

Abstract
Improvements in adult cancer survivorship can be achieved from behavioral changes and adopting screening programs. Yet, these approaches cannot be readily applied to lower the morbidity and mortality from childhood cancers. Rather, pediatric oncologists must rely on procedures and therapies to treat, rather than prevent malignancies. The systematic application of chemotherapy, radiation therapy, and surgery has led to remarkable advances in survival but these improvements have come at a cost. Children routinely receive chemotherapy agents that were designed decades ago, and these drugs have predictable side effects that result in the loss of potential for long-term survivors. The advent of targeted applications of immune-based therapies offers children with cancer a new class of oncolytic therapies that may be used to treat disease refractory to conventional approaches and lessen the toxicity of current treatment regimens without compromising remission. This review explores how 3 components of the immune system—T cells, natural killer (NK) cells, and antibodies—can be used for therapy of pediatric malignancies.
Immunotherapies have been developed for childhood cancer that range from being considered as standard practice and widely applied, to experimental and only available at specialized centers. Examples of readily available immunotherapies that have entered into clinical practice include a commercially available vaccine for the prevention of human papillomavirus (HPV) infection and associated cancers, and therapeutic monoclonal antibodies (mAbs) targeting CD20 to help treat lymphomas. The experimental immunotherapies for pediatric malignancies encompass all aspects of the immune system. Investigators have tested vaccines, infused antigen-specific T cells, and genetically modified T cells rendered specific for antigen, adoptively transferred NK cells, and administered exogenous cytokines. In this review, Dr. Grupp discusses how the adaptive immune system can be manipulated for the treatment of neuroblastoma (NBL). Dr. Verneris then shows how the innate immune system can be manipulated for the treatment of pediatric neoplasms. Finally, Dr. Sondel demonstrates how mAb, and in particular antibody-cytokine fusions, can be used to treat childhood cancer. These are 3 examples from a long list of potential immunotherapies, as many investigators have developed and are testing new immune-based treatments for pediatric malignancies.
CELL THERAPY FOR NEUROBLASTOMA
NBL is the second most common solid malignancy of childhood (after CNS tumors). Although NBL has a broad spectrum of clinical presentations and behavior, high-risk NBL is still difficult to cure [1]. Some progress in treating high-risk NBL has correlated with escalation of therapeutic intensity [2], although even with an apparent complete remission following maximal-intensity induction therapy, long-term event-free survival (EFS) with standard treatment stubbornly remains less than 40%. In this section, we describe several cell therapy-based trials and possible future approaches for patients with this disease. We will begin with the current standard (stem cell support for high-dose chemotherapy), and move to T cell-based immunotherapy.
INFUSION OF AUTOLOGOUS HEMATOPOIETIC STEM CELLS (HSCS) FOR NBL
HSCs capable of reestablishing tri-lineage hematopoiesis can be acquired from the bone marrow, but in the setting of autologous transplantation, the source of HSC has moved to collection of mobilized peripheral blood stem cells (PBSC). The harvest and storage of a patient’s own HSC followed by reinfusion of those cells after high-dose (generally myeloablative) chemotherapy is commonly referred to as autologous hematopoietic stem-cell transplantation (HSCT). A child presenting with high-risk NBL is generally considered a good candidate for autologous HSCT. Generally, NBL at presentation is a chemotherapy sensitive disease. Although most patients can achieve a complete or partial remission with induction chemotherapy, a high response rate does not translate into a high EFS rate; 80% to 85% of patients have initially chemotherapy-responsive disease, but less than 20% are long-term survivors with conventional chemotherapy.
The study that defined autologous HSCT as standard of care for high-risk NBL was Children’s Cancer Group 3891. Patients were randomized to a consolidation regimen with autologous HSCT (supported by purged bone marrow) versus continuation chemotherapy [3]. This study found that EFS was improved in the group that received autologous HSCT. In the initial report, the authors estimated a 3.7-year EFS of 38% from diagnosis in those patients who underwent autologous HSCT followed by the differentiation agent isotretinoin. An important further innovation in the use of HSC therapies for NBL was the switch to PBSC from marrow. The more rapid recovery afforded by PBSC has decreased the risk of HSCT, and allowed the concept of autologous HSCT to be extended to sequential cycles. This “tandem transplantation” approach is based on the hypothesis that further dose intensity in this setting may result in improved outcome. Several groups have tested tandem HSCT with promising results in pilot and phase II studies [4–6]. We have concluded the largest of these studies, conducted over 6 years at 4 cooperating institutions and observed a 3-year EFS of 55% in a sequentially treated group of 97 patients (Figure 1). The study was designed around early collection of PBSC, the use of CD34 selection as a method to purge NBL cells from the PBSC products, and 2 nonover-lapping myeloablative consolidation regimens. There were 3 cases of EBV lymphoproliferative disease (EBV-LPD) seen among patients treated in this fashion. EBV-LPD is uncommon following autologous HSCT, and the Mackall group has suggested that T-cell depletion that results from CD34 selection may not increase immunosuppression [7,8]. Our study experience would suggest that the combination of the use of a CD34-selected PBSC product and tandem transplant is more immunosuppressive than autologous HSCT using unpurged PBSC [9,10].
Figure 1.
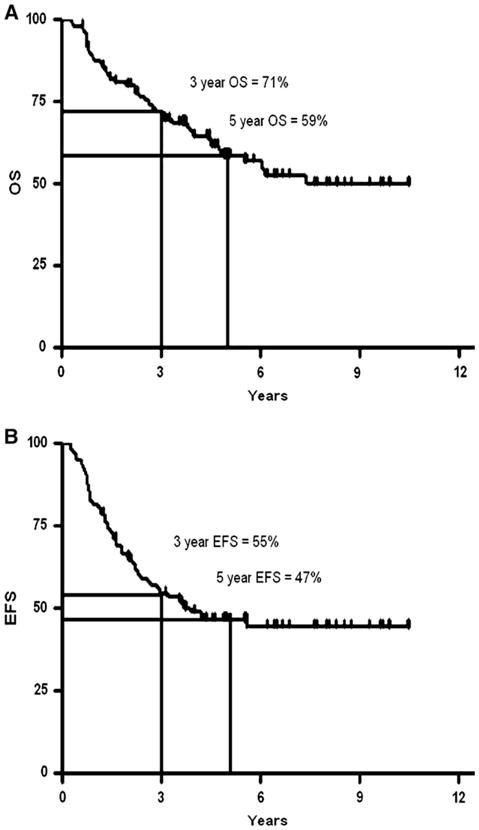
Kaplan-Meier curvesshowing the overall and event-free survival (OS, EFS) for patients undergoing tandem autologous HSCT for high-risk NBL. The patients received carboplatin/etoposide/cyclophosphamide for the first HSCT, and melphalan/TBI for the second. (A) Overall survival (OS) from diagnosis. (B) EFS from diagnosis.
T CELL AUGMENTATION FOR NBL
The issue of immunosuppression induced by autologous HSCT is important when considering alternative approaches to treating high-risk NBL. Although there is some suggestion that tandem HSCT may improve outcome in these patients, it is indisputable that we have reached the limit of dose escalation. An alternative approach is required. T cell-based therapies, possibly paired with a cancer vaccine, represent a major area to explore novel treatments [11,12]. However, T cells that may have antitumor efficacy, may not be well-suited to treating bulky disease, and thus might be most efficacious if infused at the point of minimal residual disease (MRD). This is the point reached after chemotherapy, surgery, radiation, and HSCT for NBL. Immunotherapy and/or tumor vaccines should probably be deployed as quickly as possible after completion of conventional therapy, but this is also a point where numbers of T cells and associated effector function are minimal to absent. One solution to this problem is to provide T cells to the patient in an attempt to speed immunologic recovery. This also has the potential to harness a profoundly lymphopenic environment supportive of homeostatic expansion. Unfortunately, the passenger T cells provided with a PBSC product, although large in number, do not provide this solution, as recovery of cellular immunity after standard autologous PBSC transplant (PBSCT) takes many months.
