

Abstract
Although characterization of the genotype has undergone revolutionary advances as a result of the successful genome projects, the chasm between our understanding of a fully characterized gene sequence and the phenotypic repertoire of the organism is as broad and deep as it was in the pre-genomic era. There are three fundamental unsolved problems that provide the context for the challenges in relating genotype to phenotype. We address one of these and describe a generic method for constructing a system design space in which qualitatively distinct phenotypes can be identified and counted, their relative fitness analyzed and compared, and their tolerance to change measured.
Keywords: System design principles, Biochemical systems theory, Phage lambda gene circuitry, Robustness, Tolerance, Fitness
1. Introduction
Interest in the physical characteristics that parents pass on to their offspring is as old as recorded history [1]. Although Aristotle postulated that transmission occurred through the blood, the issue eventually became the scientific question of relating genetic determinants of the parents to the phenotypic characteristics expressed by the progeny, which is recounted in the history of genetics going back to Mendel [2]. Throughout the pre-genomic era there was steady progress in our understanding of this relationship. However, the announcement of the draft sequence of the human genome in 2000 revealed the true magnitude of the problem. On the one hand, we now have a detailed genotype in the form of a sequenced genome; on the other, we know phenotypes such as hair color of cats, shape and size of flowers, height and weight of livestock, not to mention disease states in humans. The difficulty in relating these two levels of biological organization and function is hard to over-estimate. As Sydney Brenner has stated [3], “The problems faced by pre-and post-genomic genetics are … much the same -- they all involve bridging the chasm between genotype and phenotype”. Moreover, between the levels of genotype and phenotype of the organism there are many intervening levels that form a rich hierarchy of molecular subsystems. Although there are some intuitive notions of what is meant by phenotype at the level of the organism, it is far from clear what the term phenotype means at the level of the intervening biochemical systems and what the phenotypic repertoire of any given system might be.
The primary goal of this paper is to define what we mean by a phenotype at the level of a biochemical system, to enumerate the qualitatively distinct phenotypes according to this definition, and to compare their behaviors. Integral to this process is a generic method for the construction of the system design space.
To provide a context for our approach consider two fundamental problems that need to be solved in relating genotype to phenotype in a given environment: (1) the task of going from genome sequence to a model of the system and (2) the task of going from the model to the phenotype of the system in the given environment. The first task is the ongoing effort of experimental biology, and, whether considered from the bottom-up approach of molecular biology or the top-down approach of high-throughput technologies and computation, the magnitude of the problem is enormous [4]. The difficulties of arriving at such a model involve issues of parameter estimation requiring model reduction [5] and sensitivity analysis [6]. The second task, assuming that an explicit model or suite of models is in hand, is also a daunting problem. Exploring the full phenotypic potential of even a relative small nonlinear model, say with 15 parameters, by analytical methods is intractable. Furthermore, an empirical sampling of alternatives suffers from a combinatorial explosion. (If one were to sample just 10 values of each parameter in all combinations it would require 1015 simulations, and one might still have missed some important behavior beyond the sampled range, or between sampled points.)
We will address the second of these fundamental unsolved problems and focus on the notion of a “system design space” for relating the parameters of a model to its qualitatively distinct phenotypes. We will describe a generic method for constructing such a design space in which qualitatively distinct phenotypes can be identified and counted, their relative fitness analyzed and compared, and their tolerance to global change measured. The method is most easily understood in the context of a simple specific example. For this purpose we have selected one of the well-studied gene circuits of bacteriophage lambda.
It is important to realize that we could consider for our illustration any given model structure with any given values for its parameters. How closely the model reflects “reality” is not the issue here. Indeed, a model is simply our current working hypothesis at any stage in the scientific method. At any given stage in its development one must extract implications of the model in order to design experiments for testing the working model. Discrepancies between predictions from the model and the results from experimental tests are expected and used to modify the model and repeat the cycle. Regardless of how the model structure is determined and the values of the parameters estimated, one is faced with the task of revealing the phenotypic repertoire for the working model so that experiments can be designed for its testing.
2. Gene circuit controlling lambda lysogeny
The lytic/lysogenic decision of phage lambda has long been studied as a model of cell-fate decisions in higher organisms, and has also been explored by mathematical modeling [7, 8]. The regulator CII initially is a critical agent in this decision, but once the developmental path to lysogeny is established it no longer plays a role [9]. The lysogeny/induction decision has received less attention as a model system, but it has also been well studied. The classical view held that CRO and CI form a double-negative bistable switch that plays an essential role in initiation of the induction process [10]. However, there is now evidence suggesting that CRO is not required for initiation of induction [11-13], although it can influence the threshold level of DNA damage that initiates induction [12] and the subsequent progress of lytic development [13].
