

Abstract

As part of a strategy to achieve large two-photon absorptivity in fluorene-based probes, a series of donor-acceptor-donor (D-A-D) type derivatives were synthesized and their two-photon absorption (2PA) properties investigated. The synthesis of D-A-D fluorophores was achieved by efficient preparation of key intermediates for the introduction of central electron acceptor groups. To accomplish the synthesis of two of the new derivatives, a high yield method for a one-step direct dibromomethylation of phenyl sulfide was developed. The linear and nonlinear optical properties, including UV-vis absorption, fluorescence emission, fluorescence anisotropy, and two-photon absorption (2PA), of the new D-A-D compounds were measured and compared to their D-A or D-D counterparts. Fully conjugated acceptor moieties in the center of the D-A-D fluorophore led to the greatest increase in the 2PA cross section, while weakly conjugated central acceptors exhibited only a modest increase in the 2PA cross section relative to D-A diploar analogs. Encapsulation of the new probes in Pluronic™ F 108NF micelles, and subsequent incubation in HCT 116 cells, resulted in very high lysosomal colocalization (>0.98 colocalization coefficient) relative to commercial Lysotracker™ Red, making the micelle-encapsulated dyes particularly attractive as fluorescent probes for two-photon fluorescence microscopy lysosomal imaging.
Introduction
Two-photon induced fluorescence involves two photons being absorbed “simultaneously” by the molecule under investigation to excite an electron in a molecule to an electronic excited state. Subsequently, once in the S1 state (through internal conversion if a higher excited state was populated) the excited electron relaxes back to the ground state and emits a photon via identical processes as in single-photon absorption and fluorescence processes. This nonlinear process is characterized by the high spatial localization inherent in the quadratic relationship between the excitation and fluorescence intensity.1 In the early 1990's, two-photon fluorescence (2PF) imaging was pioneered by Webb et al. by using a femtosecond pulsed Ti:sapphire laser scanning microscope.1 It was soon clear that the advantageous features of two-photon fluorescence bioimaging include the reduction of photobleaching and photodamage of imaging probes and cellular structures, the capability of relatively deep penetration by exciting at longer wavelength, an enhanced image quality due to less light scattering in turbid media, such as cells, by eliminating out of focus influences and repressing background fluorescence, and the ability to bring about precise 3D spatially-localized photosensitization, photolysis, ablation, and cutting at the subcellular level. 1-3
With the decreasing cost of the two-photon fluorescence microscopy (2PFM) hardware, 2PFM imaging is increasingly being adopted by biological and biomedical researchers. However, the two-photon absorption (2PA) cross sections of most commercially available fluorescent probes, which were actually designed for one-photon fluorescence, are low. For 2PFM imaging, the two-photon excited fluorescence action cross-section ηδ, the product of the fluorescence quantum yield η and 2PA cross section δ, should be a more salient feature to characterize a fluorophore. Since η is always lower than 1, the action 2PA cross section of these probes are even lower. For example, among the commercial fluorophores, Rhodamine B has one of the highest 2PA cross section values, 200 GM (1GM = 10-50 cm4•s•photon−1) at 840 nm in MeOH. Considering the fluorescence quantum yield of Rhodamine B is 0.7 in MeOH, the 2PA action cross section is only 140 GM.4 Therefore, there is compelling need to develop efficient 2PA fluorophores with higher 2PA action cross sections, as a higher 2PA action cross section translates to less input laser power to observe 2PF, thereby reducing photobleaching and background noise, e.g., autofluorescence.
In an effort to develop 2PF probes with improved 2PA efficiencies, fluorenyl-based fluorescence probes have been prepared (Figure 1, A – C) and successfully used for fluorescence bioimaging both under one-photon and two-photon excitation.5-8 Structurally, fluorenyl derivatives have a number of advantages for use as bioimaging fluorescence probes. The conjugated planar ring system provides a well delocalized π-system, which is essential to achieve high 2PA, and the rigidity of fused rings system generally affords high fluorescence quantum yield. Importantly, fluorene derivatives are typically quite photostable under both one- and two-photon irraditation.9-12 Another advantage of using fluorene derivatives as biological probes is that the hydrophilicity/hyrdophobicity of the fluorene derivatives may be easily tuned by introducing various substituents at 9-position (Figure 1, R′). In addition to modulating solubility, the substituents at 9 position also help prevent aggregation of the dye, thereby reducing fluorescence self-quenching. These advantages have been exploited to develop a number of amine-reactive fluorene-based fluorophores for 2PFM bioimaging (Figure 1).5-8
Figure 1.

Structures of fluorene derivatives for bioimaging.
Among the fluorophores shown in Figure 1, it has been found that even hydrophobic 2-diphenylamino-7-benzothiazole-9, 9-didecylfluorene (A) is useful in staining cells such as H9c2 rat cardiomyoblasts cells for epi-fluorescence and two-photon fluorescence microscopy imaging.5 In addition, rather than the diphenylamino group in A, an isothiocyanato group was incorporated into two amino reactive analogs B and C.5,6 Amine-reactive probes B and C can be directly used for 2PFM imaging or to form bioconjugates, imparting specificity. Importantly, the cytotoxicity of fluorophores is low. However, the conjugation length of fluorene derivatives mentioned above is relatively short, leading to relatively low 2PA action cross sections, i.e. <100 GM. Recently, two hydrophilic fluorene derivatives with increased conjugation length were reported by our group for integrin-targeting, and 2PFM cell imaging was demonstrated.6-8 The probe with the most extended conjugation system showed 2PA action cross section of 680 GM at 740 nm in DMSO. Although this 2PA action cross section is already ca. five times higher than that of Rhodamine B, further efforts are warranted to both increase the 2PA cross section, the wavelength of 2PF, and the emission wavelength.
The 2PA efficiency is, in general, related to the conjugation length and the donor/acceptor strength of the chromophore's substituents. Additionally, D-A-D-type chromophores provide higher 2PA cross sections compared to those of D-A or D-D type structures with similar conjugation length and donor/acceptor groups.13,14 With the combination of a D-A-D architecture and large conjugation system, it is reasonable to expect an increase in the 2PA cross sections of a molecule. Interestingly, very few fluorenyl-based D-A-D structures have been reported,15,16 and the reported examples had short conjugation lengths, consequently leading to low 2PA cross sections.
Herein, we report the synthesis of D-A-D type chromophores with increased conjugation length, realizing a significant increase in 2PA cross section, and conducted comprehensive linear and nonlinear photophysical characterization. The D-A-D fluorene derivatives that were prepared incorporated novel bifunctional electron accepting moieties as the core. Significantly, a high yield method for a one-step direct dibromomethylation of phenyl sulfide was developed to prepare two of the new derivatives. The 2PA action cross-sections of these fluorophores was determined and compared to those of their D-A or D-D type counterparts to evaluate both the benefit of this design paradigm and a larger conjugation system on enhancement of two-photon absorptivity. Significantly, the new dyes were used in both one- and two-photon fluorescence microscopy imaging after encapsulation in Pluronic™ micelles, demonstrating outstanding lysosomal selectivity.
Lysosomes are membrane bound vesicles containing various hydrolytic enzymes necessary for digesting exogenetic macromolecules and breaking down old nonfunctioning organelles that out lived their usefulness.17 Lysosomes are involved in a number of cell life activities, including intracellular transportation, metabolism, cell membrane recycling,17 and even apoptosis.18 The malfunction of lysosomes has been implicated in several diseases, such as inflammation, tumors, silicosis, and various lysosomal storage diseases, e.g., Tay-Sachs disease, or Pompe's disease.19, 20 In addition to faciliating study of these conditions, the highly specific lysosome-targeting of the micelle delivery system described herein may find application as gene or drug carriers for lysosomal-targeting therapeutics.
Results and Discussion
Synthesis
The structures of the four pairs of 2PA fluorophores synthesized in this work are shown in Figure 2. The donor-π-donor type compound 1, where two dibutylamino groups as donor and fluorenyl as π bridge, is a reference compound to the D-A-D archetype 2. Similarly, compounds 4, 6, and 8 will be compared to 3, 5, and 7, respectively. Key to the synthesis of these D-A-D molecules was the introduction of the central acceptor group.
Figure 2.

Structures of probes 1–8.