We have recently tested an alternative approach in studies at the University of Pennsylvania and Children’s Hospital of Philadelphia (CHOP). The cell product utilized in all of these studies is ex vivo-activated and expanded autologous T cells, using an artificial “antigen-presenting cell” of anti-CD3 and anti-CD28 activating antibodies coupled to beads [13]. The cell manufacturing process operating in compliance with current good manufacturing practices (GMP) produces a highly activated polyclonal T cell population, containing numerically expanded T cells that reflect the full repertoire of the cells input into the culture [14,15]. We have referred to the infusion of these activated T cells into lymphodepleted patients as T cell augmentation (TCA). We have completed a phase I trial of TCA in adult and pediatric patients with high-risk lymphoma following autologous CD34-selected PBSC transplantation [16], demonstrating promising normalization of lymphocyte counts. In many cases, an absolute lymphocytosis was observed following TCA, suggesting that homeostatic T cell proliferation was induced.
In ongoing studies at CHOP, we have tested TCA in patients with high-risk NBL. In a series of studies, we are assessing the impact of TCA on immune reconstitution in profoundly immunodeficient patients after autologous HSCT. These patients are an informative group to study TCA, as the need for HSCT is known at diagnosis and T cells may thus be collected prior to immunosuppressive chemotherapy. Some of our preliminary data are presented in Figure 2. Patients receiving a CD34-selected PBSC product have slow recovery of CD4+ T cells, which is significantly and strikingly improved after TCA given on day +12 after PBSC infusion. Interestingly, CD4 recovery is even more rapid when the infusion time is moved to day +2, with above-normal lymphocyte and T cell counts apparent as soon at 10 days after TCA. Among patients receiving TCA day +2 after PBSC infusion, we have observed lymphocyte counts by day 12 as high at 10,000/μL.
Figure 2.

T cell recovery as assessed by peripheral blood CD4+ T cell count at indicated times after tandem autologous HSCT. The groups include: patients receiving PBSC (no T cells), patients receiving T cell augmentation at day +12 and day +2 after the second PBSC infusion, and 4 patients from the day +2 group who experienced engraftment syndrome (Eng Synd).
Four of these patients experienced an engraftment syndrome clinically indistinguishable from autologous graft-versus-host-disease (GVHD), with fever, a rash characteristic of GVHD, and, in the 2 cases where skin biopsies were performed, the pathology was consistent with GVHD. In the current study, we are assessing the impact of TCA on response to 2 vaccines— Prevnar conjugate vaccine and influenza vaccine. Preliminary analysis of the patients receiving Prevnar on day +12 after autologous HSCT shows protective antipneuomoccal antibody titers to multiple serotypes as early as day +30 (S. Grupp, unpublished data), which supports the hypothesis that TCA could be used to support an anticancer immunization strategy early after SCT and achievement of MRD. Similar results in patients with myeloma receiving TCA and Prevnar vaccination have recently been published by Carl June and coworkers [17].
A possible target for a therapeutic cancer vaccine could be the cancer antigen survivin. Survivin is expressed in NBL, with expression correlating with adverse outcome [18,19]. In our studies, we have observed high expression of survivin in all tested tumor biopsies from high-risk NBL patients [20]. Importantly, we have found that most HLA-A2+ patients with NBL have survivin-specific T cells as identified by tetramer-binding. These T cells might be expanded and to kill both allogeneic and autologous NBL in the appropriate HLA context. Support for this experimental design is provided by data demonstrating that when whole NBL RNA is transfected into antigen-presenting cells (APC) and these cells are used to expand T cells with specificity for NBL, the immunodominant epitope in the effector T cell response is survivin [20]. The Vonderheide group at the University of Pennsylvania is currently testing a multicomponent cancer vaccine containing survivin peptide in adult cancer patients. This leads to a potential study design where T cells are collected at diagnosis in NBL patients, undergo costimulated expansion and infusion on day +2, followed by a survivin-derived peptide-based cancer vaccine. All components necessary for such a study, including GMP cell manufacturing, a clinical grade vaccine, and sophisticated immunoassessment tools, are currently available.
DONOR-DERIVED NATURAL KILLER (NK) CELLS FOR IMMUNOTHERAPY OF TUMORS
NK cells are some of the earliest lymphocytes to recover after allogeneic HSCT. These innate immune effector cells recognize targets using cell-surface receptors that either positively or negatively modulate activation. Using these receptors, NK cells can detect and kill cells that have undergone viral infection or malignant transformation. NK cells also use these receptors to interact with APC. Paradoxically, after allogeneic HSCT they can either kill recipient APC that trigger graft-versus-host disease (GVHD) or NK cells can be activated by APC [21,22]. Following activation, NK cells rapidly produce IFN-γ, TNF-α, and granulocyte macrophage–colony-stimulating factor (GM-CSF), which can feed back to recruit and activate other components of the immune system. Considering these attributes, methods to improve donor-derived NK cell numbers and/or function after allogeneic HSCT may have significant impact.
High numbers of NK cells in allogeneic hematopoietic cell grafts are associated with improved transplant-associated outcomes. Recipients of allo-grafts with high NK cell content have significantly faster neutrophil recovery [23–25], a lower incidence of non-relapse mortality (NRM) [25,26], a reduction in both bacterial and viral infections, faster immune reconstitution [25], and less acute GVHD (aGVHD) [27] and chronic GVHD (cGVHD) [24,26]. Numerous studies show that NK cells recover early after allogeneic HSCT, regardless of hematopoietic cell source and/or graft manipulation such as T cell depletion (TCD) [28–30]. Although all patients show this rapid recovery, those with high numbers of NK cells in the peripheral circulation early after transplant (day +30) experience less NRM, aGVHD, acute myelogenous leukemia (AML) (but not adult acute lymphoblastic leukemia (ALL)) relapse, compared to patients with low NK cell numbers at day +30 [31].
NK CELL RECEPTORS AND THEIR FUNCTION
NK cells express a multitude of receptors that dictate whether they mediate cytotoxicity and/or cytokine secretion following contact with either tumor cells or allogeneic APC. These receptors can be functionally grouped into activating or inhibitory receptors (reviewed in [32] and [33]). The two best-characterized NK inhibitory receptors are the killer immunoglobulin receptors (KIR) and the heterodimeric complex between CD94 and NKG2A (CD94/NKG2A). Individuals can express up to 15 different KIR genes that recognize conserved determinates on HLA-A, -B, and -C [34]. In contrast, CD94/NKG2A recognizes HLA-E, which shows limited polymorphism [35]. As shown in Figure 3, binding of these inhibitory receptors with their respective ligands results in strong NK cell inhibition, whereas the lack of engagement of these receptors may allow target killing. Such a situation can occur if MHC class I is downregulated (associated with malignant transformation [36]) or after allogeneic transplant where donor NK cells may express a KIR that cannot recognize host major histocompatibility complex (MHC) (ie, KIR-ligand mismatch, Figure 3d and e, respectively) [37].
Figure 3.

