

Abstract
Carriers of germ line mutations in breast cancer susceptibility gene BRCA1 have an increased risk of developing breast and ovarian cancers; missense mutations have, however, been difficult to assess for disease association. Here we have used a biophysical approach to classify these variants. We established an assay for measuring the thermodynamic stability of the BRCA1 BRCT domains and investigated the effects of 36 missense mutations. The mutations show a range of effects. Some do not change the stability, whereas others destabilize the protein by as much as 6 kcal mol−1; one-third of the mutants could not be expressed in soluble form in Escherichia coli, and we conclude that these destabilize the protein by an even greater amount. We tested several computer algorithms for their ability to predict the mutant effects and found that by grouping them into two classes (destabilizing by less than or more than 2.2 kcal mol−1), the algorithms could predict the stability changes. Importantly, with the exception of the few mutants located in the binding site, none showed a significant reduction in affinity for phosphorylated substrate. These results indicate that despite very large losses in stability, the integrity of the structure is not compromised by the mutations. Thus, the majority of mutations cause loss of function by reducing the proportion of BRCA1 molecules that are in the folded state and increasing the proportion of molecules that are unfolded. Consequently, small molecule stabilization of the structure could be a generally applicable preventative therapeutic strategy for rescuing many BRCA1 mutations.
Keywords: Breast Cancer, Protein Denaturation, Protein Domains, Protein Folding, Protein Stability, BRCA1, BRCT, Breast Cancer Susceptibility Gene Product, Mutation
Introduction
Carriers of germ line mutations in BRCA1 have an increased lifetime risk of developing breast and ovarian cancers, and mutations in the BRCA1 gene account for 80% of all familial breast and ovarian cancer cases (1, 2). BRCA1 encodes a large protein of 1863 residues with only two small structural motifs characterized to date (3). At the C terminus is a repeat of two BRCT domains. BRCT domains are found in proteins involved in DNA repair and maintenance of genomic stability, and more recently, the BRCT repeat has been recognized as a phosphopeptide-binding domain (4, 5). At the N terminus is a RING finger domain, which, by binding to BARD1 (another RING and BRCT domain-containing protein), gives the BRCA1 protein a ubiquitin ligase activity (6, 7). BRCA1 interacts with many other proteins, and based on these associations, it has been implicated in a variety of functions, which include DNA damage response, maintenance of genomic stability, and transcription regulation (8, 9).
There are a number of databases that collate information on mutations in proteins: for example, the Single Nucleotide Polymorphism database (dbSNP) and the Breast Cancer Information database (BIC).2 Although it is easy to classify variants that result in large truncations as being deleterious to function (and therefore disease-associated), missense mutations typically remain unclassified. Thus, the BIC database currently contains 108 missense mutations in the BRCT domains of BRCA1, but only 7% of them have been classified. These missense mutations may be either polymorphisms or mutations predisposing the carrier to cancer progression.
It is important to understand the molecular basis of BRCA1 inactivation, but the functions of BRCA1 that are critical for tumor suppression are still not fully characterized, and there is therefore no single functional assay available that encompasses the breadth of BRCA1 function. Moreover, it has been predicted for proteins in general that the vast majority of disease-associated missense mutations cause loss of function in an indirect way by destabilizing the three-dimensional structure, rather than directly by disrupting a binding site or active site (an effect that would be restricted to a comparatively small number of residues (10)). It would therefore be useful to have an assay for BRCA1 that measures the structural stability of the protein so that the effects of mutations can be determined and disease risk thereby assessed. We have focused, to begin with, on the BRCT repeat of BRCA1. The reasons are: first, a number of studies have indicated that the BRCT domains are critical for tumor suppression; second, many mutations in BRCA1 are located in the BRCT domains; third, the structure of the BRCT repeat is known, and therefore, we can relate our experimental results to the location of the mutations in the structure; finally, we can look at whether computer algorithms, which require a structure, are able to predict these effects.
Here we establish a reliable assay for measuring the thermodynamic stability of the BRCA1 BRCT domain structure. We analyzed the effects on stability and on phosphopeptide binding of 36 missense mutations selected from the BIC database. Mutations were chosen so as to include different types of amino acid substitutions and at sites having a range of different local structural environments. We found that the effects of the mutations were varied, ranging from slightly stabilizing to destabilizing by up to 6 kcal mol−1. There was also a subset of mutant proteins that could not be expressed in a soluble form in Escherichia coli, and we conclude that these mutants are destabilized to an even great extent. By extrapolating our measurements of stability, made at a lower temperature, to physiological conditions, we conclude that the majority of the mutations will result in a significant amount of unfolding of the protein in the cell. Despite the large changes in stability, all of the solubly expressed mutants (with the exception of those located in the binding site) were able to bind a phosphorylated peptide with near wild-type affinities (at the lower temperature), indicating that the mutations do not induce a misfolded conformation. The finding that the native structure is preserved on mutation leads us to propose that using small molecules to stabilize the structure would be a therapeutic approach that could be applied as a preventive strategy to rescue a large number of the mutants. Finally, we compared our experimental findings with the stability changes calculated using various computer algorithms. We found that by using only two broad categories (destabilizing by less than or more than 2.2 kcal mol−1), the algorithms were able to predict the stability changes on mutation.