The core regulator of the lysogenic state is the CI protein, which as a dimer represses the early lytic transcripts from promoters pL and pR by binding to operator sites OL and OR while activating its own transcription from promoter pRM at low concentrations and repressing it at high concentrations. Following DNA damage, which leads to an SOS response, RecA protein is activated and stimulates CI monomer auto-cleavage. The lowering of CI concentration, and hence the dimer concentration, results in de-activation of cI transcription and de-repression of lytic functions.
A diagram emphasizing the core features of CI regulation is shown in Fig. 1 along with a version having symbols that simplify the mathematical notation.
Fig. 1.
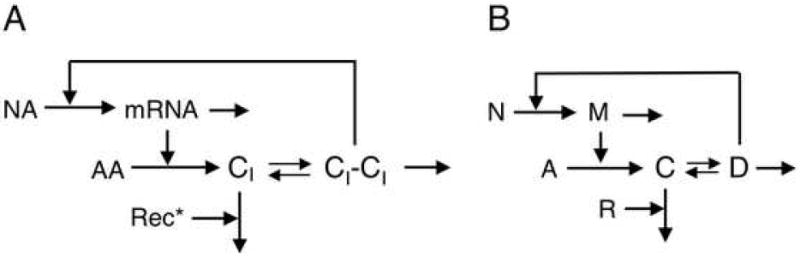
(A) Kinetic model of the cI gene circuit for phage lambda. (B) Simplified symbolic version of the kinetic model in panel A.
2.1. Kinetic model of the cI gene circuit
Early mathematical models of this system have been review by Santillan and Mackey [14]. Well-established features of these models include first-order loss of CI mRNA, first-order translation of the CI mRNA, dimerization of CI monomers, cooperative activation of pRM transcription by low concentrations of CI dimmers and repression at high concentrations. These well-established features of the cI gene circuit are included in a number of detailed kinetic [14-18] and stochastic [19-21] models that incorporate the recent experimental results of Dodd et al [22].
The following is a kinetic model that includes these conventional features of the system. It differs from existing models by the assumption of a phenomenological Hill-like function for the rate of pRM transcription and the potential for cooperative RecA* catalysis of CI monomer degradation under substrate-limited conditions. The first assumption appears to represent adequately the experimental data of Dodd et al [22], as shown below. The second assumption addresses the detailed kinetics of CI degradation, which is unknown because it has been difficult to study for technical reasons [J.W. Little, personal communication]. Nevertheless, fitting experimental data with this model yields an estimate of the capacity for regulation of CI degradation that is consistent with the limited experimental data available [23], as discussed below.
| (1) |
| (2) |
| (3) |
The behavior represented by these equations can be described as follows.
At very low concentrations of D, there is a basal rate of transcription given by γ M. As the concentration of D increases there is a cooperative activation of the rate, which is characterized by the Hill number p. When the concentration of D exceeds the intermediate value needed for half-maximal activation, the activation constant KD, the increase in rate diminishes as it approaches the maximum velocity given by γ Mmax. As the concentration of D approaches and then exceeds the inhibition constant KI, there is a cooperative repression of the rate, which is characterized by the Hill number n. The loss of mRNA, M, is first order with rate constant δ M.
The rate of translation is first order in the concentration of M with rate constant γ C. At very low concentrations of R, there is a basal rate of loss of C given by δ C. As the concentration of R increases there is a cooperative activation of the rate, which is characterized by the Hill number a. When the concentration of R exceeds the intermediate value needed for half-maximal activation, the activation constant KR, the increase in rate diminishes as it approaches the maximum velocity given by δ Cmax. The reversible dimerization of CI is characterized by the second-order rate constant γ D and the first-order rate constant β D.
2.2. Normalization about the induced state
The number of parameters and independent variables that need to be specified can be reduced without loss of generality by normalizing about a particular value, taken arbitrarily here to be the fully induced state. The result of normalizing Eqns. (1) - (3) is the following set of equations:
| (4) |
| (5) |
| (6) |
where , , , , , , , , , , , τ = δ Mt, , and . Note that d, κ and χ all have the same normalization factor (1 nM−1), so determining the factor relating D to d allows for the explicit determination of KD and KI from κ and χ, respectively.
