

Abstract
Fragile X associated tremor/ataxia (FXTAS) affects older males carrying premutation, that is, expansions of the CGG repeat (in the 55-200 range), in the FMR1 gene. The neurological changes are linked to the excessive FMR1 mRNA, becoming toxic through a ‘gain-of-function’. Since elevated levels of this mRNA are also found in carriers of the smaller expansion (grey zone) alleles, ranging from 40 to 54 CGGs, we tested for a possible role of these alleles in the origin of movement disorders associated with tremor.
We screened 228 Australian males affected with idiopathic Parkinson’s disease and other causes of parkinsonism recruited from Victoria and Tasmania, for premutation and grey zone alleles. The frequencies of either of these alleles were compared with the frequencies in a population-based sample of 578 Guthrie spots from consecutive Tasmanian male newborns (controls). There was a significant excess of premutation carriers (Fisher’s exact test P =.006). There was also a more than 2-fold increase in grey zone carriers in the combined sample of the Victorian and Tasmanian cases, with odds ratio (OR)=2.36, and 95% confidence intervals (CI):1.20-4.63, as well as in Tasmanian cases only (OR=2.33, 95% CI:1.06-5.13), compared with controls. The results suggest that the FMR1 grey zone alleles, as well as premutation alleles, might contribute to the aetiology of disorders associated with parkinsonism.
Keywords: fragile X, parkinsonism, FMR1, CGG repeat, premutation carriers, grey zone carriers
INTRODUCTION
The fragile X-associated tremor/ataxia syndrome (FXTAS), a late-onset neurodegenerative disorder1 is caused by a premutation in the FMR1 gene,2 which arises from an expansion of the trinucleotide CGG repeat in the 5’ untranslated region of this gene from the normal range of 6-~40, to the range of 55-200 repeats.3 The role of the intermediate length, or grey zone (GZ) FMR1 alleles in the aetiology of FXTAS is still unknown. GZ alleles have been relatively poorly defined with respect to repeat size, with the originally recommended range of 45- 54 CGGs,3 but extending to 35 - 60 repeats in different studies.
Typical features of FXTAS are intention tremor, ataxia and cognitive decline.1,4-6 MRI changes include white matter lesions of the middle cerebellar peduncles (MCP) in 50% of patients, widespread cerebellar and cerebral white matter T2 hyperintensities, and cortical atrophy.1,4-8 The characteristic histological changes are ubiquitin-positive intranuclear inclusions, which are most numerous in the hippocampus but also abundant in both neuronal and astrocytic nuclei throughout the cerebrum, the cerebellum (apart from Purkinje cells) and the brainstem.9
The FMR1 premutation is associated with elevated levels of mRNA,10 and the neurodegenerative changes in FXTAS have been explained by RNA becoming toxic through a ‘gain-of-function’.1,4,9 In this model, elevated levels of FMR1 transcript interfere with the binding of several RNA processing factors and generate novel forms of mRNA. Progressive neuronal death results from functional changes in, and sequestration of, the corresponding proteins.11 Drosophila and premutation mouse models have provided support for this notion.12,13
The population prevalence of male premutation carriers ranges from 1 in 250 to 1:810,14 and, with age, approximately 40% of these eventually develop FXTAS.4,6 However, the prevalence of premutation alleles amongst groups of patients with late onset movement disorders has been lower than expected. In a retrospective survey,15 the relatively highest prevalence of premutation carriers, ranging from 0 to 3.95%, was in males over 50 with cerebellar ataxia or multiple system atrophy-cerebellar type (MSA-C). Surveys of male patients originally diagnosed with atypical,16 or typical,17-19 Parkinson’s disease (PD) have failed to show a statistically significant excess of PM carriers. Three carriers were found among 776 patients with idiopatic PD,20 and although this was at least a three-fold greater rate than the normal population prevalence,14 it was not statistically significant.
Fewer studies have considered the prevalence of GZ alleles in PD,17-19 and none found them to significantly exceed of the population prevalence. However, as ‘toxic’ mRNA levels are elevated in GZ carriers, these alleles might be associated with an increased risk of developing movement or other disorders similar to those seen in PM carriers.21 Indeed, premature ovarian failure which has an increased association with PM alleles,22 is also increased in female carriers of GZ alleles.23,24 In this study we screened a sample of individuals affected with parkinsonian disorders for the prevalence of small CGG expansion FMR1 alleles, encompassing the PM and GZ range, and found a significant excess of both these alleles relative to the population based control data.