EXPERIMENTAL PROCEDURES
Protein Expression and Purification
The BRCA1 BRCT region encoding amino acids 1646–1863 of the full-length protein was cloned into a modified pRSET(A) plasmid (Invitrogen) in which the His tag has been replaced with a glutathione S-transferase tag. The mutations were introduced using the QuikChange site-directed mutagenesis kit (Stratagene) and confirmed by sequencing.
The BRCA1 BRCT construct was expressed in E. coli C41(DE3) (11) and grown in 2TY containing 50 μg/ml ampicillin at 37 °C to an optical density of ∼0.6 before overnight induction of expression with 0.1 mm isopropyl-β-d-thiogalactoside at 25 °C. The mutant proteins were expressed in an identical manner except that the temperature after induction was lowered to 20 °C to aid soluble expression. The protein was purified by affinity chromatography followed by anion exchange after cleavage of the glutathione S-transferase tag. The purified protein was supplemented with 1 mm dithiothreitol and frozen and stored at −80 °C. The proteins were judged >95% pure by SDS-PAGE and mass spectrometry. Protein concentration was determined spectrophotometrically using a theoretical extinction coefficient of 37,770 cm−1 M−1 (12). The oligomeric status was determined by analytical size-exclusion chromatography on an S200 HR10/30 (GE Healthcare). All of the solubly expressed BRCA1 BRCT variants eluted with a single symmetrical, monomeric peak at a volume expected for the molecular weight of the protein.
Equilibrium Denaturation
Aliquots of guanidinium hydrochloride (GdmCl) were prepared by dispensing the appropriate volumes of concentrated stock solutions of GdmCl in buffer (50 mm Hepes buffer, pH 8.5, 1 mm dithiothreitol) and buffer alone using a Hamilton Microlab M. Protein stock in buffer was then added to a final concentration of 0.5 μm. The samples were incubated at 10 °C for 2 h prior to measurement. Fluorescence was recorded on a PerkinElmer Life Sciences LS55 luminescence spectrophotometer. The excitation wavelength was 280 nm, and excitation and emission slit widths were 2.5 nm. Fluorescence was measured between 315 and 390 nm at a scan speed of 1 nm/s. The temperature in the cell was maintained at 10 °C using a water bath and was monitored using an external Edale thermocouple.
The denaturation curves obtained by plotting the fluorescence at 1 nm intervals between 320 and 380 nm were fitted globally. The following equation assumes a three-state model in which the fluorescence intensity of the folded state (F) and unfolded state (UN), FF and FUN, respectively, have a linear dependence on denaturant concentration, but the fluorescence intensity of the intermediate (I), FI, does not.
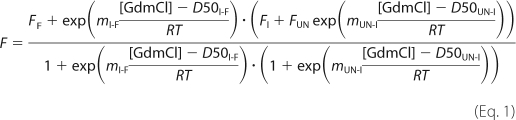 |
where m is a constant that is proportional to the increase in solvent-accessible surface area between the two states involved in the transition, D50I-F and mI-F are the midpoint and m value, respectively, for the transition between the folded state and the intermediate, and D50UN-I and mUN-I are the midpoint and m value, respectively, for the transition between I and the unfolded state, T is the absolute temperature, and R is the universal gas constant. For each protein (wild type or mutant), the data at the different wavelengths were globally fitted to this equation using GraphPad Prism 4.0 with shared m values and midpoints and no constraints on the other parameters. The data were then refitted using the average values of m determined for wild type and all mutants; these were calculated to be 4.5 ± 0.09 kcal mol−1 m−1 for the transition between the folded state and the intermediate and 1.3 ± 0.04 kcal mol−1 m−1 for the transition between the intermediate and the unfolded state.
Extrapolation of the Free Energy of Unfolding to Physiological Temperature
To determine the free energy of unfolding at 37 °C by extrapolation of measurements made at lower temperatures, each of the two unfolding transitions needs to be considered separately as each will have its own change in heat capacity on unfolding (ΔCp), melting temperature (Tm), and change in the enthalpy on unfolding (ΔH). For each transition, the plot of the free energy of unfolding versus temperature was then fitted to Equation 2
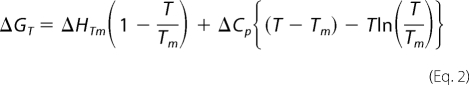 |
The thermodynamic stabilities of the mutants at 37 °C were then determined by subtracting the values for the change on mutation in the free energy of unfolding between the folded and intermediate states and between the intermediate and unfolded states (determined at the lower temperature of 10 °C) from the respective free energy change of each transition for wild type at 37 °C. It has been shown that the change in the free energy of unfolding on mutation does not change significantly with temperature (13).