2.3. Estimation of parameter values
Dodd et al. [22] produced two sets of data critical for understanding the mechanism that controls cI gene transcription in lambda lysogens. Both sets of data, which are relevant for the numerical characterization of our model, entailed the construction of Escherichia coli strains in which the structural gene for CI is fused to an IPTG-inducible promoter and the pRM promoter is fused to the structural gene lacz encoding β-galactosidase. In the first case, the pRM promoter is associated with wild-type operator sequences that allow binding of CI dimers at low concentration to the operator sites OR1-OR2 and OL1-OL2 to activate transcription, and that allow binding of CI dimers at high concentrations to the operator sites OR3 and OL3 to repress transcription. In the second case, the pRM promoter is associated with a disabled operator site OL3 that fails to bind CI dimers thereby eliminating repression at high concentrations. The concentration of CI is determined from a standard curve relating the levels of IPTG and measured levels of CI. The rate of transcription from the promoter pRM is measured in units of β-galactosidase activity as in standard reporter assays. We fit our model to these data by minimizing the root mean square deviation with constrained quasi-Newton optimization [24, 25]. We initiated the algorithm multiple times over a broad range of parameters, and consistently found that best fit was obtained by the same parameter set. The confidence intervals for the parameters were determined by repeating the process on the same data sampled with ±10% error.
The data for the strain with the mutant OL3 site are shown as blue dots in Fig. 2. Fitting the model produced the following parameter values: basal level of expression γ M = 50.1 ± 5.63 LacZ units, maximal level of expression γ Mmax = 359 ± 15.3 LacZ units, the activation constant characterizing the concentration of D for half maximal activation KD = 0.138 ± 0.0130 μM, and the Hill number characterizing activation p = 3.01 ± 0.676. We chose reasonable values γ M = 50 LacZ units, γ Mmax = 355 LacZ units, KD = 0.130 μM, and p = 3, which produced the fitted blue curve in Fig. 2 and the normalized parameter values σ = 7.1 (the capacity for regulation or ratio of maximal to basal expression) and κ = 130.
Fig. 2.
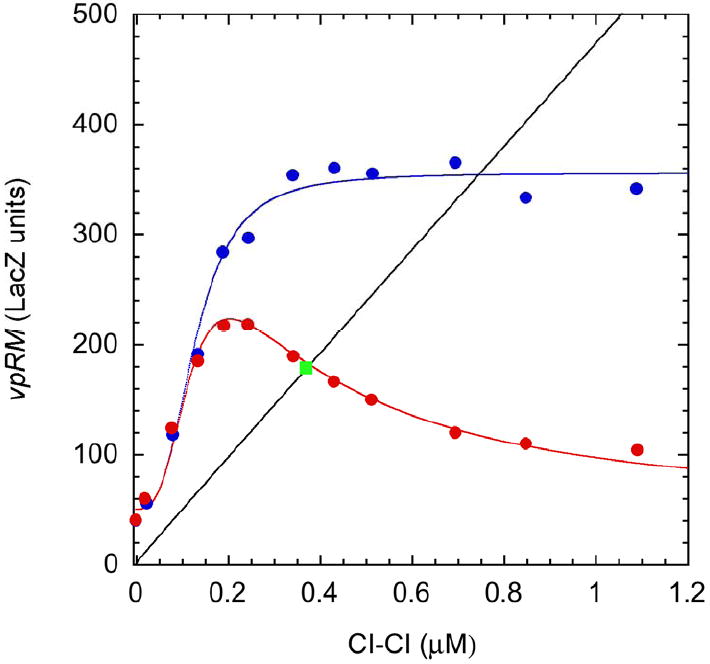
Rate of transcription from the lambda pRM promoter as a function of CI dimer concentration. Activation of transcription without repression (blue) and activation with repression (red). The symbols represent experimental data taken from Dodd et al. [22] and the curves represent the fit of the right-hand side of Eqn. (7) to the experimental data. The nominal operating point for a lysogen in wild-type E. coli grown in LB broth (estimated to be 0.35 μM in terms of the dimer) is shown in green. The black line represents the loss of CI dimers as a result of monomer cleavage, which is fit to the left-hand side of Eqn. (7). (See text for discussion.)
The data for the strain with the wild-type OL3 site are shown as red dots in Fig. 2. Using the parameters determined from data for the mutant OL3 site and fitting the model produced the remaining parameter values: the inhibition constant characterizing the concentration of D for half maximal inhibition KI = 0.316 ± 0.0359 μM and the Hill number characterizing inhibition n =1.45 ± 0.248. We chose reasonable values KI = 0.320 μM, and n = 1.5, which produced the fitted red curve in Fig. 2 and the normalized parameter value χ = 320.