SAMPLE AND METHODS
Data collection and analysis were approved by the La Trobe University, St Vincent’s Hospital and Royal Melbourne Hospital Ethics Committees, and by the Southern Tasmanian Health and Human Research Ethics and the relevant University Ethics Committees at the Royal Hobart Hospital. All individuals were asked and gave informed consent for their participation in the study. The cases and controls were of similar ethnicity, all were white Caucasians of European, mainly northern European, origin, residing in Australia. The cases were recruited between 2003-2006 from two Australian states, Victoria and Tasmania, as part of a broader (continuing) study of genetic influences on parkinsonian disorders.
Victorian Cases
92 males were recruited from three movement disorders clinics in Melbourne. Most (84 cases) met UK PDS Brain Bank Criteria (Sections I-III) for idiopathic PD (iPD), 12 of these subjects manifested some atypical features while still meeting the brain bank criteria. Eight subjects were diagnosed with other parkinsonian syndromes, including 2 MSA cases. The mean age was 66.81 (SD=9.12), ranging from 46 to 86 years.
Tasmanian Cases
136 Tasmanian participants were recruited using a questionnaire based on previously validated screening tests;25,26 107 of these were diagnosed with iPD (including 10 with atypical features) by a neurology specialist, and 29 subjects, by general practitioners. This diagnosis concurred with the questionnaire, although the authors did not verify it by examination. The mean age of Tasmanian participants was 71.12 (SD=10.14), ranging from 28 to 91 years. All Tasmanian and most Victorian cases provided saliva samples, and cheek swab samples were taken from 38 Victorian participants. In rare cases where the CGG sizing test failed to produce a result, the test was repeated using blood samples.
Control sample
The population based frequency of FMR1 allele variants was obtained by anonymous screening of 576 dry blood spots from consecutive Tasmanian male newborns,27 using DNA extracted from autoclaved Guthrie cards.28
Screening for small CGG expansions
Saliva samples were collected in an Oragene-DNA collection tubes (DNA Genotek Inc., Ontario, Canada), and genomic DNA was extracted according to the manufacturers’ instructions. Genomic DNA from check swabs was obtained by placing a buccal brush in a labelled tube containing 400 μL of 50 mM NaOH, rotating, and incubating at 95°C for 15 minutes. 80 μL of 1M Tris-HCl pH 7.5 was added, mixed and centrifuged, and then stored at 4°C. The supernatant was removed and stored at −20°C. The CGG repeat sizing was performed using a fully validated PCR assay29 using primers c and f,30 and was assessed with precision of +/- one triplet repeat across the normal and GZ ranges, using a fragment analyser (MegaBace, GE Healthcare).
For the Tasmanian newborns DNA was extracted from autoclaved Guthrie cards following the procedure described in Holden et al.28 Scoring of the CGG repeat size was as in:28 using the same primers c and f30 in a radioactive reaction. PCR products, together with allele ladders of known sizes, were run on a 6% polyacrylamide gel. The results (triplet repeat number) of this test correlated very highly with the results obtained from the saliva samples (r2=0.974, y=0.999x-0.108); of the 43 samples, 31 showed an identical repeat number, 9 differed by 1 repeat, and 3 differed by 2 repeats; this shows the robustness of the analyses.
Statistical analysis
The frequencies of PM and GZ alleles were compared between cases and controls using Fisher’s exact test. The univariate logistic regression was used to obtain odds ratio (OR) and confidence intervals (CI). The mean size of CGG repeat for cases was compared with controls using the t-test, and the Mann-Whitney U test considering the shape of the distributions of CGG repeat sizes (see Fig 1). All analyses were conducted using STATA package (STATA Statistical Software, 2004; Release 8.2, College Station, Stata Corporation, Texas).
Fig 1.
