI] [PubMed] [Google Scholar]
- 19.Carlier P. R., Han Y. F., Chow E. S., Li C. P., Wang H., Lieu T. X., Wong H. S., Pang Y. P. (1999) Bioorg. Med. Chem. 7, 351–357 [DOI] [PubMed] [Google Scholar]
- 20.Li C., Carlier P. R., Ren H., Kan K. K., Hui K., Wang H., Li W., Li Z., Xiong K., Clement E. C., Xue H., Liu X., Li M., Pang Y., Han Y. (2007) Neuropharmacology 52, 436–443 [DOI] [PubMed] [Google Scholar]
- 21.Giorgetti M., Bacciottini L., Giovannini M. G., Colivicchi M. A., Goldfarb J., Blandina P. (2000) Eur. J. Neurosci. 12, 1941–1948 [DOI] [PubMed] [Google Scholar]
- 22.Matsuyama S., Taniguchi T., Kadoyama K., Matsumoto A. (2008) Neuroreport 19, 1809–1813 [DOI] [PubMed] [Google Scholar]
- 23.Yoshiike Y., Kimura T., Yamashita S., Furudate H., Mizoroki T., Murayama M., Takashima A. (2008) PloS one 3, e3029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li W., Pi R., Chan H. H., Fu H., Lee N. T., Tsang H. W., Pu Y., Chang D. C., Li C., Luo J., Xiong K., Li Z., Xue H., Carlier P. R., Pang Y., Tsim K. W., Li M., Han Y. (2005) J. Biol. Chem. 280, 18179–18188 [DOI] [PubMed] [Google Scholar]
- 25.Cheng X. P., Qin S., Dong L. Y., Zhou J. N. (2006) Neurosci. Res. 55, 142–145 [DOI] [PubMed] [Google Scholar]
- 26.Chen H. S., Lipton S. A. (1997) J. Physiol. 499, 27–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y., Wang L., Li J., Wang X. L. (2006) J. Pharmacol. Exp. Ther. 317, 973–979 [DOI] [PubMed] [Google Scholar]
- 28.Bederson J. B., Pitts L. H., Tsuji M., Nishimura M. C., Davis R. L., Bartkowski H. (1986) Stroke 17, 472–476 [DOI] [PubMed] [Google Scholar]
- 29.Kashiwagi K., Masuko T., Nguyen C. D., Kuno T., Tanaka I., Igarashi K., Williams K. (2002) Mol. Pharmacol. 61, 533–545 [DOI] [PubMed] [Google Scholar]
- 30.Arundine M., Tymianski M. (2004) Cell. Mol. Life Sci. 61, 657–668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen H. S., Pellegrini J. W., Aggarwal S. K., Lei S. Z., Warach S., Jensen F. E., Lipton S. A. (1992) J. Neurosci. 12, 4427–4436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gilling K. E., Jatzke C., Parsons C. G. (2007) Neuropharmacology 53, 415–420 [DOI] [PubMed] [Google Scholar]
- 33.Huettner J. E., Bean B. P. (1988) Proc. Natl. Acad. Sci. U.S.A. 85, 1307–1311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clements J. D., Lester R. A., Tong G., Jahr C. E., Westbrook G. L. (1992) Science 258, 1498–1501 [DOI] [PubMed] [Google Scholar]
- 35.Parsons C. G., Stöffler A., Danysz W. (2007) Neuropharmacology 53, 699–723 [DOI] [PubMed] [Google Scholar]
- 36.Krasel C., Vilardaga J. P., Bünemann M., Lohse M. J. (2004) Biochem. Soc. Trans. 32, 1029–1031 [DOI] [PubMed] [Google Scholar]
- 37.Rogawski M. A. (2000) Amino Acids 19, 133–149 [DOI] [PubMed] [Google Scholar]
- 38.Parsons C. G., Quack G., Bresink I., Baran L., Przegalinski E., Kostowski W., Krzascik P., Hartmann S., Danysz W. (1995) Neuropharmacology 34, 1239–1258 [DOI] [PubMed] [Google Scholar]
- 39.Frankiewicz T., Potier B., Bashir Z. I., Collingridge G. L., Parsons C. G. (1996) Br. J. Pharmacol. 117, 689–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gray R., Rajan A. S., Radcliffe K. A., Yakehiro M., Dani J. A. (1996) Nature 383, 713–716 [DOI] [PubMed] [Google Scholar]
- 41.Ji D., Lape R., Dani J. A. (2001) Neuron 31, 131–141 [DOI] [PubMed] [Google Scholar]
- 42.Cavalli A., Bolognesi M. L., Minarini A., Rosini M., Tumiatti V., Recanatini M., Melchiorre C. (2008) J. Med. Chem. 51, 347–372 [DOI] [PubMed] [Google Scholar]
- 43.Youdim M. B., Buccafusco J. J. (2005) Trends Pharmacol. Sci. 26, 27–35 [DOI] [PubMed] [Google Scholar]
- 44.Zhao X., Marszalec W., Toth P. T., Huang J., Yeh J. Z., Narahashi T. (2006) Neuropharmacology 51, 1181–1191 [DOI] [PubMed] [Google Scholar]