Finally, the black line, which represents the loss of D in a stable wild-type lysogen in rich media, is obtained by assuming the first-order rate of loss to be the same for monomer and dimer (i.e., θ = 2) and the Hill number for R activation of C self cleavage to be one (i.e., a = 1, the predicted value based on maximum robustness of the local behavior, see Section 4.2), and by adjusting the parameters α and ϕ to match the values of D in the fully-induced state and in the wild-type lysogen, thus ensuring effective switching between the lytic and lysogenic states (α = 215 and ϕ =10). It is difficult to determine experimentally α, the capacity for regulation of activated RecA (i.e., its dynamic range from basal to maximal level of expression during induction), but rough estimates based on experiments with LexA stability suggest a value of ~ 100 [23a, J.W. Little, personal communication], which indicates that our estimate has the correct order of magnitude.
Other parameters that will be used in the later analysis include ψ, δ M and δ D. The value of ψ can be deduced from the previously estimated values and the equilibrium constant for the dimerization of CI (β D / γ D). The literature provides a broad range, from 0.05nM [10] to 5nM [26]. Assuming a value of ~ 1 nM yields ψ = 10. For the message that has a half-life of 6 minutes [27] the value of δ M = ln(2) / 6 min-1, and for a culture growing exponentially in a rich media with a doubling time of 20 minutes [28] the value of δ D = ln(2) / 20 min-1.
Given the assumed nonlinear structure of our model (Section 2.1), and now our fitting of its parameter values to the experimental data of Dodd et al [22], we obtain a particular model that provides an appropriate example to address the second task described in the Introduction. Namely, how can we define the qualitatively distinct phenotypes, identify their number, and compare their behaviors? Recall that this task is separable from the first (i.e., how the particular model was obtained in the first place), which may have involved extremely difficult issues including model reduction and parameter estimation that are not the focus here.
3. Generic construction of the system design space
The generic method we have proposed is based on the rate law description of chemical reactions expressed in the power-law formalism, which can be considered a canonical nonlinear formalism from four different perspectives [29]. From the fundamental perspective, it includes the generalized mass action (GMA) representation for which traditional mass action is a special case when the exponential parameters are restricted to small positive integers. From the local perspective, it provides a general representation in logarithmic space that is guaranteed by Taylor’s theory to be accurate within a well-defined neighborhood about a nominal operating state. This representation is the most tractable and appropriate for systems in a well-regulated homeostasis. From the piecewise power-law perspective, it provides a global representation constructed of local descriptions that can be made as accurate as desired, again according to Taylor’s theory. From the recast perspective, it provides an exact global representation for an enormous scope of nonlinear equations. Thus, the power-law formalism provides a nonlinear representation that is sufficiently general to describe biochemical models composed of rate laws having essentially any form of biological interest.
Our generic construction makes use of each of the different perspectives mentioned above, as will become clear in the following sections. For the purposes of outlining the generic method for construction of the system design space we have selected a particular model, which includes both mass-action and rational-function nonlinearities, with specific values of its parameters. Extracting the behaviors of such a model, which is the task we are about to address, is required for the design of experiments to test the adequacy of a model, which need not concern us here. Additional details of the construction method and examples of other system design spaces can be found elsewhere [30, 31]. It should be emphasized that construction of the system design space facilitates the characterization of the fixed points within each of the phenotypically distinct regions. Hence, the steady-state equations play an essential role.
3.1. Recasting the steady-state equations
Creating the dimensionless parameter groups (Section 2.2) is actually the first step in the construction. The second is setting the derivatives to zero in Eqns. (4) - (6) to obtain the steady-state equations. In this case, these equations can be combined to yield the following algebraic equation involving d and the independent variable r, which may be considered the environment of this system:
| (7) |
This equation can be recast [32, 29] exactly into a generalized mass action (GMA) system of equations within the power-law formalism by letting x1 = d and introducing two new variables x2 = κp + dp + χ−ndn+p and . The resulting system of equations is
| (8) |
| (9) |
| (10) |
Each side of the steady-state equations [Eqns. (8) - (10)] is, in general, a sum of several terms. When one term on each side of each equation is dominant (i.e., is the term with the greatest magnitude on its side), the system of equations can be approximated locally by a second system of equations, the S-system representation, which has a single analytical solution that is linear in the logarithms of the concentration variables and rate constants [33, 34]. The combination of several such equations, following the classical methods of Bode as adapted for biochemical systems (see Appendix B of ref. [34]), produces a third system of equations, the piecewise power-law representation [29]. The boundaries between pieces according to this method are not arbitrary, but are determined by the parameters of the original system. This third representation of the original system is the key to constructing the system design space and identifying qualitatively distinct phenotypes.