The preparation of donor-π-acceptor-π-donor (D-A-D) 2 was achieved by successfully introducing two nitro groups in the key intermediate 10 by nitration of 9 in a 1:1 mixture of fuming HNO3/concentrated H2SO4 at 0 °C.21 The reaction was clean and no by-products, except the mononitrated product, were detected. The final product 2 was then easily prepared by Horner-Emmons reaction as shown in Figure 3. Compounds 1, 3, and 4 were also synthesized by Horner-Emmons reactions using similar conditions indicated in Figure 3. The synthesis of compound 3, which has been reported previously via Heck coupling in 31% yield, was synthesized in this work by a Horner-Emmons reaction in a greatly increased yield (81%). Identical NMR spectra were observed. The yields 1-4 were relatively good (55-85%).
Figure 3.
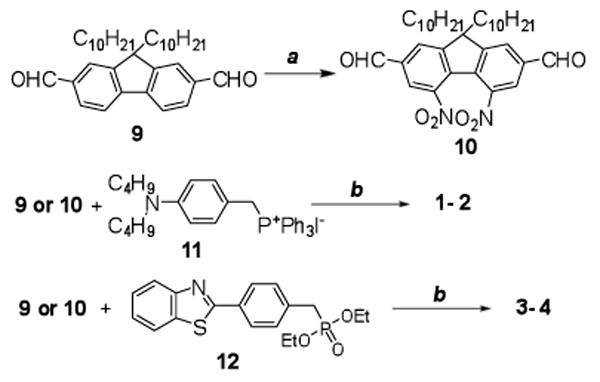
Synthesis of compounds 1 – 4. a) HNO3/H2SO4, 0 °C, 1.5 h, 41%; b) DMF, NaH, r.t., 20 h, 55– 85%.
Compounds 5 and 6 were prepared by Horner-Emmons reactions as shown in Figure 4. Aldehyde 15, used in the Horner-Emmons reaction, was synthesized by Heck coupling of bromofluorenyl aldehyde 13 with N,N-diphenylaminostryene 14 in the presence of n-Bu4NCl in 71% yield. Intermediate 16 was prepared according to a literature method in high yield.22 The development of a one-step direct dibromomethylation of phenyl sulfide in 77% yield greatly simplified the synthesis of bisbromomethylphenyl sulfide 18, a key intermediate in the preparation of 6. Oxidation of sulfide 18 to the corresponding sulfone 19 occurred in good yield. Phosphonylation of both 16 and 19, followed by reaction with 15 in dry DMF using NaH as base, afforded products 5 and 6 in yields of 53 and 37%, respectively.
Figure 4.
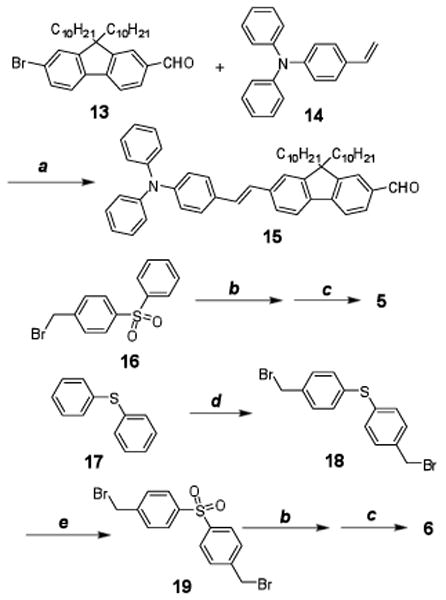
Synthesis of 5 and 6. a) Pd(AcO)2/PPh3, K2CO3, Bu4NCl, DMF, reflux, 20 h, 70%; b) P(OEt)3, reflux, 2h; c) 15, DMF, NaH, r.t., 20 h, 37-53%; d) paraformaldehyde, 33% HBr in HOAc, 70 °C, 48 h, 77%; e) H2O2, HOAc, 90 °C, 30 min, 68%.
Unsymmetrical 7 was prepared in a one-pot reaction by mixing 9 with 11 and 12 in a 1:1:1 mole ratio, resulting in an isolated yield of 43%. However, the same strategy was unsuccessful for the synthesis of 8. This compound was prepared following the same synthetic methodology used in the preparation of 5 and 6, as shown in Figure 5. Here, aldehyde 21 was again prepared via Heck coupling of bromofluorenyl aldehyde 13 with N,N-di-n-butyl-4-vinylaniline (20) in the presence of n-Bu4NCl in good yield (71%). Phosphonate 25 was prepared from the corresponding chloromethyl derivative 24. We developed a simple method preparation of 24 by reflux of commercially available 17 with 16 in NMP for a yield of 70%. Dye 8 was then obtained 43% yield. All compounds were characterized by 1H NMR, 13C NMR, and elementary analysis or high resolution mass spectroscopy.
Figure 5.
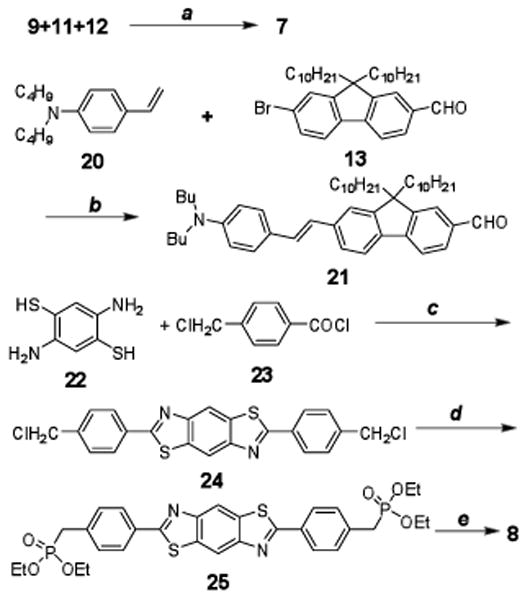
Synthesis of 7 and 8. a) DMF, NaH, r.t., 20 h, 43%; b) Pd(AcO)2/PPh3, K2CO3, Bu4NCl, DMF, reflux, 20 h, 71%; c) NMP, 110 °C, 3 h, 70%; d) P(OEt)3, o-dichlorobenzene, reflux, 16 h, 93%; e) 15, DMF, NaH, r.t., 20 h, 43%.
Linear and Nonlinear Optical Properties
Shown in Figure 6 are the UV-vis (linear) absorption, fluorescence, fluorescence anisotropy and two-photon absorption (nonlinear) spectra of aminostyrylfluorenes 1 and 2 in cyclohexane. The absorption maximum of 491 nm for dinitro-containing 2 had a bathochromic shift of 84 nm relative to 1 (parent compound). This long wavelength absorption band corresponds to a one-photon allowed S0-S1 transition (S0 and S1 are the ground and first excited electronic states, respectively), in which the fluorescence anisotropy was constant at about 3.5. As expected from quantum mechanical selection rules, for molecules with relatively high symmetry such as 1 and 2, the S0-S1 transition is a two-photon forbidden transition, a prediction borne out in the 2PA spectra where very low two-photon absorptivity was observed in the long wavelength region. As the wavelength decreased, a change in the fluorescence anisotropy values was observed at ca. 360 nm for 1 and 425 nm for 2, indicative of a different electronic transition, e.g., S0-S2. This transition is obviously a two-photon allowed transition since the maximum 2PA cross sections of both dyes were obtained at wavelengths corresponding to this transition. Similar behavior was observed for symmetrical molecules.23 The 2PA cross section of dinitrofluorene 2 at 850 nm was nearly 1200 GM, slightly higher than the 1000 GM 2PA cross section observed for 1 at 720 nm. This moderate increase of the 2PA cross section is attributed to weak conjugation of the nitro groups with the terminal amino electron-donating groups. Considering the fluorescence quantum yields of 1 and 2 are 1.0 and 0.44, respectively, the lower fluorescence quantum yield of 2 resulted in the two-photon action cross section of 2 being less than that for 1.
Figure 6.

Absorption (1ab, 2ab), fluorescence (1fl, 2fl), fluorescence anisotropy (1aniso, 2aniso) and 2PA spectra (12PA, 22PA) of 1 (grey) and 2 (black) in cyclohexane. Fluorescence anisotropy was measured in polyTHF.
For the more electron deficient compound 3, bearing terminal benzothiazole moieties, the introduction of two nitro groups in the fluorenyl bridge (4), produced only a slight bathochromic shift in the absorption maximum (28 nm) and did not exert much of an effect on the 2PA spectrum, as shown in Figure 7. This is understandable since, in general, a large 2PA cross section is considered as a result of strong intramolecular charge transfer directly related to the strength of the donor and acceptor groups. The benzothiazole group itself can be considered as a very weak electron donor (relative to a nitro group) and the intramolecular charge transfer from benzothiazole to the nitro groups is not strong enough to achieve a high 2PA cross section. When considering the absorption, 2PA, and fluorescence anisotropy data, both 3 and 4 behave like typical symmetrical molecules. The low energy one-photon allowed transition is two-photon forbidden, while the 2PA allowed transition appeared at higher energy (shorter wavelength).