Possible outcomes following NK-Cell inhibitory receptor interaction with target cells such as AML blasts. (A) Engagement of CD94/NKG2A with nonpolymorphic HLA-E (limited polymorphism) results in no killing; (B) lack of HAL-E expression on leukemia cells prevents CD94/NKG2A signaling, creating a situation where NK cells may not be inhibited; (C) KIR binding to self-HLA-A, or -B, or -C on leukemia cells results in NK inhibition; (D) a lack of HLA-A, -B, or -C on leukemia cells results in no KIR engagement and enhanced NK-cell killing, and (E) KIR that do not recognize HLA-A, -B, and -C on leukemia cells (as in the setting of KIR-ligand mismatched transplant) can result in increased killing.
Individual NK cells can display a varying number of inhibitory receptors on their surface that further complicates our understanding of NK cell function. As shown in Figure 4, most peripheral blood (PB)-derived NK cells will express CD94/NKG2A, either alone or in combination with KIR (right upper and lower quadrants). A small subset of NK cells will display KIR, but not CD94/NKG2A (Figure 4, upper left). Still another subset lacks both KIR and CD94/NKG2A (Figure 4, lower left quadrant). This constellation of expression of inhibitory receptors by individual NK cells creates a heterogeneous populations, each with differing ability to recognize subtle changes in MHC class I on target cells.
Figure 4.
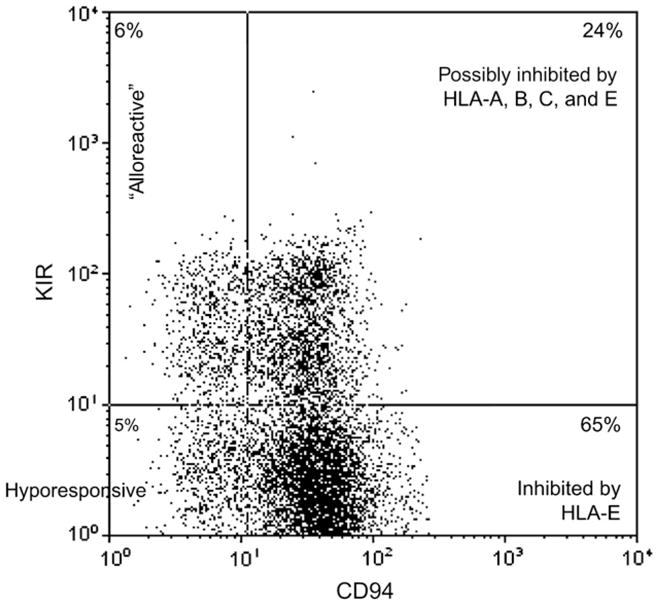
Individual NK-cells differ in inhibitory receptor expression. Shown is a FACS plot of peripheral blood NK cells (gated on CD56+CD3−) from a healthy donor. Four distinct populations of cells can be identified by staining for KIR cocktail (CD158a, CD158b, and CD158e) vs CD94. The majority of NK cells express CD94 (and NKG2A, not shown) and are inhibited by HLA-E (right upper and lower quadrant). A small fraction of cells express KIR, but not CD94 (left upper quadrant), and these cells have the potential to be “alloreactive.” The KIRnegCD94neg fraction is hyporesponsive.
The ligand for CD94/NKG2A, HLA-E shows limited polymorphism and thus, does not vary between individuals. Accordingly, NK cells that express CD94/NKG2A (either alone or in combination with KIR) are expected to be inhibited equally by donor or recipient HLA-E. In contrast, the ligands for KIR are determinants of HLA-A, -B, and -C. Such determinants are polymorphic and can vary between individuals. Following HLA mismatched transplantation there is a possibility that the KIR present on donor NK cells will not recognize HLA on recipient leukemia (Figure 3e, “KIR-ligand mismatch”). If such a situation occurs, the NK cells that express only KIR (and not CD94/NKG2A) are expected to mediate maximal graft-versus-leukemia (GVL) effects because they are not restrained by either inhibitory receptor. Such NK cells have been referred to as “alloreactive NK cells.” These cells typically make up a small fraction of PB NK cells in normal donors (Figure 4, upper left). Likewise, shortly after transplant KIR+CD94/NKG2Aneg NK cells are quite rare and increase over time [38]. Last, the NK cell subset that lacks all inhibitory receptors (KIRnegCD94/NKG2Aneg; Figure 4, lower left) is hyporesponsive and shows limited cytokine secretion and cytotoxicity. To date, it is not clear whether such cells are developmental precursors or functionally hyporesponsive [39–41].
For NK cells to fully mediate a GVL effect, NK cell activating receptors also need to be engaged. Such activating receptors include NKG2D, DNAM-1, and the natural cytotoxicity receptors (NKp30, NKp44, and NKp46) [33]. To date, less is known about the receptors that activate human NK cells than the inhibitory receptors. The ligands for some of these receptors have been identified. Examination of leukemia cell lines and freshly isolated patient samples show that the ligands for NK activating receptors are expressed. Some data also support the concept that these ligands are more abundant on AML blasts (relative to ALL blasts) [42], which may confirm the clinical observations that NK alloreacitivity may be more powerful in the myeloigenous leukemia setting [37].
STRATEGIES TO ENHANCE NK CELL FUNCTION AFTER ALLOGENEIC HSCT
Whether NK cells are fully functional after allogeneic HSCT is not entirely clear. A number of factors may impact NK cell immunobiology including conditioning regimens (ie, presence of ATG or campath H1), GVHD prophylaxis (and treatment), donor cell source, and viral reactivation. However, even within the same regimen, differences have been noted. For instance, early after allogeneic HSCT, Ruggeri et al. [37] could generate functional, alloreactive NK cell clones from recipients. In contrast, Nguyen et al. [43] found that NK cells recovering early showed poor cytotoxicity. In the above studies, no GVHD prophylaxis was used. However, immune suppressive agents are administered to most patients after HSCT in which the allo-graft is not manipulated and the impact of these medications on NK cell function is just now being understood. Wang et al. [44] recently showed that culture of NK cells with physiologic levels of cyclosporine A (CSA) results in impaired proliferation of the CD56+CD16+KIR− NK cell subset, whereas the CD56+CD16negKIRneg cells were less effected. Interestingly, this latter subset is also present at higher numbers early after HSCT, suggesting that CSA may play account for this. Exposure to CSA did not impair cytotoxicity against leukemia cell lines. In contrast, following corticosteroid treatment for GVHD, patient NK cells showed less activating receptor expression (NKp30 and NKp46), and this correlated with a reduction in cytotoxicity [45].
As described above, large numbers of NK cells in the graft or rapid NK cell recovery are associated with improved outcomes. Thus, strategies to enhance NK cell recovery after allogeneic HSCT may be indicated. Although exogenous IL-2 can increase PB-derived NK cells after autologous transplant [46], it may also increase regulatory T cells [47,48], which could negatively modulate the GVL effect. Accordingly, some studies have failed to show increased NK cell killing when posttransplant IL-2 is used [46]. Other cytokines, such as IL-12, IL-15, IL-18, and IL-21, all positively modulate NK cell function, but may also exacerbate GVHD. To date, none have been administered after allogeneic HSCT.
Increasing NK cell numbers could also be achieved by donor leukophoresis, followed by TCD, in vitro activation with IL-2, and adoptive transfer. Such studies show that haploidentical NK cell infusions can be safely administered to patients without GVHD and in some chemotherapy refractory AML patients, hematologic remissions could be achieved [49].