The fraction of molecules that are in the folded state (ff) at 37 °C was then calculated using the following equation
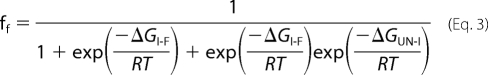 |
where ΔGI-F is the free energy change for the first unfolding transition between F and I, and ΔGUN-I is the free energy change for the second unfolding transition between I and U, T is the absolute temperature, and G is the universal gas constant. The fractions of molecules in the intermediate and denatured states can also be obtained using similar equations.
Fluorescence Anisotropy
Fluorescence anisotropy measurements were recorded at 25 °C on a PerkinElmer Life Sciences LS55 luminescence spectrophotometer equipped with a Hamilton Microlab titrator controlled by laboratory software. Excitation and emission wavelengths were 480 and 530 nm, respectively, and excitation and emission slit widths were 10 nm. The fluorescently labeled phosphopeptide VNKpSYFND-fluorescein (Pepceuticals Ltd.) was used at a concentration of 20 nm in anisotropy buffer (50 mm Tris-HCl, pH 7.5, 0.15 m NaCl, 2 mm dithiothreitol, 1 mm EDTA, 0.01% (w/v) Igepal CA-630). Wild-type or mutant BRCA1 BRCT was used as titrant in anisotropy buffer at a concentration of between 10 and 315 μm. After each addition of protein, the solution was stirred for 30 s, and 30 s later, the fluorescence and fluorescence polarization values were recorded. The data were fitted to a single-site binding model
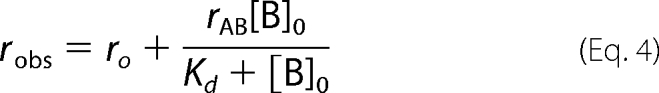 |
where ro is the anisotropy value for the free fluorescein-labeled phosphopeptide, rAB is the change in anisotropy on complex formation, and [B]0 is the concentration of BRCA1 BRCT.
RESULTS
E. coli Expression of the BRCA1 BRCT Variants
The construct used in this study corresponds to residue 1646 to residue 1863 (C terminus of the protein) of BRCA1 and comprises the two BRCT domains (referred to subsequently as BRCA1 BRCT). We expressed wild-type and mutant BRCA1 BRCT in E. coli. 13 of the 36 mutants were found to be expressed exclusively in the inclusion body fraction, even when the growth and expression conditions were varied by, for example, lowering the temperature or the concentration of isopropyl-β-d-thiogalactoside used for induction of protein expression. We also attempted to refold the inclusion bodies by a number of different methods, but we were not able to produce correctly folded, monomeric proteins. These mutants were D1692Y, A1708E, S1715C/S1715N/S1715R (i.e. mutants in which Ser at position 1715 was changed to Cys or Asn or Arg), L1764P, I1766S, L1780P, V1833M, W1837G/W1837R, and S1841N. The following observations lead us to conclude that inclusion body formation arises from low stability. First, all of the proteins, including wild type, showed some inclusion body expression (for the wild type, ∼80% of the protein was in the soluble fraction); when we measured the stabilities of the soluble fractions of the mutants, there was a correlation between the extent of inclusion body formation and reduced stability. Second, a few of the mutants expressed exclusively as inclusion bodies on initial attempts, but we were subsequently able to obtain some soluble protein by lowering the induction temperature. These mutants were more unstable than the solubly expressing mutants. We classed the 13 mutants as highly unstable, and no further work was carried out on them.
Comparison of Thermodynamic Stability of Wild-type and Mutant BRCA1 BRCT
BRCA1 BRCT has 5 tryptophan residues (2 in the first BRCT repeat and 3 in the second BRCT repeat), and their fluorescence was used to monitor the unfolding of the protein. As shown previously by Ekblad et al. (14), the BRCT domains unfold via an intermediate species that is aggregation-prone. A range of conditions was tested to minimize this effect. We found that there was no aggregation when the temperature was lowered to 10 °C and using the buffer 50 mm Hepes, pH 8.5. Although the conditions are different from those used by Ekblad et al. (14), the unfolding profiles obtained under the two conditions appear similar (Fig. 1A). Unfolding occurs in two stages, with a large increase in fluorescence associated with the transition from the folded state to an intermediate, partly folded state and a smaller decrease in fluorescence between the intermediate and the denatured state. The free energy of unfolding in the absence of guanidinium chloride was calculated to be 10.56 ± 0.27 kcal mol−1 at 10 °C (see “Experimental Procedures” for data analysis).
FIGURE 1.

Guanidinium hydrochloride-induced unfolding of BRCA1 BRCT domain curves. Measurements were made at 10 °C in 50 mm Hepes buffer, pH 8.5, containing 1 mm dithiothreitol. The protein concentration was 0.5 μm. The fluorescence emission at 340 nm is shown. A, wild type (□) and variants representative of the four groups (categorized according to their effects on the stability) are shown. ▵, group 1, not destabilizing, R1669Q; X, group 2, mildly destabilizing, D1692N; ○, group 3, moderately destabilizing, M1783T; and ●, group 4, very destabilizing, Y1853C. B, effect of phosphopeptide binding on BRCA1 BRCT stability. Wild type (■) and the moderately destabilizing mutant G1788D (●) are shown in the absence of peptide and for G1788D in the presence of 25 μm fluorescein-labeled phosphopeptide (○).