Figure 7.

Absorption (3ab, 4ab), fluorescence (3fl, 4fl), fluorescence anisotropy (3aniso, 4aniso) and 2PA spectra (32PA, 42PA) of 3 (grey) and 4 (black) in cyclohexane. Fluorescence anisotropy was measured in polyTHF.
Photophysical data for fluorenylsulfones 5 and 6 are illustrated in Figure 8. The linear absorption and emission of 6 were nearly identical to 5. The profile of the 2PA absorption spectra of 5 is consistent with a typical 2PA spectra of D-π-A dipolar chromophores. Here the one-photon allowed S0 to S1 transition, accompanied by constant fluorescence anisotropy, is accessible by 2PA due to the low molecular symmetry. Although the 2PA absorption spectrum of 6 is similar to that of 5, the situation of 6 is more complex since no clear transitions can be identified from fluorescence anisotropy data. For such a large molecule, it is not unusual that a number of electronic transitions are closely spaced, resulting in constantly changing anisotropy values. However, the 2PA cross section of 6 (1520 GM at 720 nm) was significantly larger than the smaller molecule 5 (362 GM at 720 nm). The fluorescence quantum yields of both 5 and 6 were similar (0.94 and 0.95), providing a 4-fold enhancement of the 2PA action cross section for the larger fluorenylsulfone 6 compared to 5. Moreover, at two-photon excitation wavelengths from 690-840 nm, sulfone 6 showed 2PA action cross-sections >750 GM. As expected, the 2PA cross section of 6 was somewhat lower in THF relative to cyclohexane (ca. 4000 vs. 1300 GM at 700 nm, respectively). With high 2PA action cross sections over such a broad spectral range, this derivative is particularly attractive as a probe for 2PFM imaging.
Figure 8.

Absorption (5ab, 6ab), fluorescence (5fl, 6fl), fluorescence anisotropy (5aniso, 6aniso) and two-photon absorption spectra (52PA, 62PA) of 5 (grey) and 6 (black) in cyclohexane. Fluorescence anisotropy was measured in polyTHF.
A more profound enhancement of 2PA was observed for benzobisthiazolylfluorene 8 relative to aminobenzothiazolylfluorene 7, as indicated in Figure 9. Although the absorption and fluorescence maxima of 8 exhibited only a slight bathochromic shift (14 and 10 nm, respectively) with respect to the smaller analog 7, and the fluorescence anisotropy profiles were also similar, the 2PA absorption of benzobisthiazolylfluorene 8 was substantially larger than 7 over a broad spectral range, i.e., from 550-930 nm. At its maximum 2PA wavelength of 790 nm, 8 had an impressive 2PA cross section of 2517 GM, a 10-fold enhancement relative to 7 at the same excitation wavelength (250 GM). This is not unexpected since the 2PA cross section is quadratically dependent on the stationary or transition dipole moments; a small increase in either of these will result in a significant increase in the 2PA cross section. Taking in account of the fluorescence quantum yield of 0.75 for 8 and 1.00 for 7 in cyclohexane, the 2PA action cross section 8 (1887 GM) was 7.5 times greater than that of 7. This was the largest 2PA enhancement observed for this series of dyes. The high 2PA cross sections over a broad wavelength range offers the flexibility of 2PF wavelength selection for bioimaging, depending on the nature of the biological sample.
Figure 9.

Absorption (7ab, 8ab), fluorescence (7fl, 8fl), fluorescence anisotropy (7aniso, 8aniso) and two-photon absorption spectra (72PA, 82PA) of 7 (grey) and 8 (black) in cyclohexane. Fluorescence anisotropy was measured in polyTHF.
Fluorescence Colocalization Studies and Two-photon Fluorescence Imaging
Pluronic™ materials are a series of block copolymers based on ethylene oxide (forming a hydrophilic block) and propylene oxide (forming a hydrophobic block) and widely used as carriers for drug delivery.24, 25 In this work, probes 5-8 were encapsulated in Pluronic™ F 108NF upon formation of micelles and their interaction with cells was investigated by both confocal and two-photon fluorescence imaging. Dye-encapsulated micelles were uptaken by cells without apparent difficulty, as shown in Figure 10 (where probe 6 was used). To determine whether any oganelle-selectivity occurred, colocalization studies were performed with known probes.
Figure 10.
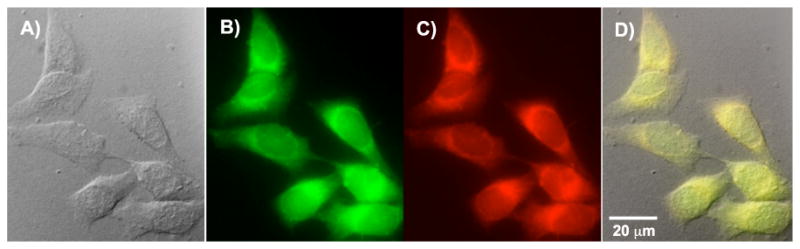
Colocalization images of HCT 116 cells co-incubated with probe 6 in Pluronic™ micelles and LysoTracker® Red. Images were taken using a 60×, oil immersion objective. A) DIC, 500 ms; B) One-photon confocal fluorescence image using a custom made filter cube (Ex:377/50; DM: 409; Em:525/40), 4ms. C) One-photon confocal LysoTracker® fluorescence image using a Texas Red filter cube (Ex:562/40; DM: 593; Em:624/40) 350ms. D) Overlapped image of A, B and C (colocalization coefficient 0.99).
In fluorescence microscopy, colocalization refers to an analysis method to characterize the degree of overlap between two different fluorescent labels, each having a separate emission wavelength, to determine if two different cellular “targets” are located in the same area. Here commercially available LysoTracker® was used for colocalization studies. Commonly used lysosomal markers include DAMP, neutral red, acridine orange, dextran, and bovine serum albumin (BSA) labeled with a fluorophore and LysoTracker probes. However, DAMP is not fluorescent, neutral red and acridine orange lack staining specificity, and fluorescent dextran and BSA have low-photostability and short-circulating life in living cells.26 The LysoTracker® probes are fluorescent acidotropic probes for labeling and tracking acidic organelles in live cells, primarily lysosomes with high selectivity and effective labeling of living cells at nanomolar concentrations, although their two-photon action cross sections (ηδ) are very low, e.g., a value of 10 GM was reported for LysoTracker® Red.27 It is important to note that cross-talk or “bleed-through” may occur if the emission spectra of the two fluorophores are similar. Accurate colocalization determination can only occur if emission spectra are sufficiently separated between fluorophores and the correct filter sets are used during the acquisition step. For this reason LysoTracker® Red was chosen since the emission wavelength of LysoTracker® Red can be detected at 624 nm, well separated from the 525 nm emission maximum of probe 6 (Figure 10).
The fluorescence images, collected from two different wavelengths, i.e., 525 nm for probe 6 and 624 nm for Lysotracker® Red, are shown in Figure 10 B and C, respectively, as well as the differential interference contrast (DIC) image (Figure 10 A), along with the overlap image of A, B, and C (Figure 10 D). Similar images were also collected for probes 5-7, and are included in the supporting information. It is obvious that the fluorescent images of probe 6 and LysoTracker® are almost identical. Quantitatively, the colocalization coefficient indicates the relative degree of overlap between signals. While there are several methods to calculate the colocalization coefficient, the one presented here is the Pearson's correlation coefficient, calculated within Slidebook 4.1, imaging processing software. The correlation coefficients of dye 5-8 relative to LysoTracker® Red are all higher than 0.97, supporting lysosomal colocalization. An additional experiment was performed for long-term tracking of lysosomes. A high colocalization coefficient (0.80) was observed after 2 h of incubation with both dye 6 and lysotracker red, followed by an additional 9 h post-incubation. The colocalization images are included in the Supporting Information.
Two-photon fluorescence microscopy (2PFM) images of the cells incubated with Pluronic™ micelle-encapsulated probes 5-8 using a modified Olympus FluoView 3000 microscope system coupled to a femtosecond Ti:sapphire femtosecond laser as excitation source. As an example, a 2PFM image of HCT 116 cells incubated with micelles containing probe 8 is shown in Figure 11 C. For comparison, DIC and one-photon fluorescence images are also presented (Figure 11 A and B). The 2PFM 3D volume view image provides higher resolution than conventional confocal imaging, supporting further investigation of these probes for bioimaging applications. Additional 2PFM images of cells incubated with probes 5, 6, and 7 can be found in the supporting information.