3.2. Number of qualitatively distinct regions in design space
Since each term on each side of Eqns. (8) - (10) is potentially dominant, there are as many potential solutions as there are combinations of terms in the piecewise power-law representation; hence a bound on the total number of steady-state solutions (T) is provided by
| (11) |
where n is the number of equations and Li and Ri are the number of terms on the left- and right-hand sides of the ith equation. (In the case of Eqns. (8) - (10), the bound is T = (3 * 3) * (1 * 3) * (1 * 2) = 54.)
However, not all potential solutions are necessarily valid. The conditions for any given term to be dominant are provided by a set of linear inequalities in log space. A test of each potential solution against the inequalities necessary for its validity will determine whether or not a potential solution is in fact a valid solution. Substituting the valid solution into the corresponding dominance conditions yields a set of linear inequalities that defines the boundaries of the region in which the solution is valid. By following this strategy for the lambda cI gene circuit, and selecting the normalized parameters r and κ (see Section 2.3) for the axis of a 2-D plot that represents a slice through the design space, we obtain the result depicted in Fig.3. Note that only 10 of the 54 possibilities are valid solutions for this particular design space. Regions on the top of this design space (1,37,38) correspond to stable steady-state operating points with low values for the rate of transcription from the promoter pRM; we will henceforth refer to the qualitative nature of these as lytic-like or simply lytic states. Regions on the bottom of this design space (11,47,46,45) correspond to stable steady-state operating points with high values; we will henceforth refer to the qualitative nature of these as lysogen-like or simply lysogenic states. The regions in the middle (7,43,44) correspond to unstable steady-state operating points with intermediate values.
Fig. 3.
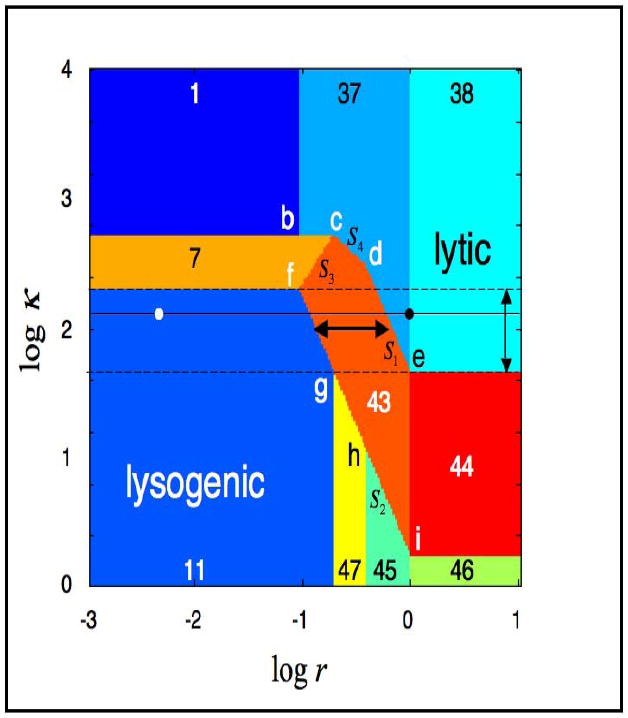
System design space for the model in Fig. 1. The units of the axes are log base 10. The regions of different color represent qualitatively different phenotypes. The regions are numbered arbitrarily. The letters b through i represent the coordinates of the intersections, and the slopes are labeled S1 through S4. The white dot represents the location of the wild-type system in the lysogenic state; the black dot represents the location of the system in the lytic state. Induction, caused by an increase in logr, corresponds to rightward movement along the horizontal black line. Regions 7, 43 and 44 represent hysteretic behavior with three steady states, one unstable and two stable, corresponding to the two neighboring stable regions. Wild-type induction occurs across the critical hysteretic region (horizontal arrow). The value of logκ must lie between the dashed lines (vertical arrow) in order to maintain the temperate life cycle operating across the critical hysteretic region.
In each region of Fig. 3, the steady-state equations for the concentration of monomer CI (C), dimer CI (D) and mRNA (M) follow analytically from the form of the original nonlinear equations, independent of the particular numerical values for the parameters. Thus, one can in principle characterize the various regions and compare their behaviors analytically without knowing the values of the parameters. This has been done for simple systems [30, 31], but in practice it becomes intractable for large systems. The region of design space in which the system operates and the numerical positioning of the boundaries are determined when specific values for the parameters of the model are given.