All of the mutant proteins displayed denaturation profiles that were similar in appearance to that of the wild-type protein, and the data could be fitted in the same way as that described for the wild type. None of the mutations resulted in a change in the m value (a constant that is proportional to the increase in solvent-exposed surface area upon unfolding) of either of the unfolding transitions by more than 10%, suggesting that the unfolding mechanism was unchanged. We therefore refitted the data for each mutant, fixing the m values of the two transitions to the average values calculated using wild type and all of the mutants (4.5 ± 0.09 kcal mol−1 m−1 for the first transition and 1.3 ± 0.04 kcal mol−1 m−1 for the second transition).
The effects of the mutations are shown in Fig. 2 and supplemental Table S1. The majority of the solubly expressed mutant proteins (17 of the 23) were found to be destabilizing. Only two mutations stabilized the protein by more than 1 kcal mol−1, and seven mutations changed the stability by less than 1 kcal mol−1 (Fig. 2). The m value is larger for the first transition than for the second, indicating that the loss of structure is greater in the first unfolding transition than in the second; the changes in the free energy of unfolding on mutation are much larger for the first transition than for the second (Fig. 2), consistent with a greater loss of structure.
FIGURE 2.

Effect of mutations on the thermodynamic stability of BRCA1 BRCT domains. The dark gray bars show the change upon mutation in the free energy of unfolding between the folded and unfolded states. The change upon mutation in the free energy of unfolding between the folded state and the intermediate is shown in light gray, and the change upon mutation between the intermediate and the unfolded state is shown in white. Error bars are ± S.D. from repeat measurements.
The mutants were classified into four groups according to the effect on the stability: 1) “not destabilizing,” when the free energy of unfolding is either the same as or greater than that of wild type; 2) “mildly destabilizing,” mutations that decrease the free energy of unfolding by up to 2.2 kcal mol−1; 3) “moderately destabilizing,” mutations that decrease the free energy of unfolding by 2.2–5 kcal mol−1; 4) “very destabilizing,” mutations that decrease the free energy of unfolding by more than 5 kcal mol−1 or that were insoluble when expressed in E. coli. The mutants in group 1 (not destabilizing) are M1663K, M1663L, A1669S, R1699Q, R1699L, C1787S, and P1806A. The majority of these mutants are conservative changes in terms of charge and size and are located at sites on the surface of the protein. However, two conservative mutations (A1669S and C1787S) are at sites that are buried in the hydrophobic core (Fig. 3A). The results suggest that regions of the BRCT core contain sufficient flexibility to accommodate small changes in the side chain without having a destabilizing effect.
FIGURE 3.

Location of the mutations in the BRCA1 BRCT structure, grouped according to their effects on thermodynamic stability. The Protein Data Bank (PDB) code 1JNX (37) was used. The N-terminal BRCT is colored olive, the C-terminal BRCT is colored blue, and the linker is colored purple. The sites at which we have analyzed a mutation are shown in red. The mutations are as follows: A, not destabilizing; in B, mildly destabilizing; C, moderately destabilizing; D, very destabilizing.
Fig. 3B shows the locations of the mildly destabilizing (group 2) mutants M1652I, L1664P, V1665M, D1692N, G1706A, R1751Q, T1773S, M1775R, D1778N, and G1788V. With the exception of G1706A and M1652I, all are at solvent-exposed positions that are likely to have the flexibility to accommodate changes in side-chain size or changes in charge.
The majority of the moderately destabilizing (group 3) mutants (R1699W, V1736A, M1783T, G1788D, V1808A, and A1843P) are non-conservative changes in the side chain and are buried in the interface between the N- and C-terminal BRCT domains, indicating that the interface does not have the flexibility to accommodate large changes in side-chain size and charge (Fig. 3C). V1736A and V1808A are more conservative mutations than the others, but they are located at sites buried in the hydrophobic core and therefore would be expected to have a significant destabilizing effect.
The very destabilizing (group 4) mutants are D1692Y, A1708E, and S1715C/S1715N/S1715R (in the N-terminal BRCT) and L1764P, I1766S, L1780P, V1833M, W1837G/W1837R, S1841N, and Y1853C (in the C-terminal BRCT). All except for D1692Y are at sites buried in the hydrophobic core (Fig. 3D), and therefore, it is not surprising that these non-conservative mutations cause the greatest destabilization of the structure. Two variants, A1708E and L1780P, are buried in the interface of the BRCT domains. The equivalent positions to the C-terminal sites Val-1833, Trp-1837, Ser-1841, and Tyr-1853 in the N-terminal BRCT domain are Trp-1718, Ser-1722, Val-1713, and Phe-1734, and they should be likewise important for stability; indeed mutations at all of these sites have been recorded in the BIC database. Ser-1715 is a buried residue in β-strand 4, the same structural element in which Val-1833 is located in the C-terminal BRCT domain. All of the three mutations in the BIC data base at Ser-1715 resulted in expression in inclusion bodies, even the relatively conservative mutant S1715C, suggesting that Ser-1715 is crucial for the packing of the hydrophobic core. Therefore, it might be expected that mutation of the analogous position in the C-terminal BRCT domain, Arg-1835, would also be detrimental; however, currently there are no reported variants in BIC at this position.