Figure 11.
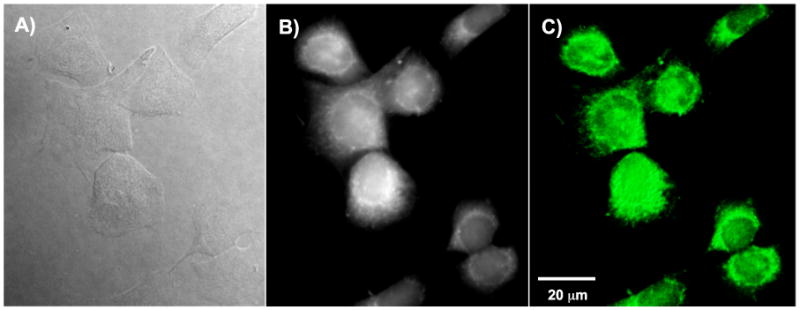
Images of HCT 116 cells incubated with fluorescence probe 8 in Pluronic™ micelles (30 μM, 3 h) taken with 60×, oil immersion objective. a) DIC, 400 ms. b) One-photon fluorescence image (filter cube Ex: 377/50 DM: 409 Em: 525/40). c) 3D reconstruction from overlaid 2PFM images (Ex: 700 nm; Em. long-pass filter 690 nm).
Conclusion
Synthetic methodology was developed to prepare a series of donor-acceptor-donor (D-A-D) type fluorene derivatives. The efficient synthesis of the precursor of the central acceptor 10, 19, and 25 was key for the synthesis of 2, 4, 6, and 8. Direct nitration of 2, 7-diformylfluorene introduced two nitro groups into the fluorenyl ring at the 4 and 5 positions (10). The development of one-step direct dibromomethylation of phenyl sulfide, followed by oxidation, facilitated the synthesis of sulfone 19. An efficient method was developed in this work for preparation of bisphosphonate 25 from commercially available starting materials. In general, D-A-D compounds exhibited high fluorescence quantum yields and improved 2PA cross sections relative to their D-A or D-D counterparts with similar donor/acceptor groups and conjugation length. The conjugated acceptor core of fluorophore 8 led to the greatest 2PA cross section enhancement, while the poorly conjugated central acceptor core in 2 resulted in only a modest increase in the 2PA cross section. The strength of the terminal donor moiety also played a role in the D-A-D molecules; with the weak electron donor in 4 having little effect on 2PA efficiency.
By encapsulation of the dyes in Pluronic™ F 128 NF micelles, probes 5-8 were uptaken by cells, presumably through endocytosis, resulting in high lysosomal selectivity as demonstrated through colocalization (>0.98 coefficients) experiments with Lysotracker® Red. Preliminary results using these dyes as fluorescence probes for 2PFM cell imaging indicating they afford high resolution and lysosomal selectivity when encapsulated in micelles. The high specificity and 2PA cross section of our probes suggest a strategy to overcome limitations for currently used lysosomal trackers for 2PFM applications. Recently, hydrophobic 2PF probes encapsulated in Pluronic™ micelles were also used to image mice brain vasculature in vivo by 2PFM,28 a further testament to the potential of this class of probes and micelle-encapsulation strategy.
Experimental Section
General
9,9-Didecyl-9H-fluorene-2,7-dicarbaldehyde (9),29 (4-(dibutylamino)benzyl) triphenylphosphonium iodide (11),30 diethyl 4-(benzo[d]thiazol-2-yl)benzylphosphonate (12),31 7-bromo-9,9-didecyl-9H-fluorene-2-carbaldehyde (13),32 4-vinyltriphenylamine (14),33 1-(bromomethyl)-4-(phenylsulfonyl)benzene (16),22 and N,N-di-n-butyl-4-vinylaniline (20),34 were prepared according to literature methods. A new route was used for the synthesis of 3 which was different from that previously reported,35 and the 1H NMR spectrum was consistent with the previously reported spectrum. 2, 5-Diaminobenzene-1, 4-dithiol (22) was a gift from the Air Force Research Laboratory/Polymer Branch at Wright-Patterson Air Force Base. 4-(Chloromethyl)benzoyl chloride (17) was purchased from Acros Organics. Pluronic™ F 108NF was obtained from BASF. LysoTracker® Red was purchased from Invitrogen. All other reagents and solvents were used as received from commercial suppliers. Melting points are uncorrected. 1H NMR and 13C NMR spectra were recorded at either 300 or 500 MHz and at 75 or 125 MHz, respectively. Absorption spectra were measured with an UV-visible spectrophotometer. Steady state fluorescence spectra were obtained at room temperature with a spectrofluorimeter using 10 mm quartz cuvettes. Fluorescence anisotropy spectra were measured using three polarizers in the L-format method, with correction for background signals, in the high viscosity solvent polytetrahydrofuran (pTHF) at room temperature. Experimental details of anisotropy measurements were previously reported.36, 37 Fluorescence quantum yields were measured for all compounds by a standard method,38 relative to Rhodamine 6G in ethanol at room temperature.
Preparation of 9, 9–didecyl-4, 5–dinitro-9H–fluorene-2, 7-dicarbaldehyde (10)
A mixture of fuming HNO3 (8 mL, d = 1.42 g/cm3) and 98% H2SO4 was cooled to 0 °C. 9, 9-Didecyl-9H-fluorene-2, 7-dicarbaldehyde (9, 2.0 g, 3.4 mmol) was then added slowly and the resulting mixture was stirred at 0 °C for 1.5 h. The reaction was terminated by addition of 20 g ice. The organic phase was separated, and the aqueous phase was extracted with hexane. The combined organic phase was then washed with water and dried over MgSO4. Solvent was removed under reduced pressure, and the crude product was purification via column chromatography (4:3 hexanes/CH2Cl2), affording 0.96 g of off-white solid (41% yield) m.p. 57–58 °C. 1H NMR (300 MHz, CDCl3) δ 10.09 (s, 2H), 8.36 (s, 2H), 8.09 (s, 2H), 2.10 (m, 4H), 1.16-0.95 (m, 28H), 0.76 (t, J = 6.8 Hz, 6H), 0.46 (m, 4H). 13C NMR (75MHz, DMSO-D6) δ 187.7, 155.3, 145.9, 135.9, 133.0, 124.5, 124.4, 55.7, 38.7, 30.7, 28.5, 28.3, 28.1, 28.0, 22.7, 21.5, 13.0. Anal. Calcd. for C35H48N2O6 (592.77): C, 70.92; H, 8.16; N, 4.73. Found: C, 71.20; H, 8.12; N, 4.83.
General procedure for preparation of dyes 1-5
9,9-Didecyl-9H-fluorene-2,7-dicarbaldehyde (9) or 9,9-didecyl-4,5-dinitro-9H-fluorene-2,7-dicarbaldehyde (10, 0.5 mmol), 4-(N,N-dibutylaminobenzyl) triphenylphosphonium iodide (11) and/or diethyl 4-(benzo[d]thiazol-2-yl) benzylphosphonate (12, 1.0 mmol) in 10 mL of dry DMF was degassed with Ar for 30 min. NaH (0.24 g, 10.0 mmol) was then added, and the mixture was stirred at room temperature under Ar for 20–24 h. The crude product precipitated upon slow addition of water, and was collected by filtration, followed by column chromatographic purification to afford pure compounds 1-4 and 7 (characterization details below).
4,4′-(1E,1′E)-2, 2′-(9, 9-Didecyl-9H-fluorene-2, 7-diyl)bis(ethane-2, 1-diyl)bis(N, N-dibutylaniline) (1)
3:1 Hexanes/CH2Cl2 as eluent (62% yield) m.p. 95 – 96 °C. 1H NMR (300 MHz, CDCl3) δ 7.56 (d, J = 8.1 Hz, 2H), 7.41–7.36 (m, 8H), 7.06 (d, J = 16.2 Hz, 2H), 6.94 (d, J = 15.9 Hz, 2H), 6.61 (d, J = 9.0 Hz, 4H), 3.27 (t, J = 7.5 Hz, 8H), 1.97 (m, 4H), 1.58 (m, 8H), 1.35 (m, 8H), 1.25–1.05 (m, 28H), 0.95 (t, J = 7.2 Hz, 12H), 0.82 (t, J = 6.6 Hz, 6H), 0.69 (m, 4H). 13C NMR (75MHz, CDCl3) δ 151.3, 147.6, 139.8, 137.0, 128.0, 127.7, 125.0, 124.8, 124.4, 120.1, 119.6, 111.7, 55.1, 51.0, 40.9, 32.2, 30.4, 30.0, 29.9, 29.8, 29.6, 24.1, 23.0, 20.7, 14.5, 14.4. Anal. Calcd. for C65H96N2 (905.47): C, 86.22; H, 10.69; N, 3.09. Found: C, 86.18; H, 10.83; N, 3.16.