Other approaches may include the use of pharmacologic agents to “sensitize” leukemia to NK cell attack. Recently, Rohner [50] has shown that a cocktail of differentiation agents (5-aza-2′-deoxycytidine, tri-chostatin A, vitamin D3, bryostatin-1) increases NKG2D ligand expression on AML cell lines. Similarly, using the HDAC inhibitor trichostatin, Kato et al. [51] could increase NK killing of patient-derived ALL blasts by increasing NKG2D ligand expression. Other “off-the-shelf” approaches may include use of mAb to block NK cell inhibitory receptor (KIR or CD94/NKG2A) signaling. Such antibodies against CD94 or KIR have been shown to enhance in vitro cytotoxicity against HLA expressing targets [52,53], such as ALL cell lines.
ANTIBODY FOR IMMUNOTHERAPY OF TUMOR
Since the original description of antibodies potentially functioning as “magic bullets” by Paul Erlich at the turn of the last century, immunologists have sought ways to utilize the immune system and particularly antibodies as selective antitumor therapeutics. Not until 1975, when Kohler and Milstein described the technology for production of monoclonal, mono-specific antibodies, was the technology available to potentially realize Erlich’s dream in the setting of cancer treatment. Initial efforts to immunize mice with human tumors to generate tumor-specific mAbs resulted in a myriad of antibodies that recognized species specific antigenic determinants expressed both on tumors and normal cells. Many of these target antigens are tissue specific, and have been of great help in identifying important structures on the surfaces of normal human cells. This has included characterization of numerous normal membrane components of distinct subsets of immune cells based on the initial distinction of the “T4” (now CD4) and T8 (now CD8) determinants on helper versus cytotoxic T cells. With diligent screening, a somewhat small set of target antigens recognizable by mouse antibodies were identified that were either specific to tumors of certain histology, or were highly overexpressed by certain tumors and expressed at low levels on most normal tissues or only on a small subset of potentially “expendable” normal tissues. The underlying principle of utilizing such reagents for therapy is based on the selective recognition of tumor cells by tumor reactive monoclonals but not of most normal cells by the putative therapeutic antibody [54–58].
Initial antigens of interest that were described included the CD10, CD20, CD19, and CD5 molecules expressed on subsets of human B and T cells, including leukemic cells, the GD2 molecule on NBL and melanoma, as well as molecules expressed on malignancies more common in adults such as HER-2 seen in breast cancers and the epithelial cell adhesion molecule (EP-CAM) seen on adult epithelial cancers.
Although the above targets (and several more) have been used in clinical testing for cancer treatment, a variety of other mAbs have also been tested, but not themselves as “anticancer therapeutics.” This category includes the battery of “immunosuppressive” mAbs that have been used in several clinical settings of auto-immune disease and to prevent graft rejection or GVHD, particularly in the setting of allogeneic HSCT. These include mAbs against distinct lymphoid subsets or against triggering receptors on the surface of allo-activated lymphocytes (such as the IL-2 receptor).
ANTIBODY ENGINEERING
Once the genes for mAbs were cloned and placed in expression plasmids, it was possible to genetically engineer these genes for therapeutic purposes [57,59,60]. This has included grafting the variable regions of mouse immunoglobulin genes onto the constant regions of human immunoglobulin genes to create “chimeric” mAbs. When the small complementarity determining region (CDR) of the murine antibodies (which determines antigen binding) are grafted into the appropriate CDR locations of human immunoglobulin genes, the resulting protein is a “humanized” antibody. Such chimeric or humanized antibodies should be recognized as less “foreign” to the human immune system and thus be neutralized less actively. In addition, modification of the Fc region of the immunoglobulin molecule either through amino acid substitution or by modifying the glycosylation can influence the effector functions of the antibody. Finally, genetic manipulation can graft entirely distinct molecules onto immunoglobulins to provide fusion proteins with multiple specificities and functions (see below).
MECHANISMS OF ACTION OF TUMOR-REACTIVE MAB THERAPEUTICS
mAbs that recognize targets selectively expressed on tumors may have antitumor effects through a variety of mechanisms. First is activation of the complement cascade, which results in membrane damage and osmotic lysis to the target cell. Second involves activation of antibody dependent cellular cytotoxicity (ADCC). This involves activation of effector cells (normally NK cells, neutrophils, or monocytes/macrophages) that express activating Fc receptors [61,62]. Once the mAb forms multipoint binding to the target antigen on the tumor cells, the Fc receptor lattice on the effector cells recognizes the pattern of expressed immunoglobulins on the tumor cell and results in effector cell activation and the destruction of target cells by molecular pathways distinct for NK cells (granzyme, perforin, and/or Fas ligand) or for neutrophils and macrophagesmonocytes (reactive oxygen species, nitric oxide, tumor necrosis factor, and other pathways) [63–65]. mAb can also cause direct antitumor effects by binding to and “blocking” growth factor receptors expressed on tumor cells, provided that these antibodies bind to the receptor without activating it (inhibitory/antagonistic mAbs). For example, antibodies to the epidermal growth factor receptor (EGFR) block its ability to be stimulated by its selective ligand (EGF), resulting in tumor growth inhibition. In addition, some agonistic mAbs bind to cell membrane receptors and transmit an active signal. If the membrane receptor that is recognized is a “death receptor” such as Fas, the antibody can result in cell death. Although this has theoretic implications, most such antibodies do not show tumor specificity.
Of the panel of tumor reactive mAbs that have had clinical testing and activity, a few have been FDA approved for general clinical use [54,55]. The mAb against the CD20 determinant on B cells and their precursors (Rituximab) has demonstrated greatest efficacy and is in the widest clinical use [66,67]. Although this mAb may have some signaling capabilities against B cells, and can also mediate complement dependent killing (CDC), it seems that its in vivo effect is most largely mediated through ADCC. This was demonstrated by independent clinical studies evaluating the Fc receptor polymorphisms associated with antitumor activity. Patients that have inherited an NK cell receptor phenotype or a neutrophil/monocyte receptor phenotype for their Fc receptors associated with high affinity binding to the Rituximab IgG show a far greater likelihood of antitumor effects in vivo than those that have inherited Fc receptor alleles less active at ADCC with IgG [68]. Analogous data have recently been observed for treatment of NBL with tumor-specific mAb and GM-CSF [69].
ANTIBODY-MEDIATED ENHANCEMENT OF EFFECTOR-CELL FUNCTION FOR TREATMENT OF NBL
If the activity of mAbs is mediated, at least in part, through ADCC, then mechanisms to augment ADCC by activating the effector function of cells mediating ADCC should augment antitumor effects. Preclinical studies have confirmed this by using IL-2 to augment NK-mediated ADCC or GM-CSF to augment neutrophil/monocyte ADCC [65,66]. Cheung et al. [69] have recently shown that patients receiving treatment with an anit-GD2 mouse mAb (clone 3F8) together with GM-CSF show a greater likelihood of disease free survival if their neutrophil/macrophage Fc receptor phenotype is of high affinity for the 3F8 molecule. This same correlation was not observed when patients were given 3F8 but not GM-CSF, suggesting that the GM-CSF treatment was important for enabling neutrophil/monocyte mediated in vivo ADCC. In keeping with this concept, the Children’s Oncology Group (COG) is in the midst of a large Phase III trial for children with high-risk NBL that have achieved CR by multiagent induction chemotherapy with autologous bone marrow transplant consolidation. They are being randomized to cis retinoic acid alone versus cis retinoic acid together with a chimeric anti-GD2 monoclonal antibody (Ch14.18) together with GM-CSF and with IL-2 [70]. Patients are still being accrued and the comparative analysis will be in several years prior to provide sufficient follow-up.