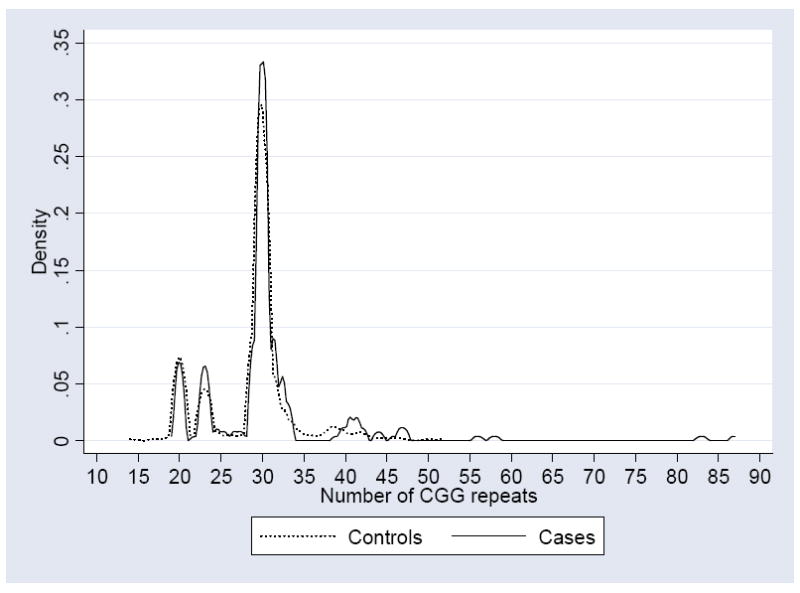
Kernel density estimate of the distributions of CGG repeats in the sample with parkinsonism, and in the control sample of Tasmanian newborns.
RESULTS
Four premutation carriers (56, 58, 83 and 87 CGGs, respectively) were identified among 228 cases, compared with none in 576 controls (Table 1), representing a significant excess (P value from the Fisher’s exact both one-sided and two-sided test=0.006). All four were originally diagnosed as iPD, one with atypical features.
Table 1.
Comparison of the relative frequencies of the FMR1 alleles with CGG repeat expansions within the PM range (55-200 CGGs), and GZ range (40-54 CGGs), between samples of combined Victorian and Tasmanian cases1 and Tasmanian cases alone,2 and control sample, using Fisher’s exact test. The odds ratio (OR) and confidence intervals (CI) estimated using univariate logistic regression are also shown.
| Cases | Controls | Fisher’s test | Logistic regression | |||||
|---|---|---|---|---|---|---|---|---|
| CGG category | n | (%) | n | % | P-value | OR | 95% CI | P-value |
| 1Premutation- | 224 | (98.50) | 576 | (100) | ||||
| Premutation+ | 4 | (1.75) | 0 | (0) | 0.006 | NA | NA | NA |
| 1Grey zone- | 211 | (92.54) | 557 | (96.70) | ||||
| Grey zone+ | 17 | (7.46) | 19 | (3.30) | 0.014 | 2.36 | 1.20-4.63 | 0.012 |
| 2Grey zone- | 126 | (92.95) | 557 | (96.70) | ||||
| Grey zone+ | 10 | (7.35) | 19 | (3.30) | 0.049 | 2.33 | 1.06-5.13 | 0.036 |
All P-values reported in the Table were 2-sided. 1-sided P-value of Fisher’s test for PM category was 0.006; for GZ category it was 0.011 for combined sample, and 0.034, for Tasmanian sample only.
Tasmanian and Victorian cases combined.
Tasmanian cases only
There were also significantly more carriers of GZ alleles (40-54 CGG repeats range) in the combined Victorian and Tasmanian cases compared to controls (Table 1). These carriers are more likely to occur in PD sample than in control sample (OR= 2.36; 95% CI: 1.20-4.63, P=0.012). Because the control subjects were Tasmanian newborns, their frequency of GZ alleles was separately compared to that for the Tasmanian cases which were derived from the same (Tasmanian) population, with a similar result (OR= 2.33; 95% CI:1.06-5.13, P=0.036). P-values from Fisher’s exact test (two-sided) were 0.011 for the combined sample, and 0.034, for Tasmanian sample only (Table 1). In 9 GZ carriers the original diagnosis was iPD (3 with atypical features), and in the tenth case the diagnosis was MSA.
The 40 repeat allele was adopted as the lower boundary of GZ range, based on earlier finding that increased transcriptional activity was observed when FMR1 alleles exceeded 39 repeats.21 Notably, adopting 41 repeats as the lower boundary did not change the results.