4, 4′-(1E, 1′E)-2, 2′-(9, 9-didecyl-4, 5-dinitro-9H-fluorene-2, 7-diyl)bis(ethene-2, 1-diyl) bis(N,N-dibutylaniline) (2)
2:1 Hexanes/CH2Cl2 as eluent (55% yield) m.p. 68 – 69 °C. 1H NMR (300 MHz, CDCl3) δ 7.98 (s, 2H), 7.56 (s, 2H), 7.42 (d, 4H, J = 8.7 Hz), 7.18 (d, J = 15.9 Hz, 2H), 6.94 (d, J = 16.2 Hz, 2H), 6.65 (d, J = 9.0 Hz, 4H), 3.32 (t, J = 7.2 Hz, 8H), 2.07 (m, 4H), 1.61 (m, 8H), 1.36 (m, 8H), 1.25–1.00 (m, 28H), 0.97 (t, J = 7.4 Hz, 12H), 0.82 (t, J = 6.8 Hz, 6H), 0.63 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 154.8, 148.6, 146.5, 139.8, 132.5, 129.6, 128.5, 128.5, 123.4, 123.2, 120.9, 111.7, 56.1, 51.2, 40.9, 32.3, 32.1, 30.2, 30.0, 29.9, 29.7, 29.6, 24.0, 23.1, 20.8, 14.6, 14.5. Anal. Calcd. for C65H94N4O4 (995.47): C, 78.43; H, 9.52; N, 5.63. Found: C, 78.06; H, 9.57; N, 5.44.
2, 2′-(4, 4′-(1E, 1′E)-2, 2′-(9, 9-Didecyl-9H-fluorene-2, 7-diyl)bis(ethene-2, 1-diyl)bis(4, 1-phenylene)) dibenzo[d]thiazole35 (3)
1:2 Hexanes/CH2Cl2 as eluent (81% yield) m.p. 154 – 155 °C (lit. m.p 143 – 144 °C).35 1H NMR (500 MHz, CDCl3) δ 8.12 (d, J = 8.0 Hz, 4H), 8.09 (d, J = 8.0 Hz, 2H), 7.93 (d, J = 8.0 Hz, 2H), 7.69 (d + d, 6H), 7.56 (d, J = 8.0 Hz, 2H), 7.52 (m, 4H), 7.41 (t, J = 8.3 Hz, 2H), 7.35 (d, J = 16.0 Hz, 2H), 7.23 (d, J = 16.0 Hz, 2H), 2.05 (t, J = 7.5 Hz, 4H), 1.22–1.08 (m, 28H), 0.82 (t, J = 7.0 Hz, 6H), 0.68 (m, 4H).
2, 2′-(4, 4′-(1E, 1′E)-2, 2′-(9, 9-Didecyl-4, 5-dinitro-9H-fluorene-2, 7-diyl) bis(ethene-2, 1-diyl) bis(4, 1-phenylene)) dibenzo[d]thiazole (4)
1:3 Hexanes/CH2Cl2 as eluent (82% yield) m.p. 171 – 172 °C. 1H NMR (300 MHz, CDCl3) 8.15 – 8.06 (m, 8H), 7.91 (d, 2H, J = 8.4 Hz), 7.69 (m, 6H), 7.50 (d, J = 7.5 Hz, 2H), 7.39 (d, J = 7.5 Hz, 2H), 7.32 (s, 4H), 2.10 (m, 4H), 1.15–1.07 (m, 28H), 0.81 (t, J = 6.6 Hz, 6H), 0.62 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 167.3, 155.2, 154.2, 146.8, 138.9, 138.6, 135.2, 133.7, 131.4, 129.8, 128.2, 127.8, 127.6, 126.6, 125.5, 124.3, 123.4, 121.8, 56.4, 40.8, 32.3, 30.2, 29.9, 29.7, 29.6, 24.0, 23.1, 14.6. Anal. Calcd. for C63H66N4O4S2 (1007.35): C, 75.11; H, 6.60; N, 5.56. Found: C, 75.24; H, 6.78; N, 5.42.
[4-(2-{7-[2-(4-Benzothiazol-2-yl-phenyl)-vinyl]-9, 9-didecyl-9H-fluoren-2-yl}-vinyl)-phenyl]-dibutyl-amine (7)
1:3 Hexanes/CH2Cl2 as eluent, (43% yield) m.p. 95 – 96 °C. 1H NMR (300 MHz, CDCl3) δ 8.07 (m, 3H), 7.89 (d, J = 7.8 Hz, 1H), 7.63 (m, 4H), 7.51–7.38 (m, 8H), 7.31 (d, J = 16.8 Hz, 2H), 7.17 (d, J=16.2 Hz, 2H), 7.08 (d, J=16.2 Hz, 2H), 6.95 (d, J=16.2 Hz, 2H), 6.62 (d, J=8.1 Hz, 2H), 3.29 (m, 4H), 2.02 (m, 4H), 1.58 (m, 4H), 1.37 (m, 4H), 1.15–1.06 (m, 28H), 0.97 (t, J = 7.1 Hz, 6H), 0.82 (t, J = 5.9 Hz, 6H), 0.68 (m, 4H). 13C NMR (75 MHz, CDCl3) δ 154.3, 151.6, 147.9, 141.6, 140.5, 139.5, 137.7, 135.5, 135.1, 132.4, 131.3, 128.5, 128.1, 127.9, 127.0, 126.7, 126.5, 126.1, 125.3, 124.8, 124.4, 123.3, 121.8, 121.1, 120.3, 120.1, 119.9, 111.8, 55.3, 51.2, 41.0, 32.3, 32.1, 30.5, 30.1, 30.0, 30.0, 29.7, 24.3, 23.1, 20.9, 14.6, 14.5. Anal. Calcd. for C64H82N2S (911.42): C, 84.34; H, 9.07; N, 3.07. Found: C, 84.34; H, 9.28; N, 3.08.
Preparation of (E)-9, 9-didecyl-7-(4-(diphenylamino)styryl)-9H-fluorene-2-carbaldehyde 15
7-Bromo-9,9-didecyl-9H-fluorene-2-carbaldehyde (13) (1.384 g, 2.5 mmol), 4-vinyltriphenylamine (14, 0.678 g, 2.5 mmol), PPh3 (65.56 mg, 0.25 mmol), and K2CO3 (6.25 mmol, 0.864g) in DMF were degassed with Ar for 30 min, then Pd(OAc)2 (84.20 mg, 0.125 mmol) and n-Bu4NCl (0.695 g, 2.5 mmol) were added, followed by refluxing the mixture under Ar for 20 h. Water and hexanes were added and the organic phase was concentrated. The crude product was purified by column chromatography (20:1 hexanes/EtOAc), affording 1.31g of 15 as yellow sticky oil (70% yield). 1H NMR (500 MHz, CDCl3) δ 10.05 (s, 1H), 7.86 (m, 2H), 7.80 (d, J = 7.5 Hz, 1H), 7.74 (d, J = 7.5 Hz, 1H), 7.48–7.53 (m, 2H), 7.42 (d, J = 9.0 Hz, 2H), 7.25–7.29 (m, 4H), 7.11–7.18 (m, 6H), 7.03–7.08 (m, 4H), 2.03 (m, 4H), 1.03–1.25 (m, 28H), 0.83 (t, J = 7.0 Hz, 6H), 0.62 (m, 4H).13C NMR (125 MHz, CDCl3) δ 14.1, 22.7, 23.8, 29.3, 29.5, 29.6, 30.0, 31.9, 40.3, 55.2, 119.8, 120.7, 121.2, 123.0, 123.1, 123.4, 124.6, 125.7, 127.1, 127.4, 128.6, 129.3, 130.7, 131.3, 135.1, 138.4, 138.9, 147.4, 147.5, 147.5, 151.7, 152.8, 192.4. HRMS-(APPI) for C54H65NO: theoretical m/z [M+H]+ = 744.5139, found [M+H]+ = 744.5157.