ANTIBODY CONJUGATES AND FUSION PROTEINS
To optimize direct antibody-mediated effects together with effector cell activation, Sondel and Gillies [66] have created fusion proteins consisting of tumor reactive mAbs genetically linked to human cytokines such as IL-2. These fusion proteins have been designated immunocytokines. The initially described immunocytokine consists of the humanized anti-GD2 molecule (Hu14.18) linked to human IL-2 and designated Hu14.18-IL2 (Figure 5). This fusion protein mediates striking antitumor effects against GD2-positive malignancies in preclinical mouse models with far greater efficacy than molar equivalent amounts of IL-2, together with Hu14.18 mAb, given as separate reagents. Clinical Phase I trials of this reagent in adults with melanoma and children with neuroblastoma through the COG are complete, and Phase II studies are near completion [71,72]. In vivo immune activation using surrogate endpoints have been observed. Other conjugates include linking mAbs to radionuclides to deliver “radioimmunotherapy.” This has been particularly explored using anti-CD20 monoclonals related to Rituximab. Other mAbs have been linked directly to toxins. Gemtuzumab is an anti-CD33 monoclonal that recognizes AML cells linked to the potent toxin calicheamicin. This agent is U.S. Food and Drug Administration (FDA) approved for its use in treating adults with AML, and is currently being tested through the COG for its potential use in children with AML.
Figure 5.
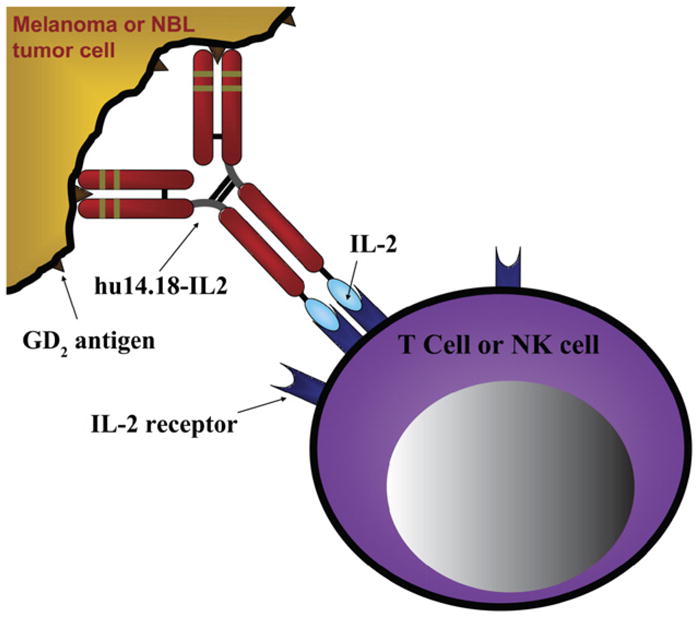
Schematic of hu14.18-IL2 (EMD 273063) immunocytokine linking GD2+ tumor cells and IL-2R+ lymphocyte [75].
NOVEL “NONTUMOR” TARGETS FOR POTENTIAL CANCER IMMUNOTHERAPY
Although most ongoing clinical testing involves mAbs that recognize antigenic determinants selectively expressed by tumor cells themselves, several more recently described target antigens are found on nontumor elements, yet may be appropriate targets for mAb administration as cancer therapy. One main category involves determinants selectively expressed on the stroma of tumors in vivo. This can include determinants selectively expressed by the stromal fibroblasts found within tumors, which may have a distinct phenotype from fibroblasts in nonneoplastic sites. Potentially more important are the pattern of integrins and growth factor receptors expressed on the neo angiogenic blood vessels found within the tumor microenvironment. These express a variety of growth factor receptors and integrin molecules (such as VEGFR, and avβ3 integrin) that are the targets of mAbs currently in clinical trials. In this setting, the tumor blood vessels and stroma are not neoplastic cells themselves, but are the target for potentially destructive actions of mAbs directed against them [56].
Other distinct targets for cancer immunotherapy include antigens that are not expressed on the tumor cells or within the tumor microenvironment. These are molecules that are expressed on immune cells that downregulate the immune system. In particular, T regulatory cells appear to inhibit antitumor immune responses and, at least preclinically, there elimination using monoclonal antibody can augment tumor reactive immune responses. Somewhat analogously, the CTLA4 molecule found on antigen-specific T cells transmits an inhibitory signal to the T cells upon contact with the appropriate costimulatory molecule on target cells or APC. The use of an antagonistic anti-CTLA4 antibody can block this immunosuppressive signal. The result is a greater population of activated tumor-specific lymphocytes. A side effect is the induction of autoimmunity with a wide variety of normal tissue targets.
CLINICAL SETTING FOR TUMOR-REACTIVE mAbs
Although the striking success of the anti-CD20 mAb, Rituximab, includes activity in the face of “bulky, clinically evident” disease [73], most preclinical trials of immunotherapy (including mAbs) demonstrate the greatest and most long-lasting effects are obtained when immunotherapies are applied in the setting of MRD. Thus, clinical efficacy for mAbs will likely require integrating antibody treatment regimens into the standard multimodality approach toward management of children with cancers [74]. The best timing for these treatments may be influenced by the schedule required for remission induction utilizing standard therapies, and the need to retain or activate some degree of endogenous immune function to facilitate the immune-mediated antitumor effects.
PROVIDING ANTIBODY RECOGNITION MECHANISMS TO EFFECTOR CELLS
The most straightforward clinical application of mAbs (or their genetic derivatives) involves their intravenous administration directly to patients as “anti-tumor drugs” with their storage and administration by a standard hospital/clinic pharmacy. More technically complex, but novel approaches, have involved genetically engineering the antigen binding site of tumor reactive mAbs onto triggering structures that can be specifically transfected into T cells or into NK cells. Such an approach confers upon the transfected effector cells tumor specific “artificial receptors” that utilize antibody recognition of the tumor antigens. Preclinical testing of these concepts, using transfected T or NK cells can demonstrate efficacy; clinical testing of such genetically engineered cells, including testing in the pediatric setting, is underway.
The capability to screen potentially thousands of tumor-reactive mAbs to identify the targets of greatest utility, coupled with genetic engineering of these molecules to optimize their clinical efficacy has already resulted in several cancer selective mAbs being approved by the FDA for standard treatment of certain malignancies. Ongoing testing is underway in the pediatric setting, particularly for lymphoid and myeloid malignancies, as well as NBL. Integrating these treatments into standard multimodality therapy will require close analysis of timing and potential synergistic versus antagonistic effects on the mechanisms involved, yet will likely result in combined regimens with greater efficacy.
CONCLUSION
Immunotherapy for pediatric cancers has been more than 85 years in the making if one starts in 1922 with Dr. William Coley, at the Sloan-Kettering Institute, who demonstrated that the growth of some cancers can be controlled, and a few advanced cancers cured, with injections of a mixed vaccine of streptococcal and staphylococcal bacteria (Coley’s toxin). Pediatric oncologists recognize that although the application of current conventional cytotoxic agents may be effective in the short run, they lead to unacceptable toxicities in the long run. As a result, centers caring for children with cancer are testing immune-based therapies since these biologics are exquisitely capable of differentiating normal from malignant cells. As demonstrated in this review, immunotherapies based on T cells, NK cells, and mAbs, are already being tailored for the treatment of pediatric malignancies. Thus, after decades in development, immunotherapy is set to enter the mainstream of pediatric oncology practice as an adjunct to other multimodal therapies.