The mean number of CGG repeats in the combined sample of cases (30.3, SD=7.78) was significantly different from the mean for the control sample (28.8, SD= 5.05). The P value from the t-test was 0.0014, and from the Mann-Whitney test it was 0.0089.
DISCUSSION
There were significantly more FMR1 premutation carriers among older males affected with parkinsonian disorders, most of whom being diagnosed with iPD, than in our controls. The frequency of carriers was 1.75%, compared with the population frequency ranging ~0.1% -0.4%,14 representing, on average, ten-fold increase. The range of CGG repeats in the affected carriers is similar to that seen in FXTAS patients ascertained through fragile X families either in the US,4,5 or in Australia, where 85 was the highest repeat size in our earlier study.6 There were also more GZ alleles (7.4%) in the affected males than in the controls (~ 3%). Although this finding is unprecedented, a similar trend was recently reported in another smaller study of 137 male cases affected with PD, where the frequency of GZ alleles (41-54 CGGs) was 4.9%, compared to 1.7% in 310 normal individuals of comparable age,19 and a significant association between the allele size and cognitive decline in PD cases was reported. Although the frequency of GZ alleles in both these studies represents only a 2 to 3 - fold increase relative to controls, the high prevalence of these alleles in the general population implies that even small potential effect can be borne by a large number of individuals.
Our results may be biased by both the relatively small size of the samples, and that controls were not age-matched: the effect of the PM/GZ carrier status on an individual’s life span is currently unknown. However, anonymous newborns provided an unselected, local population based data. Moreover, the prevalence of GZ carriers in our control sample was similar to frequencies reported in populations of similar (northern European) origin of all ages (reviewed in 27), and was only slightly higher than in an older age group.19 For example, the frequency of GZ carriers in our PD group (mostly of northern European origin) is significantly higher compared (using Fisher’s exact test) with the frequency in similar aged control Germans and Norwegians from the Kurz et al.19 study (14/207 with 5/310).
Our preliminary results suggest that both PM and GZ alleles may contribute to the risks of parkinsonism, and that the ‘toxic’ effect of expanded mRNA linked to premutation may also occur when the expansions are smaller. It is possible that the modest elevation of ‘toxic’ RNA, especially in the GZ and lower-end PM carriers21 may present an additional risk for neurodegeneration by synergizing with other PD susceptibility genes. This notion is supported by the occurrence of parkinsonian manifestations amongst patients primarily diagnosed as FXTAS,5,6 with mild resting tremor in nearly half of the patients, and bradykinesia and mild rigidity, in 57% and 71%.4 Furthermore, four PD patients with the FMR1 premutation were recently described,31 and one PM carrier affected by parkinsonism, who was also heterozygous for PARKIN mutation, was reported in another study.32
There is always a possibility of ascertainment bias when cases are selected from speciality clinics, by inclusion or exclusion of specific categories, such as carriers of PARKIN genes (reviewed in:33), or selective inclusion of only typical iPD.17-19 This is in contrast with broader recruitment criteria applied in our study, where the participants had not been rigorously pre-selected for either any specific diagnosis within the parkinsonism spectrum, or for the background genotype.
We conclude that both PM and GZ alleles may contribute to the acquisition of the parkinsonian phenotype, possibly through the cytotoxic effect of elevated FMR1mRNA,11,12 acting in concert with other genetic and non-genetic factors that increase susceptibility to parkinsonism. Detailed neurological and molecular testing of the carriers of small CGG expansions for the level of FMR1 transcript and its effect on cell survival, as well as for the known parkinson susceptibility factors, are required to provide confirmatory evidence for this notion.
Acknowledgments
This study was supported by the National Institutes of Child Health and Human Development Grant HD 36071, and NHMRC project grant No 330400, to Dr DZ Loesch. We thank Ms Paige Simpson for contacting, and collecting swabs from patients of the Movement Disorders clinics. We also thank Ms Elaine Prenter for assistance with the collection of data from Tasmanian PD cases.