Mutant BRCA1 BRCT Domains Bind to Phosphopeptide with Near Wild-type Affinity
Tandem BRCT domains have been shown to bind phosphopeptides at a site located between the two repeats. The BRCT domains of BRCA1 have been found to bind the phosphorylated proteins BACH1 and CtIP (15, 16). Rodriguez et al. (17) showed for tandem BRCT domains from several proteins that there was selectivity in phosphopeptide substrate binding. The phosphopeptide used here, VNKpSYFND, was the one that was optimized for binding to BRCA1 BRCT in the Rodriguez study; it was labeled at its C terminus with fluorescein to monitor binding by fluorescence anisotropy. The dissociation constant, Kd, for the binding of wild-type BRCA1 BRCT to the phosphopeptide was 112 ± 6 nm. A competition experiment using unlabeled peptide gave a similar value for the Kd, indicating that the binding affinity was not affected significantly by the presence of the fluorescein label.
We characterized one binding-site residue in our study, Arg-1699. The crystal structure shows that the side chain of Arg-1699 forms part of the binding pocket for the phosphopeptide and that it interacts with the +3 phenylalanine of the peptide in a charge-dependent manner (16). The mutants R1699L and R1699Q, which are both stabilizing, either remove or reverse the charge of the side chain, and both reduce the peptide binding affinity by about 100-fold. The destabilizing mutant R1699W results in an increase in bulk of the side chain as well as removing the charge, and the peptide binding affinity is reduced ∼300-fold when compared with wild type (Fig. 4A). These changes in affinity indicate that the mutations R1699L/R1699Q/R1699W would all be detrimental to BRCA1 function.
FIGURE 4.

The effects of mutation on phosphopeptide binding. Kd for wild-type and mutant BRCA1 BRCT binding to phosphorylated peptide determined by fluorescence anisotropy measurements. A, effect of mutations at residue Arg-1699 located in the phosphopeptide-binding site. B, effect of mutations at other sites. The mutations are shaded according to their effects on stability: white, not destabilizing; gray, mildly destabilizing; black, moderately destabilizing. Error bars are ± S.D. from repeat measurements.
When the other mutations are viewed according to the four groups based on their thermodynamic stability, the majority of those classed as not destabilizing have Kd values similar to that of wild type. The mutations that were mildly destabilizing decrease the affinity ∼1.4-fold. One of these is the conservative mutation T1773S, located at a site that forms part of the wall of the hydrophobic binding pocket. Two others, R1751Q and G1706A, are at the interface between the two BRCT domains, which forms the binding pocket for the peptide. D1692N is located distant from the binding site. D1778N, in the interface between repeats but not directly in contact with the peptide, increased the affinity for the peptide ∼1.5-fold (Fig. 4B).
The mutants that fell into the moderately destabilizing class did not reduce the binding affinity more than 1.5-fold, with one exception. V1736A reduced the binding affinity ∼5-fold. This residue, although located distant from the binding site, is in the linker region, which is known to be important for binding, and so changes in stability at this site may have a long range effect on the binding site (Fig. 4B).
The Majority of Missense Mutations Will Result in Large Proportions of Unfolded Protein under Physiological Conditions
To gain a better indication of the effect of mutation-induced destabilization in the context of the cell, we need to determine the thermodynamic stability under conditions that are close to the physiological ones. Previous studies have shown, however, that at near physiological temperatures, BRCA1 BRCT is aggregation-prone and unfolds irreversibly, which therefore prevents direct measurement of stability at 37 °C (14). Instead, we determined the stability of wild-type BRCA1 BRCT at five temperatures between 10 and 25 °C (supplemental Fig. S1), and the temperature dependence of the free energy changes for each of the two unfolding transitions was fitted to Equation 2 (see “Experimental Procedures”). By extrapolation of these curves, the stability of wild type at 37 °C was determined to be 2.9 kcal mol−1. The knowledge of the effect on stability of the missense mutations measured at 10 °C allows us to estimate the stabilities of the mutants, and the fraction of molecules that are folded (using Equation 3), at physiological temperature (Fig. 5). For the variants classified as mildly destabilizing, a significant proportion of molecules are not in the folded state at 37 °C (between 25 and 80%). For the moderately and very destabilizing mutants, less than 10% of molecules are folded at 37 °C.
FIGURE 5.