References
- 1.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 2.Cheung NV, Heller G. Chemotherapy dose intensity correlates strongly with response, median survival, and median progression-free survival in metastatic neuroblastoma. J Clin Oncol. 1991;9:1050–1058. doi: 10.1200/JCO.1991.9.6.1050. [DOI] [PubMed] [Google Scholar]
- 3.Matthay KK, Villablanca JG, Seeger RC, et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radio-therapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N Engl J Med. 1999;341:1165–1173. doi: 10.1056/NEJM199910143411601. [DOI] [PubMed] [Google Scholar]
- 4.Grupp SA, Stern JW, Bunin N, et al. Rapid-sequence tandem transplant for children with high-risk neuroblastoma. Med Pediatr Oncol. 2000;35:696–700. doi: 10.1002/1096-911x(20001201)35:6<696::aid-mpo46>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 5.Grupp SA, Stern JW, Bunin N, et al. Tandem high-dose therapy in rapid sequence for children with high-risk neuroblastoma. J Clin Oncol. 2000;18:2567–2575. doi: 10.1200/JCO.2000.18.13.2567. [DOI] [PubMed] [Google Scholar]
- 6.Kletzel M, Katzenstein HM, Haut PR, et al. Treatment of high-risk neuroblastoma with triple-tandem high-dose therapy and stem-cell rescue: results of the Chicago Pilot II Study. J Clin Oncol. 2002;20:2284–2292. doi: 10.1200/JCO.2002.06.060. [DOI] [PubMed] [Google Scholar]
- 7.Mackall CL, Fleisher TA, Brown MR, et al. Lymphocyte depletion during treatment with intensive chemotherapy for cancer. Blood. 1994;84:2221–2228. [PubMed] [Google Scholar]
- 8.Mackall CL, Stein D, Fleisher TA, et al. Prolonged CD4 depletion after sequential autologous peripheral blood progenitor cell infusions in children and young adults. Blood. 2000;96:754–762. [PubMed] [Google Scholar]
- 9.Kanold J, Yakouben K, Tchirkov A, et al. Long-term results of CD34 (+) cell transplantation in children with neuroblastoma. Med Pediatr Oncol. 2000;35:1–7. doi: 10.1002/1096-911x(200007)35:1<1::aid-mpo1>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 10.Powell JL, Bunin NJ, Callahan C, Aplenc R, Griffin G, Grupp SA. An unexpectedly high incidence of Epstein-Barr virus lymphoproliferative disease after CD34+ selected autologous peripheral blood stem cell transplant in neuroblastoma. Bone Marrow Transplant. 2004;33:651–657. doi: 10.1038/sj.bmt.1704402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.June CH. Adoptive T cell therapy for cancer in the clinic. J Clin Invest. 2007;117:1466–1476. doi: 10.1172/JCI32446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.June CH. Principles of adoptive T cell cancer therapy. J Clin Invest. 2007;117:1204–1212. doi: 10.1172/JCI31446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levine BL, Ueda Y, Craighead N, Huang ML, June CH. CD28 ligands CD80 (B7–1) and CD86 (B&-2) induce long-term auto-crine growth of CD4+ T cells and induce similar patterns of cytokine secretion in vitro. Int Immunol. 1995;7:891–904. doi: 10.1093/intimm/7.6.891. [DOI] [PubMed] [Google Scholar]
- 14.Levine BL, Bernstein W, Craighead N, et al. Ex vivo replicative potential of adult human peripheral blood CD4+T cells. Transplant Proc. 1997;29:2028. doi: 10.1016/s0041-1345(97)00216-9. [DOI] [PubMed] [Google Scholar]
- 15.Levine BL, Cotte J, Small CC, et al. Large-scale production of CD4+ T cells from HIV-1-infected donors after CD3/CD28 costimulation. J Hematother. 1998;7:437–448. doi: 10.1089/scd.1.1998.7.437. [DOI] [PubMed] [Google Scholar]
- 16.Laport GG, Levine BL, Stadtmauer EA, et al. Adoptive transfer of costimulated T cells induces lymphocytosis in patients with relapsed/refractory non-Hodgkin lymphoma following CD34+-selected hematopoietic cell transplantation. Blood. 2003;102:2004–2013. doi: 10.1182/blood-2003-01-0095. [DOI] [PubMed] [Google Scholar]
- 17.Rapoport AP, Stadtmauer EA, Aqui N, et al. Restoration of immunity in lymphopenic individuals with cancer by vaccination and adoptive T-cell transfer. Nat Med. 2005;11:1230–1237. doi: 10.1038/nm1310. [DOI] [PubMed] [Google Scholar]
- 18.Islam A, Kageyama H, Takada N, et al. High expression of Survivin, mapped to 17q25, is significantly associated with poor prognostic factors and promotes cell survival in human neuroblastoma. Oncogene. 2000;19:617–623. doi: 10.1038/sj.onc.1203358. [DOI] [PubMed] [Google Scholar]
- 19.Azuhata T, Scott D, Takamizawa S, et al. The inhibitor of apoptosis protein survivin is associated with high-risk behavior of neuroblastoma. J Pediatr Surg. 2001;36:1785–1791. doi: 10.1053/jpsu.2001.28839. [DOI] [PubMed] [Google Scholar]
- 20.Coughlin CM, Fleming MD, Carroll RG, et al. Immunosurveillance and survivin-specific T-cell immunity in children with high-risk neuroblastoma. J Clin Oncol. 2006;24:5725–5734. doi: 10.1200/JCO.2005.05.3314. [DOI] [PubMed] [Google Scholar]
- 21.Walzer T, Dalod M, Robbins SH, Zitvogel L, Vivier E. Natural-killer cells and dendritic cells: “l’union fait la force”. Blood. 2005;106:2252–2258. doi: 10.1182/blood-2005-03-1154. [DOI] [PubMed] [Google Scholar]
- 22.Shlomchik WD, Couzens MS, Tang CB, et al. Prevention of graft versus host disease by inactivation of host antigen-presenting cells. Science. 1999;285:412–415. doi: 10.1126/science.285.5426.412. [DOI] [PubMed] [Google Scholar]
- 23.Kim DH, Sohn SK, Jeon SB, et al. Prognostic significance of platelet recovery pattern after allogeneic HLA-identical sibling transplantation and its association with severe acute GVHD. Bone Marrow Transplant. 2006;37:101–108. doi: 10.1038/sj.bmt.1705203. [DOI] [PubMed] [Google Scholar]
- 24.Larghero J, Rocha V, Porcher R, et al. Association of bone marrow natural killer cell dose with neutrophil recovery and chronic graft-versus-host disease after HLA identical sibling bone marrow transplants. Br J Haematol. 2007;138:101–109. doi: 10.1111/j.1365-2141.2007.06623.x. [DOI] [PubMed] [Google Scholar]
- 25.Kim DH, Won DI, Lee NY, Sohn SK, Suh JS, Lee KB. Non-CD34+ cells, especially CD8+ cytotoxic T cells and CD56+ natural killer cells, rather than CD34 cells, predict early engraftment and better transplantation outcomes in patients with hematologic malignancies after allogeneic peripheral stem cell transplantation. Biol Blood Marrow Transplant. 2006;12:719–728. doi: 10.1016/j.bbmt.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 26.Kim DH, Sohn SK, Lee NY, et al. Transplantation with higher dose of natural killer cells associated with better outcomes in terms of non-relapse mortality and infectious events after allogeneic peripheral blood stem cell transplantation from HLA-matched sibling donors. Eur J Haematol. 2005;75:299–308. doi: 10.1111/j.1600-0609.2005.00514.x. [DOI] [PubMed] [Google Scholar]