3.3. Landmarks in design space
The geometry of design space is determined algebraically and consists of straight-line boundaries with well-defined slopes and intersections in log space. These features are determined by the structure of the original model. In the case of Eqns. (1) - (3), these key landmarks are indicated in Fig. 3, and the x, y coordinates (log(r), log(κ) respectively) of the intersections and values of the slopes are summarized below.
3.3.1. Intersections
| (12) |
| (13) |
| (14) |
| (15) |
| (16) |
| (17) |
| (18) |
| (19) |
3.3.2. Slopes
| (20) |
| (21) |
| (22) |
4. Evaluation of Local Performance Within Phenotypic Regions
Within each region there is a qualitatively distinct solution that is fully characterized [30] by well-established methods that quantify signal amplification (logarithmic gains [35]), robustness (parameter insensitivity [36]), and stability (eigenvalues [37]) involving local (small) changes in variables and parameters. Logarithmic gains, defined as L(Xi, Xk) = ∂ ln Xi / ∂ ln Xk, represent the amplification or attenuation of biochemical signals as they propagate through the system from input variables to output variables. Parameter sensitivities, defined as S(Xi, pj) = ∂ ln Xi / ∂ ln pj, represent the amplification or attenuation of changes in system variables in response to changes in the parameters that define the structure of the system. The eigenvalues, defined by the linearized system, are only a function of the system’s kinetic orders and turnover numbers. Each of these characteristics can be readily calculated and compared against a set of quantitative performance criteria to determine the relative fitness of systems represented in each region. These qualitatively distinct solutions we define as molecular phenotypes.
There are other measures of local robustness that include second-order parameter sensitivities [38, 6] and give a better indication of the synergy between parameters. These could be used to examine particular fixed points of the original equations, but at an increased computational expense. However, these second-order approaches are not relevant in the phenotypic regions we have defined because the equations in each region are already explicitly expressed in terms of linear functions in the logarithms of the independent variables and rate constants. Thus, the first-order sensitivities mentioned above are the relevant expressions and a second-order term would be zero.
4.1. Quantitative criteria for effective local performance
Two criteria often found to correlate with the natural selection of a particular system design are the maximization (minimization) of a particular steady-state function and the response time following change. Moreover, when these values are optimal they often are robustly so (e.g., see [31]). Several criteria for effective local performance that reflect these expectations can be summarized as follows.
The lysogenic state should be locally robust to perturbations in the values of the N parameters that define the system, and this robustness can be quantified by the parameter sensitivities S(D, pi). It should not be influenced by fluctuations in the input signal (the level of DNA damage), which can be quantified by the logarithmic gain L(D, R). The robustness of the logarithmic gain in the face of perturbations in parameter values is also expected to be small, and this robustness can be quantified by the parameter sensitivities S[L(D, R), pi]. The dimer form of CI is responsible for the primary regulatory actions, but similar criteria hold for the monomer form of CI and the mRNA as well.
It is important to minimize the response time for restoring the system to its nominal steady state following small changes in the variables of the system, and this can be quantified in terms of the dominant eigenvalue λdominant. One could of course consider other criteria, but these will suffice for our purposes here.
4.2. Analysis of local performance
The local robustness of the system in each of the phenotypic regions of system design space can be calculated analytically for the model in Fig. 1. From these results, one can predict maximum local robustness in most cases when the Hill numbers p and n are large, although tradeoffs are involved in determining the optimum values; whereas local robustness is maximal when a is small (data not shown). By inserting the parameter values estimated in Section 2.3 into these analytical results one can convert the analytical results into the numerical results summarized in Table 1. Small values imply superior performance for each of the criteria.
Table 1.