References
- 1.Hagerman RJ, Leehey M, Heinrichs W, et al. Intention tremor, Parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. [DOI] [PubMed] [Google Scholar]
- 2.Verkerk AJ, Pieretti M, Sutcliffe JS, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- 3.Maddalena A, Richards CS, McGinniss MJ, et al. Technical standards and guidelines for fragile X: The first of series of disease specific supplements to the Standard Guidelines for Clinical Genetics Laboratories of the American College of Medical Genetics. Quality Assurance Subcommittee of the Laboratory Practice Committee. Genet Med. 2001;3:200–205. doi: 10.1097/00125817-200105000-00010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jacquemont S, Hagerman RJ, Leehey MD, et al. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA. 2004;291(4):460–469. doi: 10.1001/jama.291.4.460. [DOI] [PubMed] [Google Scholar]
- 5.Jacquemont S, Hagerman RJ, Leehey M, et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am J Hum Genet. 2003;72:869–878. doi: 10.1086/374321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Loesch DZ, Churchyard A, Brotchie P, et al. Evidence for, and a spectrum of, neurological involvement in fragile X premutation: FXTAS and beyond. Clin Genet. 2005;67:412–417. doi: 10.1111/j.1399-0004.2005.00425.x. [DOI] [PubMed] [Google Scholar]
- 7.Loesch DZ, Litewka L, Brotchie P, et al. Magnetic resonance imaging study in older fragile X premutation male carriers. Ann Neurol. 2005;58:326–330. doi: 10.1002/ana.20542. [DOI] [PubMed] [Google Scholar]
- 8.Brunberg JA, Jacquemont S, Hagerman RJ, et al. Fragile X premutation carriers: characteristic MR imaging findings in adult males with progressive cerebellar and cognitive dysfunction. Am J Neuroradiol. 2002;23:1757–1766. [PMC free article] [PubMed] [Google Scholar]
- 9.Greco CM, Hagerman RJ, Tassone F, et al. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain. 2002;125:1760–1771. doi: 10.1093/brain/awf184. [DOI] [PubMed] [Google Scholar]
- 10.Tassone F, Hagerman RJ, Taylor AK, et al. Elevated levels of FMR1 mRNA in carrier males: a new mechanism of involvement in fragile X syndrome. Am J Hum Genet. 2000;66:6–15. doi: 10.1086/302720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Galvao R, Mendes-Soares L, Camara J, Jaco I, Carmo-Fonseca M. Triplet repeats, RNA secondary structure and toxic gain-of-function models for pathogenesis. Brain Res Bull. 2001;56:191–201. doi: 10.1016/s0361-9230(01)00651-7. [DOI] [PubMed] [Google Scholar]
- 12.Jin P, Zarnescu DC, Zhang F, et al. RNA-mediated neurodegeration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39:739–747. doi: 10.1016/s0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- 13.Willemsen R, Hoogeveen-Westerveld M, Reis S, et al. The FMR1 CGG repeat mouse displays ubiquitin-positive intranuclear neuronal inclusions; implications for the cerebellar tremor/ataxia syndrome. Hum Mol Genet. 2003;12:949–959. doi: 10.1093/hmg/ddg114. [DOI] [PubMed] [Google Scholar]
- 14.Song FJ, Barton P, Sleightholme V, et al. A screening for fragile X syndrome : a literature review and modeling study. Health Technol Assess. 2003;7(16):1–106. doi: 10.3310/hta7160. [DOI] [PubMed] [Google Scholar]
- 15.Jacquemont S, Leehey MA, Hagerman RJ, et al. Size bias of fragile X premutation alleles in late-onset movement disorders. J Med Genet. 2006;43:804–809. doi: 10.1136/jmg.2006.042374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tan EK, Zhao Y, Puong KY, et al. Fragile X premutation alleles in SCA, ET and parkinsonism in an Asian cohort. Neurology. 2004;63:362–363. doi: 10.1212/01.wnl.0000130199.57181.7b. [DOI] [PubMed] [Google Scholar]
- 17.Deng H, Le W, Jankovic J. Premutation alleles associated with Parkinson disease and essential tremor. JAMA. 2004;292(14):1685–1686. doi: 10.1001/jama.292.14.1685-b. [DOI] [PubMed] [Google Scholar]