- 27.Yamasaki S, Henzan H, Ohno Y, et al. Influence of transplanted dose of CD56+ cells on development of graft-versus-host disease in patients receiving G-CSF-mobilized peripheral blood progenitor cells from HLA-identical sibling donors. Bone Marrow Transplant. 2003;32:505–510. doi: 10.1038/sj.bmt.1704165. [DOI] [PubMed] [Google Scholar]
- 28.Chklovskaia E, Nowbakht P, Nissen C, Gratwohl A, Bargetzi M, Wodnar-Filipowicz A. Reconstitution of dendritic and natural killer-cell subsets after allogeneic stem cell transplantation: effects of endogenous flt3 ligand. Blood. 2004;103:3860–3868. doi: 10.1182/blood-2003-04-1200. [DOI] [PubMed] [Google Scholar]
- 29.Abu-Ghosh A, Goldman S, Slone V, et al. Immunological reconstitution and correlation of circulating serum inflammatory mediators/cytokines with the incidence of acute graft-versus-host disease during the first 100 days following unrelated umbilical cord blood transplantation. Bone Marrow Transplant. 1999;24:535–544. doi: 10.1038/sj.bmt.1701921. [DOI] [PubMed] [Google Scholar]
- 30.Lamb LS, Jr, Gee AP, Henslee-Downey PJ, et al. Phenotypic and functional reconstitution of peripheral blood lymphocytes following T cell-depleted bone marrow transplantation from partially mismatched related donors. Bone Marrow Transplant. 1998;21:461–471. doi: 10.1038/sj.bmt.1701110. [DOI] [PubMed] [Google Scholar]
- 31.Savani BN, Mielke S, Adams S, et al. Rapid natural killer cell recovery determines outcome after T-cell-depleted HLA-identical stem cell transplantation in patients with myeloid leukemias but not with acute lymphoblastic leukemia. Leukemia. 2007;21:2145–2152. doi: 10.1038/sj.leu.2404892. [DOI] [PubMed] [Google Scholar]
- 32.Lanier LL. NK cell receptors. Annu Rev Immunol. 1998;16:359–393. doi: 10.1146/annurev.immunol.16.1.359. [DOI] [PubMed] [Google Scholar]
- 33.Moretta A, Bottino C, Vitale M, et al. Activating receptors and coreceptors involved in human natural killer cell-mediated cytolysis. Annu Rev Immunol. 2001;19:197–223. doi: 10.1146/annurev.immunol.19.1.197. [DOI] [PubMed] [Google Scholar]
- 34.Parham P. MHC class I molecules and KIRs in human history, health and survival. Nat Rev. 2005;5:201–214. doi: 10.1038/nri1570. [DOI] [PubMed] [Google Scholar]
- 35.Braud VM, Allan DS, O’Callaghan CA, et al. HLA-E binds to natural killer cell receptors CD94/NKG2A, B and C. Nature. 1998;391:795–799. doi: 10.1038/35869. [DOI] [PubMed] [Google Scholar]
- 36.Garrido F, Ruiz-Cabello F, Cabrera T, et al. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol Today. 1997;18:89–95. doi: 10.1016/s0167-5699(96)10075-x. [DOI] [PubMed] [Google Scholar]
- 37.Ruggeri L, Capanni M, Urbani E, et al. Effectiveness of donor natural killer cell alloreactivity in mismatched hematopoietic transplants. Science. 2002;295:2097–2100. doi: 10.1126/science.1068440. [DOI] [PubMed] [Google Scholar]
- 38.Ruggeri L, Mancusi A, Capanni M, et al. Donor natural killer cell allorecognition of missing self in haploidentical hematopoietic transplantation for acute myeloid leukemia: challenging its predictive value. Blood. 2007;110:433–440. doi: 10.1182/blood-2006-07-038687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cooley S, Xiao F, Pitt M, et al. A subpopulation of human peripheral blood NK cells that lacks inhibitory receptors for self-MHC is developmentally immature. Blood. 2007;110:578–586. doi: 10.1182/blood-2006-07-036228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anfossi N, André P, Guia S, et al. Human NK cell education by inhibitory receptors for MHC class I. Immunity. 2006;25:331–342. doi: 10.1016/j.immuni.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 41.Grzywacz B, Kataria N, Sikora M, et al. Coordinated acquisition of inhibitory and activating receptors and functional properties by developing human natural killer cells. Blood. 2006;108:3824–3833. doi: 10.1182/blood-2006-04-020198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pende D, Spaggiari GM, Marcenaro S, et al. Analysis of the receptor-ligand interactions in the natural killer-mediated lysis of freshly isolated myeloid or lymphoblastic leukemias: evidence for the involvement of the Poliovirus receptor (CD155) and Nectin-2 (CD112) Blood. 2005;105:2066–2073. doi: 10.1182/blood-2004-09-3548. [DOI] [PubMed] [Google Scholar]
- 43.Nguyen S, Dhedin N, Vernant JP, et al. NK-cell reconstitution after haploidentical hematopoietic stem-cell transplantations: immaturity of NK cells and inhibitory effect of NKG2A override GvL effect. Blood. 2005;105:4135–4142. doi: 10.1182/blood-2004-10-4113. [DOI] [PubMed] [Google Scholar]
- 44.Wang H, Grzywacz B, Sukovich D, et al. The unexpected effect of cyclosporin A on CD56+CD16− and CD56+CD16+natural killer cell subpopulations. Blood. 2007;110:1530–1539. doi: 10.1182/blood-2006-10-048173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vitale C, Chiossone L, Cantoni C, et al. The corticosteroid-induced inhibitory effect on NK cell function reflects down-regulation and/or dysfunction of triggering receptors involved in natural cytotoxicity. Eur J Immunol. 2004;34:3028–3038. doi: 10.1002/eji.200425418. [DOI] [PubMed] [Google Scholar]
- 46.Perillo A, Pierelli L, Battaglia A, et al. Administration of low-dose interleukin-2 plus G-CSF/EPO early after autologous PBSC transplantation: effects on immune recovery and NK activity in a prospective study in women with breast and ovarian cancer. Bone Marrow Transplant. 2002;30:571–578. doi: 10.1038/sj.bmt.1703687. [DOI] [PubMed] [Google Scholar]
- 47.Sosman JA, Stiff P, Moss SM, et al. Pilot trial of interleukin-2 with granulocyte colony-stimulating factor for the mobilization of progenitor cells in advanced breast cancer patients undergoing high-dose chemotherapy: expansion of immune effectors within the stem-cell graft and post-stem-cell infusion. J Clin Oncol. 2001;19:634–644. doi: 10.1200/JCO.2001.19.3.634. [DOI] [PubMed] [Google Scholar]
- 48.Shah MH, Freud AG, Benson DM, Jr, et al. A phase I study of ultra low dose interleukin-2 and stem cell factor in patients with HIV infection or HIV and cancer. Clin Cancer Res. 2006;12:3993–3996. doi: 10.1158/1078-0432.CCR-06-0268. [DOI] [PubMed] [Google Scholar]