Effect of missense mutations on the amount of protein that is folded at 37 °C. The fractions of BRCA1 BRCT molecules in the folded (light gray), intermediate (white), and unfolded states (dark gray) at 37 °C are given. The data are grouped according to their effects on stability: group 1 (not destabilizing); group 2 (mildly destabilizing); group 3 (moderately destabilizing); group 4 (very destabilizing). Error bars are ± S.D. from repeat measurements.
Comparison of the Effects of Mutation on Thermodynamic Stability Measured by Experiment and by Prediction Programs
A number of computational methods have been developed to predict the effects of missense mutations on the structural stability of proteins. Here we have compared the predictions made by the programs FoldX (18), SDM (19), I-Mutant 2.0 (20), ERIS (21), PolyPhen (22), SIFT (23), and DFIRE (24) with those obtained from our experimental measurements (supplemental Table S1). All of the programs except PolyPhen and SIFT generate results as free energies. Several of the methods are trained on a data set of site-directed mutants from protein-folding studies, and these are usually conservative mutations, unlike the non-conservative mutations that frequently result from single nucleotide polymorphisms. Other methods such as SDM use substitution tables produced by sequence alignments. It has been suggested, from the study of missense mutations for which the phenotypes are known, that a decrease in free energy of unfolding of ∼2 kcal mol−1 will not impair the ability of the protein to function in vivo (25). We therefore divided the experimental and computational data into two broad classes, those that destabilized the protein by more than or less than 2.2 kcal mol−1, and then counted the number of mutations that were correctly classified by each program (Fig. 6A). For the two programs that do not generate results as free energies, the mutants were grouped according to whether they were predicted to be benign (SIFT)/neutral (PolyPhen) or deleterious (SIFT)/possibly or probably damaging (PolyPhen). DFIRE and I-Mutant 2.0 were better at predicting the mildly destabilizing mutations than the highly destabilizing mutations, whereas the opposite was the case for SDM, PolyPhen, and SIFT (Fig. 6A). In Fig. 6B, we divided the mutations into a greater number of groups (stabilizing, destabilizing by 0–1, 1–2, 2–3, 3–4, 4–5, 5–6, and >6 kcal mol−1) and plotted the number of mutations that fell into each group. The plot shows that for the mutants in our study, I-Mutant 2.0 tends to underestimate the destabilizing effects of the mutations and that SDM and DFIRE underestimate the effects of the most destabilizing mutations.
FIGURE 6.

Comparison of experimental results and prediction methods. A, the mutations are divided into two groups according to whether they were destabilizing by less than or more than 2.2 kcal mol−1 (as determined by experiment). The prediction methods (FoldX, DFIRE, SDM, ERIS, I-Mutant 2.0, SIFT, and PolyPhen) were then scored for each mutant depending on whether they fell into the correct category. B, the mutations are grouped according to the changes in stability: stabilizing mutations are in gray; all the other bands are destabilizing by: 0–1 kcal mol−1 (vertical stripes), 1–2 kcal mol−1 (black), 2–3 kcal mol−1 (horizontal stripes), 3–4 kcal mol−1 (hatched), 4–5 kcal mol−1 (right diagonal stripes), 5–6 kcal mol−1 (left diagonal stripes), and >6 kcal mol−1 (including those variants that were expressed in the insoluble fraction) (white).
Phosphopeptide Binding Increases the Stability of the BRCA1 BRCT Domains
We measured the stability of the BRCA1 BRCT domains in the presence of an excess of phosphopeptide and found that the midpoint of unfolding was shifted to a higher denaturant concentration (Fig. 1B). A similar effect was observed for the moderately destabilizing variant G1788D, and the stability of the protein was increased to close to the wild-type value (Fig. 1B). The R1699L variant was used as a negative control. The mutation disrupts binding to the phosphate group of the phosphopeptide, and the presence of the peptide was found to have no effect on its stability.
DISCUSSION
Here we have developed a quantitative assay of the thermodynamic stability of the BRCT domains of BRCA1. We selected 36 mutations from the BIC data base, covering a range of different sites in the structure including a founder mutation, G1708E (26), and a common polymorphism, M1652I (27), and we measured the effects on structural stability under low temperature conditions that permit quantitative analysis. We found for the majority of the mutations that the protein was destabilized to such an extent that a significant proportion of molecules would be unfolded under physiological conditions. Using the low temperature conditions in which the mutant proteins are folded, we observed that all can bind to phosphopeptide substrate with affinities close to that of the wild type. These results indicate that the destabilizing effects of the mutations do not cause the protein to adopt a misfolded conformation but rather that the mutations simply shift the equilibrium between folded and unfolded molecules toward the latter. Therefore, we expect that small molecules that bind preferentially to the folded state would be able to rescue a large number of BRCA1 missense mutations. We show that the presence of phosphopeptide does indeed increase the stability of wild-type BRCA1 BRCT and that it restores the stability of an unstable mutant to the wild-type level. Finally, we have looked at the ability of a number of computer algorithms to predict the effects of the mutations on stability. We find that by grouping the mutations into two broad classes, destabilizing by less than or more than 2.2 kcal mol−1, the algorithms have some success in predicting the changes in stability.