Summary of local robustness for each phenotypic region of the system design space in Fig. 3. Results are calculated based on the parameter values determined by the fit to the experimental data of Dodd et al. [22a]. In regions where robustness is a function of r or κ the following typical values are used. In regions 7, 43 and 44 the values of κ are 316, 130, and 10. In regions 37, 43, 45, and 47 the values of r are .0.32, 0.32, 0.63, and 0.32.
| Region | Criteria | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Σ | S(•,pi) | / N | L(•,R) | Σ | S[L(•,R),pi] | / N | |||||||
| D | C | M | D | C | M | D | C | M | |
| Lysogenic regions (stable steady states) | |||||||||
| 11 | 0.161±0.209* | 0.141±0.164 | 0.187±0.247 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| 47 | 0.292±0.255 | 0.180±0.144 | 0.318±0.309 | -0.500 | -0.250 | 0.750 | 0.117±0.301 | 0.117±0.301 | 0.083±0.253 |
| 45 | 0.862±0.878 | 0.364±0.464 | 0.133±0.340 | -2.000 | -1.000 | 0.000 | 0.067±0.249 | 0.067±0.249 | 0.000 |
| 46 | 0.667±0.863 | 0.267±0.442 | 0.133±0.340 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Hysteretic regions (unstable steady states) | |||||||||
| 7 | 0.277±0.411 | 0.205±0.227 | 0.411±0.586 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| 43 | 0.291±0.300 | 0.212±0.204 | 0.488±0.499 | 0.400 | 0.200 | 1.200 | 0.147±0.376 | 0.147±0.376 | 0.080±0.251 |
| 44 | 0.238±0.310 | 0.186±0.216 | 0.319±0.407 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| Lytic regions (stable steady states) | |||||||||
| 1 | 0.267±0.442 | 0.194±0.238 | 0.133±0.340 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
| 37 | 0.952±0.946 | 0.409±0.502 | 0.133±0.340 | -2.000 | -1.000 | 0.000 | 0.067±0.249 | 0.067±0.249 | 0.000 |
| 38 | 0.667±0.863 | 0.267±0.442 | 0.133±0.340 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 |
Mean ± standard deviation
The local robustness in each of the phenotypic regions reveals that on average the influence of perturbations in parameters is attenuated (sensitivities less than one in the columns 2-4 in Table 1). The logarithmic gains (columns 5-7) describe the influence that changes in the environmental input signal (RecA levels) have on the output concentrations of the circuit (CI dimer, CI monomer, and mRNA). Fluctuations in the level of DNA damage have no influence on the phenotype when the circuit operates on the basal (regions 11, 7, 1) or maximal (regions 46, 44, 38) portions of the curve relating RecA activity to DNA damage. These fluctuations have an attenuated influence (logarithmic gain less than one in magnitude) on the phenotype when the circuit operates on the repressing portion (region 47) of the curve that relates rate of transcription to dimer concentration. They have an amplified influence (logarithmic gain greater than one in magnitude) on dimer concentration when the circuit operates on the upper plateau (region 45) or the lower plateau (region 37) of the curve relating rate of transcription to dimer concentration, or on the mRNA level when the circuit operates on the activating portion (region 43) of the curve relating rate of transcription to dimer concentration. In all cases, the influence that perturbations in parameters have upon the logarithmic gains (columns 8-10) is on average attenuated (sensitivities less than one). Overall, there is tendency for the local behavior to be more robust in regions with high values of the input variable (46, 44, 38) or regions with low values (11, 47, 7, 1), and to be less robust in the regions with intermediate values (45, 43, 37).
The local response times in each of the phenotypic regions of system design space are readily calculated. The half-times for the restoration of the steady states range from ~ 5 min (the fastest) to ~ 30 min (the slowest). Regions that correspond to basal levels of DNA damage (1, 11) are slower, while regions that correspond to high levels of DNA damage (38, 46) are faster. The response times shift from slow to fast over the intervening regions (37, 45, 47) as DNA damage increases. The same pattern is seen in the response time for divergence from the unstable steady states: faster in 44, slower in 7 and shifting from slow to fast in 43. On the whole, lysogenic regions (11, 47, 45, 46) are faster than their lytic counterparts (1, 37, 38), which is consistent with the experimental evidence for the local robustness of the lysogenic state [39].
5. Evaluation of Global Performance Across Phenotypic Regions
Characterizing local (small) changes in performance is important for characterizing the qualitatively distinct phenotypes in each region, but it does not address the overall behavior when phenotypes change as a result of large environmental stimuli or mutation in components of the system. The partitioning of system design space into qualitatively different phenotypic regions provides a means, based on the boundaries, for calculating tolerances to global (large) changes in parameter values [31]. These tolerances are defined for each parameter as the ratio of its value at the nominal steady state (normal operating point for the system) to its value on the boundary to an adjacent phenotypic region (or the inverse, depending on which value is larger). The geometry of system design space in Fig. 3 itself suggests other criteria that are relevant for assessing global behavior of the system.