- 49.Miller JS, Soignier Y, Panoskaltsis-Mortari A, et al. Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105:3051–3057. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 50.Rohner A, Langenkamp U, Siegler U, Kalberer CP, Wodnar-Filipowicz A. Differentiation-promoting drugs up-regulate NKG2D ligand expression and enhance the susceptibility of acute myeloid leukemia cells to natural killer cell-mediated lysis. Leuk Res. 2007;31:1393–1402. doi: 10.1016/j.leukres.2007.02.020. [DOI] [PubMed] [Google Scholar]
- 51.Kato N, Tanaka J, Sugita J, et al. Regulation of the expression of MHC class I-related chain A, B (MICA, MICB) via chromatin remodeling and its impact on the susceptibility of leukemic cells to the cytotoxicity of NKG2D-expressing cells. Leukemia. 2007;21:2103–2108. doi: 10.1038/sj.leu.2404862. [DOI] [PubMed] [Google Scholar]
- 52.Borrego F, Ulbrecht M, Weiss EH, Coligan JE, Brooks AG. Recognition of human histocompatibility leukocyte antigen (HLA)-E complexed with HLA class I signal sequence-derived peptides by CD94/NKG2 confers protection from natural killer cell-mediated lysis. J Exp Med. 1998;187:813–818. doi: 10.1084/jem.187.5.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gumperz JE, Paterson JC, Litwin V, et al. Specificity of two anti-class I HLA monoclonal antibodies that block class I recognition by the NKB1 killer cell inhibitory receptor. Tissue Antigens. 1996;48(4 Pt 1):278–284. doi: 10.1111/j.1399-0039.1996.tb02646.x. [DOI] [PubMed] [Google Scholar]
- 54.Cersosimo RJ. Monoclonal antibodies in the treatment of cancer, Part 1. Am J Health Syst Pharm. 2003;60:1531. doi: 10.1093/ajhp/60.15.1531. [DOI] [PubMed] [Google Scholar]
- 55.Cersosimo RJ. Monoclonal antibodies in the treatment of cancer, Part 2. Am J Health Syst Pharm. 2003;60:1631. [PubMed] [Google Scholar]
- 56.Bicknell R. Targeting angiogenesis with monoclonal antibodies. Ann Oncol. 2006;(Suppl 10):x76–x78. doi: 10.1093/annonc/mdl241. [DOI] [PubMed] [Google Scholar]
- 57.Mohindru M, Verma A. Engineered antibodies act as targeted therapies in cancer treatment. Indian J Pediatr. 2005;72:943–947. doi: 10.1007/BF02731669. [DOI] [PubMed] [Google Scholar]
- 58.Lin MZ, Teitell MA, Schiller GJ. The evolution of antibodies into versatile tumor-targeting agents. Clin Cancer Res. 2005;11:129–138. [PubMed] [Google Scholar]
- 59.Goldenberg DM, Sharkey RM. Targeted therapy of cancer: new prospects for antibodies and immunoconjugates. CA Cancer J Clin. 2006;56:226–243. doi: 10.3322/canjclin.56.4.226. [DOI] [PubMed] [Google Scholar]
- 60.Sondel PM, DeSantes KB. Cell, cytokine, monoclonal antibody and gene therapy. In: Bathan Orkin, Cook Ginsburg., editors. The Hematology of Infancy and Childwood. 6. Chap 31. Philadelphia: Saunders; 2003. pp. 1307–1332. [Google Scholar]
- 61.Treon SP, Mitsiades C, Mitsiades N, et al. Tumor cell expression of CD59 Is associated with resistance to CD20 serotherapy in patients with B-cell malignancies. J Immunother. 2001;24:263. [PubMed] [Google Scholar]
- 62.Clynes RA, Towers TL, Presta LG, et al. Inhibitory Fc receptors modulate in vivo cytoxicity against tumor targets. Nat Med. 2000;6:443. doi: 10.1038/74704. [DOI] [PubMed] [Google Scholar]
- 63.Albertini MR, Hank JA, Sondel PM. Native and genetically engineered anti-disialoganglioside monoclonal antibody treatment in melanoma. In: Giaccone G, Schilsky R, Sondel PM, editors. Cancer Chemotherapy and Biological Response Modifiers. Vol. 22. London: Elsevier Press; 2005. pp. 789–797. Chap 31. [DOI] [PubMed] [Google Scholar]
- 64.Sondel PM, Hank JA. Antibody directed, effector cell mediated, tumor destruction. Heme/Oncol Clini North Am. 2001;15:703–721. doi: 10.1016/s0889-8588(05)70243-4. [DOI] [PubMed] [Google Scholar]
- 65.Cheung NK, Sondel PM. Neuroblastoma immunology and immunotherapy. In: Cheung Cohen., NIS, editor. Neuroblastoma. New York: Springer Press; 2005. pp. 223–247. [Google Scholar]
- 66.Sondel PM, Gillies SD. Immunocytokines for cancer immunotherapy. In: Morse M, Clay TM, Lyerle HM, editors. Handbook of Cancer Vaccines. Totowa NJ: Humana Press; 2004. pp. 341–348. [Google Scholar]
- 67.Blaes AH, Peterson BA, Bartlett N, Dunn DL, Morrison VA. Rituximab therapy is effective for posttransplant lymphoproliferative disorders after solid organ transplantation: results of a phase II trial. Cancer. 2005;104:1661–1667. doi: 10.1002/cncr.21391. [DOI] [PubMed] [Google Scholar]
- 68.Weng WK, Levy R. Two immunoglobulin G fragment C receptor polymorphisms independently predict response to rituximab in patients with follicular lymphoma. J Clin Oncol. 2003;21:3940. doi: 10.1200/JCO.2003.05.013. [DOI] [PubMed] [Google Scholar]
- 69.Cheung NK, et al. FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with Anti-GD2 antibody and granulocyte macrophage colony-stimulating factor. J Clin Oncol. 2006;24:2885–2890. doi: 10.1200/JCO.2005.04.6011. [DOI] [PubMed] [Google Scholar]
- 70.Gilman AL, Ozkaynak F, Matthay K, et al. Phase I study of ch14.18 with GM-CSF and IL-2 in children with neuroblastoma following autologous bone marrow transplant or stem cell rescue: A Report from the Children’s Oncology Group. Submitted. [Google Scholar]
- 71.Osenga KL, Hank JA, Albertini MR, et al. A phase I clinical trial of the Hu14.18-IL2 (EMD 273063) as a treatment for children with refractory or recurrent neuroblastoma and melanoma: a study of the children’s oncology group. Clinical Cancer Res. 2006;12:1750–1759. doi: 10.1158/1078-0432.CCR-05-2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.King DM, Albertini MR, Schalch H, et al. A phase I clinical trial of the immunocytokine EMD 273063 (hu14, 18-IL2) in patients with melanoma. J Clin Oncol. 2004;22:4463–4473. doi: 10.1200/JCO.2004.11.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Multani P, White CA. Rituximab. In: Giaccone G, Schilsley R, Sondel PM, editors. Cancer Chemotherapy and Biological Response Modifiers. Vol. 21. London: Elsevier Press; 2003. pp. 235–258. [PubMed] [Google Scholar]
- 74.Mackall CL, Sondel PM. Tumor immunology and pediatric cancer. In: Pizzo PA, Poplack DG, editors. Principles and Practice of Pediatric Oncology. Philadelphia: Lippincott Raven; 2005. pp. 118–144. [Google Scholar]
- 75.Gillies SD, Reilly EB, Lo KM, Reisfeld RA. Antibody-targeted interleukin 2 stimulates T-cell killing of autologous tumor cells. Proc Natl Acad Sci USA. 1992;89:1428–1432. doi: 10.1073/pnas.89.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]