Preparation of bis(4-(bromomethyl) phenyl) sulfane 18
Phenyl sulfide 17 (1.86 g, 0.01 mmol) and paraformaldehyde (1.2 g, 0.04 mmol) in 33% HBr/HOAc (10 mL) were heated at 70 °C for 48 h. Water and 2:1 hexanes/EtOAc were added. White solid precipitated and was collected by filtration to afford 2.86 g of 18 (77% yield), m. p. 129 – 130.5 °C (lit. 135.3 – 136.3 °C).39 1H NMR (500 MHz, CDCl3) δ 7.29–7.34 (m, 8H), 4.47 (s, 4H). 13C NMR (125 MHz, CDCl3) δ 32.9, 129.9, 131.3, 135.8, 136.8. HRMS(DIP-CI) for C14H12Br2S: theoretical m/z [M]+ = 369.9026, found [M]+ = 369.9008.
Preparation of 4, 4′-sulfonylbis((bromomethyl)benzene) 19
Bis(4-(bromomethyl) phenyl)sulfane 18 (0.5 g, 1.34 mmol) was dissolved in HOAc (4 mL) at 70 °C. 30 % H2O2 (1 mL) was slowly added and the mixture was heated at 90 °C for 30 min. The product precipitated after cooling and was collected by filtration, washed with water, and dried, providing 0.37 g of 19 (68% yield), m. p. 145 – 146 °C (lit. 134 – 135 °C).40 1H NMR (500 MHz, CDCl3) δ 7.92 (d, J = 5.0 Hz, 4H), 7.53 (d, J = 5.0 Hz, 4H), 4.46 (s, 4H). 13C NMR (125 MHz, CDCl3) δ 31.3, 128.3, 130.0, 141.1, 143.3. HRMS(DIP-CI) for C14H12Br2O2S: theoretical m/z [M+H]+ = 402.9003, found [M+H]+ = 402.9010.
Preparation of dye 5
1-(Bromomethyl)-4-(phenylsulfonyl) benzene (16, 36.2 mg, 0.116 mmol) was refluxed with triethyl phosphite (1 mL) for 2 h. Excess triethyl phosphite was evaporated and the residue was dried in vacuo. (E)-9,9-didecyl-7-(4-(diphenylamino)styryl)-9H-fluorene-2-carbaldehyde (15, 86.5 mg, 0.116 mmol) was then mixed with above product in dry DMF (2 mL) under Ar. NaH (27.8 mg, 1.16 mmol) was added and the reaction was stirred at room temperature for 20 h. Water was added to terminate the reaction, and the product was extracted with EtOAc. Solvent was removed and the crude product was purified by column chromatography (1:1 hexanes/CH2Cl2), producing 58.6 mg of 5 (53% yield) as a soft glassy material, which turned into viscous liquid upon heating. No defined melting point was observed. 1NMR (500 MHz, CDCl3) δ 7.88 (m, 4H), 7.57 (m, 4H), 7.52–7.33 (m, 8H), 7.23–7.17 (m, 6H), 7.07–6.95 (m, 11H), 1.93 (m, 4H), 1.18–0.97 (m, 28H), 0.73 (t, J = 5.0 Hz, 6H), 0.58 (m, 4H). 13C NMR (125 MHz, CDCl3) δ 151.6, 147.5, 147.3, 141.8, 140.0, 139.5, 133.2, 133.1, 131.6, 129.3, 127.6, 127.5, 127.3, 126.9, 126.2, 125.6, 125.5, 124.5, 123.6, 123.0, 121.1, 120.6, 120.1, 119.9, 55.0, 40.5, 31.9, 29.6, 29.6, 29.3, 29.3, 23.8, 22.7, 14.1. HRMS (MALDI-TOF) for C67H75NO2S: theoretical m/z [M+H]+ = 958.5591, found [M+H]+ = 958.5587.
Preparation of dye 6
4, 4′-Sulfonyl bis((bromomethyl)benzene) (19, 0.1 g, 0.247 mmol) was refluxed with triethyl phosphite for 2 h. Excess triethyl phosphite was evaporated, and the residue was dried in vacuo. (E)-9,9–didecyl-7-(4-(diphenylamino) styryl)-9H-fluorene-2-carbaldehyde (15, 0.37 g, 0.5 mmol) was then mixed with the above product in dry DMF (5 mL) under Ar. NaH was added, and the reaction was stirred at room temperature for 20 h. Water was added, and the product was extracted with EtOAc. Solvent was removed, and the crude product was purified by column chromatography (1:1 hexanes/CH2Cl2), providing 0.16 g of 6 (37% yield) as a glassy material, which turned into viscous liquid upon heating. No defined melting point was observed. 1NMR (500 MHz, CDCl3) δ 7.95 (d, J = 10.0 Hz, 4H), 7.65 (m, 8H), 7.41–7.51 (m, 12H), 7.29 (m, 12 H), 7.02–7.13 (m, 20H), 2.01 (m, 8H), 1.04–1.26 (m, 56H), 0.79 (t, J = 5.0 Hz, 12H), 0.65 (m, 8H). 13C NMR (125 MHz, CDCl3) δ 151.7, 147.6, 147.3, 141.8, 140.0, 139.8, 137.0, 133.1, 131.6, 129.3, 128.1, 127.7, 127.6, 127.3, 126.9, 126.2, 125.7, 125.5, 124.5, 123.6, 123.1, 121.1, 120.6, 120.1, 120.0, 55.0, 40.5, 31.9, 29.6, 29.6, 29.3, 29.3, 23.8, 22.7, 14.1. HRMS(MALDI-TOF) for C122H140N2O2S: theoretical m/z [M]+ = 1697.0636, found [M]+ = 1697.0663.
Preparation of (E)-9, 9-didecyl-7-(4-(dibutylamino)styryl)-9H-fluorene-2-carbaldehyde (21)
7-Bromo-9,9-didecyl-9H-fluorene-2-carbaldehyde 13 (0.554 g, 1.0 mmol), N,N-di-n-butyl-4-vinylaniline (20, 0.231 g, 1.0 mmol), PPh3 (26.2 mg, 0.10 mmol), and K2CO3 (0.346g, 2.5 mmol) in DMF were degassed for 30 min, then Pd(OAc)2 (33.7 mg, 0.05 mmol) and n-Bu4NCl (0.278 g, 1.0 mmol) were added. The mixture was then refluxed for 20 h under Ar. Water and hexane were added, the organic phase was concentrated, and the crude product was purified by column chromatography using 1.5:1 hexanes/CH2Cl2 as eluent, affording 0.50 g of 21 as yellow oil (71% yield). 1H NMR (300 MHz, CDCl3) δ 9.95 (s, 1H), 7.77–7.68 (m, 3H), 7.62 (d, 1H), 7.40 (d, J=8.1 Hz, 1H), 7.35–7.32(m, 3H), 7.05(d, J=16.2 Hz, 1H), 6.88(d, J=16.2 Hz, 1H), 6.57 (d, J=8.7 Hz, 2H), 3.23 (t, J=7.5 Hz, 4H), 1.95(t, J=8.0 Hz, 4H), 1.51(m, 4H), 1.29(m, 4H), 1.20–0.88 (m, 28H), 0.83–0.74(m, 12H), 0.54(m, 4H). 13C NMR (74.5 MHz, CDCl3) δ 13.0, 13.1, 19.3, 21.6, 22.7, 28.2, 28.4, 28.4, 28.5, 28.9, 30.5, 30.8, 39.2, 49.8, 54.0, 107.2, 110.5, 118.4, 119.0, 119.9, 121.7, 122.6, 124.0, 126.6, 128.1, 129.4, 133.6, 136.8, 137.9, 146.3, 146.5, 150.3, 151.4, 191.0. HRMS (ESI) for C50H73NO: theoretical m/z [M+H]+ = 704.5765. Found [M+H]+ = 704.5751.