- 18.Toft M, Aasly J, Bisceglio G, et al. Parkinsonism, FXTAS, and FMR1 premutation. Mov Disord. 2005;20:230–233. doi: 10.1002/mds.20297. [DOI] [PubMed] [Google Scholar]
- 19.Kurz MW, Schlitter AM, Klenk Y, et al. FMR1 alleles in Parkinson’s disease: Relation to cognitive decline and hallucinations, a longitudinal study. J Geriatr Psychiatry Neurol. 2007;20(2):89–92. doi: 10.1177/0891988706297737. [DOI] [PubMed] [Google Scholar]
- 20.Kraff J, Tang HT, Cilia R, et al. Screen for excess FMR1 premutation slleles among males with parkinsonism. Arch Neurol. 2007;64(7):1002–1006. doi: 10.1001/archneur.64.7.1002. [DOI] [PubMed] [Google Scholar]
- 21.Loesch DZ, Quang BM, Huggins RM, et al. Transcript levels of the intermediate size or grey zone alleles are elevated, and correlate with the number of CGG repeats. J Med Genet. 2007;44:200–204. doi: 10.1136/jmg.2006.043950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Allingham-Hawkins DJ, Babul-Hirji R, Chitayat D, et al. Fragile x premutation is a significant risk factor for premature ovarian failure: the International Collaborative POF in Fragile X study-a preliminary data. Am J Med Genet. 1999;83:322–325. [PMC free article] [PubMed] [Google Scholar]
- 23.Bodega B, Bione S, Dalpra L, et al. Influence of intermediate and uninterrupted FMR1 CGG expansions in premature ovarian failure manifestation. Hum Reprod. 2006;21(4):952–957. doi: 10.1093/humrep/dei432. [DOI] [PubMed] [Google Scholar]
- 24.Bretherick KL, Fluker MR, Robinson WP. FMR1 repeat sizes in the gray zone and high end of the normal range are associated with premature ovarian failure. Hum Genet. 2005;117:376–382. doi: 10.1007/s00439-005-1326-8. [DOI] [PubMed] [Google Scholar]
- 25.Calne DB, Snow BJ, Lee C. Criteria for diagnosing Parkinson’s disease. Ann Neurol. 1992;32(Suppl):S125–127. doi: 10.1002/ana.410320721. [DOI] [PubMed] [Google Scholar]
- 26.Duarte J, Claveria IE, de Pedro-Cuesta J, et al. Screening Parkinson’s disease: a validate questionnaire of high specificity and sensitivity. Mov Disord. 1995;10:643–649. doi: 10.1002/mds.870100518. [DOI] [PubMed] [Google Scholar]
- 27.Mitchell RJ, Holden JJA, Zhang C, et al. FMR1 (fragile X) alleles in Tasmania: A screening study of the special educational needs (SEN) population. Clin Genet. 2005;67:38–46. doi: 10.1111/j.1399-0004.2004.00344.x. [DOI] [PubMed] [Google Scholar]
- 28.Holden JJA, Chalifoux M, Wing M, et al. A rapid, reliable, and inexpensive method for the detection of di-and trinucleotide repeat markers and disease loci from dried blood spots. Am J Med Genet. 1996;64:313–318. doi: 10.1002/(SICI)1096-8628(19960809)64:2<313::AID-AJMG16>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 29.Khaniani MS, Kalitsis P, Burgess T, Slater HR. An improved Diagnostic PCR Assay for identification of Cryptic Heterozygosity for CGG Triplet Repeat Alleles in the Fragile X Gene (FMR1) Mol Cytogenet. 2008;1(1):5. doi: 10.1186/1755-8166-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fu YH, Kuhl DPA, Pizzuti A, et al. Variation of the CGG repeat X site results in genetic instability: Resolution of Sherman paradox. Cell. 1991;67:1047–1058. doi: 10.1016/0092-8674(91)90283-5. [DOI] [PubMed] [Google Scholar]
- 31.Hall DA, Howard K, Hagerman R, Leehey MA. Parkinsonism and FMR1 premutation carriers may be indistiguishable from Parkinson’s disease. Parkinsonism and Related Disorders. 2009;15:156–159. doi: 10.1016/j.parkreldis.2008.04.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hedrich K, Pramstaller PP, Stubke K, et al. Premutation in the FMR1 gene as a modifying factor in Parkin-associated Parkinson’s Disease? Mov Disord. 2005;20(8):1060–1062. doi: 10.1002/mds.20512. [DOI] [PubMed] [Google Scholar]
- 33.Yang YX, Wood NW, Latchman DS. Molecular basis of Parkinson’s disease. Neuroreport. 2009;20(2):150–156. doi: 10.1097/WNR.0b013e32831c50df. [DOI] [PubMed] [Google Scholar]