Other assays, namely a transcription activation assay (28), a yeast small colony phenotype assay (29), and a protease sensitivity assay (30), have been devised to assess the effect of missense mutations in the BRCT domains. There was agreement between our biophysical assay and the cell-based assays (transcription activation and small colony phenotype) (28, 29, 31, 32). Thus, those mutations that we found to be destabilizing by more than 2.2 kcal mol−1 and for which the folded state would therefore be significantly depopulated at 37 °C were inactive in the cell-based assay. It has been suggested that loss of function of missense BRCA1 variants could arise from cellular mislocalization, and it is possible that mutation-induced unfolding or misfolding of the BRCT domains could be the cause (33). Those mutations that we found to be destabilizing by less than 2.2 kcal mol−1 were active in the cell-based assays, with one exception, G1788V. G1788V had reduced activity in the transcription activation assay, and NMR spectroscopy indicates that this residue is essential for the stability of the BRCT fold (34). However, in our experiments, the thermodynamic stability and phosphopeptide binding affinity of G1788V were similar to those of the wild type.
A number of the variants recorded in the BIC database, M1663L, M1663K, R1699L, S1715C, D1778N, C1787S, S1841N, and A1843P, have not been analyzed in any assays, although in some cases, predictions have been made (35, 36). Our results suggest that M1663L, M1663K, D1778N, and C1787S will not be deleterious to function, whereas R1699L, S1715C, S1841N, and A1843P will confer a susceptibility to breast and ovarian cancer, most of them as a result of reduced protein stability.
Supplementary Material
Acknowledgments
We thank Stephen McLaughlin (Department of Biochemistry, University of Cambridge) for help with data analysis, Joost Schymkowitz and Frederic Rousseau (University of Brussels, Belgium) for help with using FoldX, and Catherine Worth (Department of Biochemistry, University of Cambridge) for use of the SDM program.

The on-line version of this article (available at http://www.jbc.org) contains supplemental text, Fig. S1, and Table S1.
- BIC
- Breast Cancer Information database
- GdmCl
- guanidinium hydrochloride.
REFERENCES
- 1.Easton D. F., Ford D., Bishop D. T. (1995) Am. J. Hum. Genet. 56, 265–271 [PMC free article] [PubMed] [Google Scholar]
- 2.Antoniou A., Pharoah P. D., Narod S., Risch H. A., Eyfjord J. E., Hopper J. L., Loman N., Olsson H., Johannsson O., Borg A., Pasini B., Radice P., Manoukian S., Eccles D. M., Tang N., Olah E., Anton-Culver H., Warner E., Lubinski J., Gronwald J., Gorski B., Tulinius H., Thorlacius S., Eerola H., Nevanlinna H., Syrjäkoski K., Kallioniemi O. P., Thompson D., Evans C., Peto J., Lalloo F., Evans D. G., Easton D. F. (2003) Am. J. Hum. Genet. 72, 1117–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Miki Y., Swensen J., Shattuck-Eidens D., Futreal P. A., Harshman K., Tavtigian S., Liu Q., Cochran C., Bennett L. M., Ding W., et al. (1994) Science 266, 66–71 [DOI] [PubMed] [Google Scholar]
- 4.Glover J. N. (2006) Fam. Cancer 5, 89–93 [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez M. C., Songyang Z. (2008) Front. Biosci. 13, 5905–5915 [DOI] [PubMed] [Google Scholar]
- 6.Starita L. M., Parvin J. D. (2006) Cancer Biol. Ther. 5, 137–141 [DOI] [PubMed] [Google Scholar]
- 7.Wu W., Koike A., Takeshita T., Ohta T. (2008) Cell Div. 3, 1–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pujana M. A., Han J. D., Starita L. M., Stevens K. N., Tewari M., Ahn J. S., Rennert G., Moreno V., Kirchhoff T., Gold B., Assmann V., Elshamy W. M., Rual J. F., Levine D., Rozek L. S., Gelman R. S., Gunsalus K. C., Greenberg R. A., Sobhian B., Bertin N., Venkatesan K., Ayivi-Guedehoussou N., Solé X., Hernández P., Lázaro C., Nathanson K. L., Weber B. L., Cusick M. E., Hill D. E., Offit K., Livingston D. M., Gruber S. B., Parvin J. D., Vidal M. (2007) Nat. Genet. 39, 1338–1349 [DOI] [PubMed] [Google Scholar]