Preparation of 2, 6-bis[4-(chloromethyl)phenyl] benzo[1, 2-d:4, 5-d']bisthiazole (24)
2, 5-Diaminobenzene-1, 4-dithiol (22, 0.39 g, 2.26 mmol) and 4-(chloromethyl)benzoyl chloride (23, 0.86g, 4.55 mmol) in 5 mL anhydrous NMP was heated at 110 °C for 3 h. Upon cooling, water was added and the precipitate was collected by filtration. Recrystallization from toluene/o-dichlorobenzene afforded 0.71g of 24 (70% yield), m.p. >500 °C (decomp.). 1H NMR (300 MHz, DMSO–d6) δ 8.87 (s, 2H), 8.15 (d, J = 7.8 Hz, 4H), 7.66 (d, J = 7.8 Hz, 4H), 4.88 (s, 4H). 13C NMR could not be collected due to the limited solubility. HRMS (ESI) for C22H14Cl2N2S2: theoretical m/z [M+H]+ = 441.0052. Found [M+H]+ = 441.0061. Anal. Calcd. for (C22H14Cl2N2S2): C, 59.86; H, 3.20; N, 6.35; S, 14.53. Found: C, 60.01; H, 3.26; N, 6.43; S, 14.59.
Preparation of tetraethyl 4, 4′-(benzo[1, 2-d:4, 5-d']bisthiazole-2, 6-diyl) bisbenzylphosphonate (25)
A solution of 24 (0.78 g, 1.77 mmol) and triethyl phosphite (1.17 g, 7.08 mmol) in 5 mL o-dichlorobenzene was heated at reflux for 16 h. The product was then precipitated from hexane, collected by filtration, and purified by column chromatography using 20:1 CH2Cl2/MeOH as eluent, providing 1.06 g of 24 (93% yield), m.p. 289.5 – 290 °C. 1H NMR (300 MHz, CDCl3) δ 8.52 (s, 2H), 8.06 (d, J = 8.1 Hz, 4H), 7.45 (d, J = 8.1 Hz, 4H), 4.05 (m, 8H), 3.23 (d, J = 22.2 Hz, 4H), 1.27(t, J = 7.1 Hz, 12H). 13C NMR (75 MHz, CDCl3) δ 16.8, 33.4, 35.2, 62.7, 115.6, 128.0, 130.7, 132.3, 134.6, 135.5, 152.3, 168.7. Anal. Calcd. for (C30H34N2O6P2S2): C, 55.89; H, 5.32; N, 4.35; S, 9.95. Found: C, 56.11; H, 5.36; N, 4.41; S, 10.04.
Preparation of dye 8
A solution of 21 (58 mg, 0.082 mmol) and 25 (26 mg, 0.041 mmol) in 1 mL DMF was degassed by Ar for 30 min. NaH (14 mg, 0.82 mmol) was then added and the mixture was stirred at room temperature under Ar for 20 h. The product was precipitated by addition of water and collected by filtration. The crude product was purified by column chromatography using 1:2 CH2Cl2/hexanes as eluent, affording 31 mg of 6 (43% yield), m. p. 196 – 197 °C. 1H NMR (300 MHz, CDCl3) δ 8.37 (s, 2H), 7.99 (d, J = 8.0 Hz, 4H), 7.54–7.40 (m, 18H), 7.33–7.27 (m, 4H), 7.10 (d, J = 16.0 Hz, 2H), 7.09 (d, J = 16.0 Hz, 2H), 7.00 (d, J = 16.0 Hz, 2H), 6.65 (d, J = 16.0 Hz, 4H), 3.32 (t, J = 7.5 Hz, 8H), 2.03 (m, 8H), 1.62 (m, 8H), 1.39 (m, 8H), 1.23–0.98 (m, 68H), 0.83 (m, t, J = 7.2 Hz), 0.73 (m, 8H). 13C NMR (125 MHz, CDCl3) δ 168.5, 152.3, 151.6, 151.5, 147.8, 141.5, 140.5, 139.4, 137.6, 135.3, 134.5, 132.1, 128.0, 127.9, 127.7, 127.1, 126.9, 124.7, 121.3, 121.0, 120.6, 120.3, 120.0, 119.9, 115.4, 115.2, 111.6, 55.0, 50.8, 40.6, 31.9, 30.1, 29.5, 29.3, 23.9, 22.7, 20.6, 20.4, 20.3, 14.3, 14.1, 14.0. HRMS (ESI–TOF) for C122H158N4S2: theoretical m/z [M]2+ = 871.5958, found [M]2+ = 871.5959.
Two-photon absorption measurements
2PA spectra of compounds 1–8 were determined over a broad spectral region by the typical two-photon induced fluorescence (2PF) method,41 relative to Rhodamine B in methanol and Fluorescein in water (pH 11) as standards. A PTI QuantaMaster spectrofluorimeter and femtosecond Clark-MXR CPA-2010 laser-pumped an optical parametric generator/amplifier (TOPAS, Light Conversion), with pulse duration, 140 fs, (FWHM), tuning range 580–940 nm, pulse energies 0.15 mJ, and 1 kHz repetition rate. Two-photon fluorescence measurements were performed in 10 mm fluorometric quartz cuvettes with dye concentrations ∼3×10-5 M in cyclohexane. The experimental fluorescence excitation and detection conditions were conducted with the negligible reabsorption processes to ensure no effect on 2PA measurements. The quadratic dependence of two-photon induced fluorescence intensity on the excitation power was verified for each excitation wavelength by systematically varying the excitation power.
General procedure for preparation of dye encapsulated micelles
A solution of dye in CH2Cl2 (5 mL) was mixed with Pluronic™ F 108NF (30 mg) in water (5 mL). The resulting mixture was stirred at room temperature for 48 h to slowly evaporate the CH2Cl2. The dye concentration was approximately 0.5 mM in water, as estimated by absorption spectra.
General procedure for HCT 116 cells incubation with fluorescent probes
An epithelial colorectal carcinoma cell line HCT 116, purchased from ATCC (America Type Culture Collection, Manassas, VA, USA), was used. All cells were incubated in Dulbecco's modified Eagle's Medium (DMEM, Invitrogen, Carlsbad, CA, USA), supplemented with 10% fetal bovine serum (FBS, Atlanta Biologicals, Lawrenceville, GA, USA), 100 units/mL penicillin-streptomycin (Atlanta Biologicals, Lawrenceville, GA, USA), and incubated at 37 °C in a 95% humidified atmosphere containing 5% CO2.
HCT 116 cells were placed onto poly-D-lysine coated coverslips placed into 24-well glass plates (40,000 cells per well) and incubated for 36 h before incubating with the fluorescent probes. A 0.5 mM stock solution of the fluorescent probe in Pluronic™ F 108NF micelles was dissolved in water. Diluted solutions of probe (30 μM) by complete growth medium, Dulbecco's modification of Eagle's medium (DMEM), were then freshly prepared and placed over the cells for a 3 h period. For colocalization experiments, a diluted mixture of dye (30 μM) and LysoTracker® Red (75 nM) in the growth medium was used. After the incubation, cells were washed with PBS (pH=7.2, Fischer Scientific) three to five times, fixed using 3.7% formaldehyde solution at 37 °C for 15 min, and incubated with 0.5 mL/well NaBH4 (1 mg/mL) solution in PBS (pH=8.0, prepared by adding few drops of 6N NaOH solution into PBS) at 37 °C for 15 min. The last step was then repeated. The plates were washed twice with PBS and once with water. Finally, the glass coverslips were mounted using Prolong Gold mounting media for microscopy imaging.
One-photon fluorescence imaging
An inverted microscope (Olympus IX70) equipped with a Q Imaging cooled CCD (Model Retiga EXi) was used for conventional fluorescence imaging, where the output of a filtered 100 W mercury lamp was used as the excitation source. For probes 5–8, a customized filter cube (Ex 377/50, DM 409, Em 460/50) was used for fluorescence imaging, while for LysoTracker® Red, a Texas Red filter cube (Ex:562/40; DM: 593; Em:624/40) was used.
Two-photon fluorescence microscopy (2PFM) imaging
2PFM images were collected on a modified Olympus Fluoview FV300 microscope system coupled to a tunable Coherent Mira 900F Ti:sapphire, 76 MHz, modelocked, femtosecond laser tuned to 700 nm. An emission long-pass filter (cutoff 690 nm) was placed in the microscope scanhead to avoid background irradiance from the excitation source. Consecutive layers, separated by approximately 0.15 μm, were recorded to create a 3D reconstruction from overlaid 2PFM images. The two-photon induced fluorescence was collected with a 60× microscope objective (UPLANSAPO 60×, NA= 1.35, Olympus).
Supplementary Material
Acknowledgments
The authors wish to acknowledge the National Science Foundation (CHE-0832622 and ECCS-0621715) and the National Institutes of Health (1 R15 EB008858-01) for support of this work.
Footnotes
Supporting Information Available: Copies of 1H and 13C NMR spectra, fluorescence images of the colocalization of probes 5, 7, and 8 with Lysotracker® Red in HCT 116 cells, and 2PFM images of HCT 116 cells incubated with probes 5-7 are included. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Denk W, Strickler JH, Webb WW. Science. 1990;248:73–76. doi: 10.1126/science.2321027. [DOI] [PubMed] [Google Scholar]
- 2.Konig K. J Microsc. 2000;200:83–104. doi: 10.1046/j.1365-2818.2000.00738.x. [DOI] [PubMed] [Google Scholar]
- 3.Piston DW. Trends Cell Biol. 1999;9:66–69. doi: 10.1016/s0962-8924(98)01432-9. [DOI] [PubMed] [Google Scholar]
- 4.Xu C, Williams RM, Zipfel W, Webb WW. Bioimaging. 1996;4:198–207. [Google Scholar]
- 5.Schafer-Hales KJ, Belfield KD, Yao S, Frederiksen PK, Hales JM, Kolattukudy PE. J Biomed Opt. 2005;10:051402-1, 8. doi: 10.1117/1.2104528. [DOI] [PubMed] [Google Scholar]
- 6.Morales AR, Schafer-Hales KJ, Marcus AI, Belfield KD. Bioconjugate Chem. 2008;19:2559–2567. doi: 10.1021/bc800415t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Morales AR, Yanez CO, Schafer-Hales KJ, Marcus AI, Belfield KD. Bioconjugate Chem. 2009;20:1992–2000. doi: 10.1021/bc900302t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Morales AR, Luchita G, Yanez CO, Bondar MV, Przhonska OV, Belfield KD. Org Biomol Chem. 2010;8 doi: 10.1039/b925934a. in press. [DOI] [PubMed] [Google Scholar]
- 9.Corredor CC, Belfield KD, Bondar MV, Przhonska OV, Yao S. J Photochem Photobiol A Chem. 2006;184:105–112. [Google Scholar]
- 10.Belfield KD, Bondar MV, Przhonska OV, Schafer KJ. J Photochem Photobiol A Chem. 2004;162:489–496. doi: 10.1039/b307426a. [DOI] [PubMed] [Google Scholar]
- 11.Belfield KD, Bondar MV, Przhonska OV, Schafer KJ. J Photochem Photobiol A Chem. 2004;162:569–574. doi: 10.1039/b307426a. [DOI] [PubMed] [Google Scholar]
- 12.Belfield KD, Bondar MV, Przhonska OV, Schafer KJ. Photochem Photobiol Sci. 2004;3:138–141. doi: 10.1039/b307426a. [DOI] [PubMed] [Google Scholar]
- 13.Albota M, Beljonne D, Bredas JL, Ehrlich JE, Fu JY, Heikal AA, Hess SE, Kogej T, Levin MD, Marder SR, McCord-Maughon D, Perry JW, Rockel H, Rumi M, Subramaniam G, Webb WW, Wu XL, Xu C. Science. 1998;281:1653–1656. doi: 10.1126/science.281.5383.1653. [DOI] [PubMed] [Google Scholar]
- 14.Beljonne D, Kogej T, Marder SR, Perry JW, Bredas JL. MCLC S&T, Sect B: Nonlinear Opt. 1999;21:461–480. [Google Scholar]
- 15.He GS, Lin TC, Prasad PN. Opt Express. 2002;10:566–574. doi: 10.1364/oe.10.000566. [DOI] [PubMed] [Google Scholar]
- 16.He GS, Lin TC, Prasad PN, Kannan R, Vaia RA, Tan LS. J Phys Chem B. 2002;106:11081–11084. [Google Scholar]
- 17.deDuve C. Eur J Biochem. 1983;137:391–397. doi: 10.1111/j.1432-1033.1983.tb07841.x. [DOI] [PubMed] [Google Scholar]
- 18.Stoka V, Turk B, Schendel SL, Kim TH, Cirman T, Snipas SJ, Ellerbyi LM, Bredeseni D, Freeze H, Abrahamson M, Bromme D, Krajewski S, Reed JC, Yin XM, Turk V, Salvesen GS. J Biol Chem. 2001;276:3149–3157. doi: 10.1074/jbc.M008944200. [DOI] [PubMed] [Google Scholar]
- 19.Jedeszko C, Sloane BF. Biol Chem. 2004;385:1017–1027. doi: 10.1515/BC.2004.132. [DOI] [PubMed] [Google Scholar]
- 20.Fehrenbacher N, Jaattela M. Cancer Res. 2005;65:2993–2995. doi: 10.1158/0008-5472.CAN-05-0476. [DOI] [PubMed] [Google Scholar]
- 21.Yao S, Belfield KD. J Org Chem. 2005;70:5126–5132. doi: 10.1021/jo0503512. [DOI] [PubMed] [Google Scholar]
- 22.Yanez CO, Andrade CD, Belfield KD. Chem Commun. 2009:827–829. doi: 10.1039/b815831b. [DOI] [PubMed] [Google Scholar]
- 23.Hales JM, Hagan DJ, Van Stryland EW, Schafer KJ, Morales AR, Belfield KD, Pacher P, Kwon O, Zojer E, Bredas JL. J Chem Phys. 2004;121:3152–3160. doi: 10.1063/1.1770726. [DOI] [PubMed] [Google Scholar]
- 24.Kabanov AV, Batrakova EV, Alakhov VY. J Controlled Release. 2002;82:189–212. doi: 10.1016/s0168-3659(02)00009-3. [DOI] [PubMed] [Google Scholar]
- 25.Marin A, Sun H, Husseini GA, Pitt WG, Christensen DA, Rapoport NY. J Controlled Release. 2002;84:39–47. doi: 10.1016/s0168-3659(02)00262-6. [DOI] [PubMed] [Google Scholar]
- 26.Shi H, He X, Yuan Y, Wang K, Liu D. Anal Chem. 2010;82:2213–2220. doi: 10.1021/ac902417s. [DOI] [PubMed] [Google Scholar]
- 27.Kim HM, An MJ, Hong JH, Jeong BH, Kwon O, Hyon JY, Hong SC, Lee KJ, Cho BR. Angew Chem. 2008;120:2263–2266. doi: 10.1002/anie.200704586. [DOI] [PubMed] [Google Scholar]
- 28.Maurin M, Vurth L, Vial JC, Baldeck P, Marder Seth R, Van der Sanden B, Stephan O. Nanotechnology. 2009;20:235102. doi: 10.1088/0957-4484/20/23/235102. [DOI] [PubMed] [Google Scholar]
- 29.Belfield KD, Yao S, Morales AR, Hales JM, Hagan DJ, Van Stryland EW, Chapela VM, Percino J. Polym Adv Technol. 2005;16:150–155. [Google Scholar]
- 30.Bredereck H, Simchen G, Griebenow W. Chem Ber. 1973;106:3732–42. [Google Scholar]
- 31.Yoshino K, Kohno T, Uno T, Morita T, Tsukamoto G. J Med Chem. 1986;29:820–825. doi: 10.1021/jm00155a037. [DOI] [PubMed] [Google Scholar]
- 32.Kannan R, Tan LS, Reinhardt BA, Vaia RA. US Patent 6730793. 2004
- 33.Xia H, He J, Xu B, Wen S, Li Y, Tian W. Tetrahedron. 2008;64:5736–5742. [Google Scholar]
- 34.Cho MJ, Lim JH, Hong CS, Kim JH, Lee HS, Choi DH. Dyes Pigm. 2008;79:193–199. [Google Scholar]
- 35.Belfield KD, Morales AR, Kang BS, Hales JM, Hagan DJ, Van Stryland EW, Chapela VM, Percino J. Chem Mater. 2004;16:4634–4641. [Google Scholar]
- 36.Lakowicz JR. Principles of Fluorescence Spectroscopy. Kluwer Academic/Plenum; New York: 1999. pp. 291–316. [Google Scholar]
- 37.Belfield KD, Bondar MV, Przhonska OV, Schafer KJ. J Fluoresc. 2002;12:449–454. [Google Scholar]
- 38.Fischer M, Georges J. Chem Phys Lett. 1996;260:115–118. [Google Scholar]
- 39.Oi F, Yanase N, Yamamoto M. JP Patent 2005154379. 2005
- 40.Jung HK, Lee JK, Kang MS, Kim SW, Kim JJ, Park SY. Polym Bull. 1999;43:13–20. [Google Scholar]
- 41.Xu C, Webb WW. J Opt Soc Am B: Opt Phys. 1996;13:481–491. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.