- 9.Boulton S. J. (2006) Biochem. Soc. Trans. 34, 633–645 [DOI] [PubMed] [Google Scholar]
- 10.Wang Z., Moult J. (2001) Hum. Mutat. 17, 263–270 [DOI] [PubMed] [Google Scholar]
- 11.Miroux B., Walker J. E. (1996) J. Mol. Biol. 260, 289–298 [DOI] [PubMed] [Google Scholar]
- 12.Gill S. C., von Hippel P. H. (1989) Anal. Biochem. 182, 319–326 [DOI] [PubMed] [Google Scholar]
- 13.Dalby P. A., Oliveberg M., Fersht A. R. (1998) J. Mol. Biol. 276, 625–646 [DOI] [PubMed] [Google Scholar]
- 14.Ekblad C. M., Wilkinson H. R., Schymkowitz J. W., Rousseau F., Freund S. M., Itzhaki L. S. (2002) J. Mol. Biol. 320, 431–442 [DOI] [PubMed] [Google Scholar]
- 15.Botuyan M. V., Nominé Y., Yu X., Juranic N., Macura S., Chen J., Mer G. (2004) Structure 12, 1137–1146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Varma A. K., Brown R. S., Birrane G., Ladias J. A. (2005) Biochemistry 44, 10941–10946 [DOI] [PubMed] [Google Scholar]
- 17.Rodriguez M., Yu X., Chen J., Songyang Z. (2003) J. Biol. Chem. 278, 52914–52918 [DOI] [PubMed] [Google Scholar]
- 18.Guerois R., Nielsen J. E., Serrano L. (2002) J. Mol. Biol. 320, 369–387 [DOI] [PubMed] [Google Scholar]
- 19.Worth C. L., Bickerton G. R., Schreyer A., Forman J. R., Cheng T. M., Lee S., Gong S., Burke D. F., Blundell T. L. (2007) J. Bioinform. Comput. Biol. 5, 1297–1318 [DOI] [PubMed] [Google Scholar]
- 20.Capriotti E., Fariselli P., Casadio R. (2005) Nucleic Acids Res. 33, W306–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yin S., Ding F., Dokholyan N. V. (2007) Nat. Methods 4, 466–467 [DOI] [PubMed] [Google Scholar]
- 22.Ramensky V., Bork P., Sunyaev S. (2002) Nucleic Acids Res. 30, 3894–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ng P. C., Henikoff S. (2003) Nucleic Acids Res. 31, 3812–3814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou H., Zhou Y. (2002) Proteins 49, 483–492 [DOI] [PubMed] [Google Scholar]
- 25.Yue P., Li Z., Moult J. (2005) J. Mol. Biol. 353, 459–473 [DOI] [PubMed] [Google Scholar]
- 26.Ferla R., Calò V., Cascio S., Rinaldi G., Badalamenti G., Carreca I., Surmacz E., Colucci G., Bazan V., Russo A. (2007) Ann. Oncol. 18, Suppl. 6, vi93–vi98 [DOI] [PubMed] [Google Scholar]
- 27.Deffenbaugh A. M., Frank T. S., Hoffman M., Cannon-Albright L., Neuhausen S. L. (2002) Genet. Test 6, 119–121 [DOI] [PubMed] [Google Scholar]
- 28.Hayes F., Cayanan C., Barillà D., Monteiro A. N. (2000) Cancer Res. 60, 2411–2418 [PMC free article] [PubMed] [Google Scholar]
- 29.Coyne R. S., McDonald H. B., Edgemon K., Brody L. C. (2004) Cancer Biol. Ther. 3, 453–457 [DOI] [PubMed] [Google Scholar]
- 30.Williams R. S., Chasman D. I., Hau D. D., Hui B., Lau A. Y., Glover J. N. (2003) J. Biol. Chem. 278, 53007–53016 [DOI] [PubMed] [Google Scholar]
- 31.Carvalho M. A., Marsillac S. M., Karchin R., Manoukian S., Grist S., Swaby R. F., Urmenyi T. P., Rondinelli E., Silva R., Gayol L., Baumbach L., Sutphen R., Pickard-Brzosowicz J. L., Nathanson K. L., Sali A., Goldgar D., Couch F. J., Radice P., Monteiro A. N. (2007) Cancer Res. 67, 1494–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Vallon-Christersson J., Cayanan C., Haraldsson K., Loman N., Bergthorsson J. T., Brøndum-Nielsen K., Gerdes A. M., Møller P., Kristoffersson U., Olsson H., Borg A., Monteiro A. N. (2001) Hum. Mol. Genet. 10, 353–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rodriguez J. A., Au W. W., Henderson B. R. (2004) Exp. Cell Res. 293, 14–21 [DOI] [PubMed] [Google Scholar]
- 34.Gaiser O. J., Ball L. J., Schmieder P., Leitner D., Strauss H., Wahl M., Kühne R., Oschkinat H., Heinemann U. (2004) Biochemistry 43, 15983–15995 [DOI] [PubMed] [Google Scholar]
- 35.Karchin R., Monteiro A. N., Tavtigian S. V., Carvalho M. A., Sali A. (2007) PLoS Comput. Biol. 3, e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Abkevich V., Zharkikh A., Deffenbaugh A. M., Frank D., Chen Y., Shattuck D., Skolnick M. H., Gutin A., Tavtigian S. V. (2004) J. Med. Genet. 41, 492–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Williams R. S., Green R., Glover J. N. (2001) Nat. Struct. Biol. 8, 838–842 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.