5.1. Quantitative criteria for effective global performance
Two criteria often found to correlate with the natural selection of a particular system design are large tolerances to a change in phenotype when the change represents dysfunction and fast responses to a change in phenotype when it is critical for survival. Moreover, when these values are optimal they often are robustly so (again, see [31]). Several criteria for effective global performance that reflect these tendencies can be summarized as follows.
The hysteretic region 43 in the design space of Fig. 3 provides a type of “safety factor”. Once a switch from lysogenic to lytic growth has been initiated, the phage becomes committed in the following sense: if there is a decrease in the SOS signal, it must be large enough to move the operation of the system completely back across the hysteretic region, any lesser decrease is insufficient to prevent completion of the switch. The hysteretic region also acts as a buffer to prevent inappropriate switching when there is only a transient small SOS signal, one that is insufficient to cause the system to cross the hysteretic region. These functions of the hysteretic region are enhanced by maximizing the horizontal distance between the inclined lines in Fig. 3, which can be quantified by the value of ΔH, which is given by σ[(2p−1)/(2pa)], or in terms of the original parameters
Again, this safety factor should be robust to parameter variations, and this robustness can be quantified by the parameter sensitivities S[ΔH, pi].
For long-term survival of the temperate life cycle, the phage must be capable of repeatedly switching from lysogenic to lytic growth and back again across the critical hysteretic region. This is facilitated by having a design that places the operating point of the system between the dashed lines in Fig. 3 and by having a large distance between these limits, which can be quantified by the value of ΔS, which is given by α2θ−2ϕ−2σ−(2p−1)/p, or in terms of the original parameters
Furthermore, this zone of operation should be robust to parameter variations, and this robustness can be quantified by the parameter sensitivities S[ΔS, pi].
The speed with which the phage can switch lysogeny OFF should be a maximum, given that this is a response to a critically deteriorating state of the host. This can be quantified by the half-time in going from a steady state with high CI concentration to one representing low CI concentrations, tOFF. Similarly, the speed with which the phage can switch lysogeny ON should be a maximum, although this may not be as critical as the need for speed of induction. This can be quantified by the half-time in going from a steady state with low CI concentration to one representing high CI concentrations, tON.
Finally, a system that has been selected to have good local as well as global performance is expected to reside within a phenotypic region of system design space far from the boundaries to other phenotypic regions in which the local performance is poorer. This corresponds to large global tolerances to parameter change. Global tolerances are quantified (according to the definition given at the beginning of Section 5) by calculating [Tlow,Thigh], where Tlow is the tolerance to a decrease and Thigh to an increase in a parameter value.
5.2. Analysis of global performance
The size of the hysteretic buffer and the robustness of this buffer can be calculated analytically. The numerical results using the values of the parameters in Section 2.3 show that the breadth of this zone is represented by a 5-fold change in the normalized RecA activity. The parameter sensitivities of this ΔH value are attenuated with an average value of 0.242 ± 0.468.
The aspects of global behavior pertaining to the maintenance of a broad zone of switching across the hysteretic buffer, and the robustness of this zone in response to perturbations in parameter values, also can be calculated analytically. The numerical results, using the values of the parameters in Section 2.3, show that the breadth of this zone is represented by a 4-fold change in the normalized Michaelis constant for activation. The parameter sensitivities of this ΔS value are attenuated with an average value of 0.654 ± 0.627.
The global switching times are determined by solving the normalized equations [Eqns. (4) - (6)]. The results, using the values of the parameters in Section 2.3 and increasing the value of r from 0.00465 to 1.0, show that the half-time to switch lysogeny OFF is approximately 8 minutes for a culture growing exponentially in a rich media. Alternatively, when decreasing the value of r from 1.0 to 0.00465, the half-time to switch lysogeny ON is approximately 54 minutes, about 7-fold longer, under the same conditions.
One could use the local equations in each region to solve for the transient from an initial condition within one region until a boundary is reached, use the final values within the first region as the initial values for the solution within the second region, and solve for the transient behavior until a new steady state is achieved, or until another boundary is reached and the process repeated as necessary. Indeed, there are well-established precedents for this in the modeling hybrid systems [40]. However, there are errors near the boundaries where the switching occurs that may be an issue is some cases. In any case, It is just as easy to solve the original equations when exploring the dynamics between the phenotypic regions that have been identified by the design space approach.
The global tolerances to large changes in parameter values are calculated from knowledge of their wild-type value within the region and their value on the boundary of the region in the system design space (Fig. 3). The values of tolerances for the combination of the best lysogenic regions (11 and 47) are summarized in Table 2. The most stringent tolerances are ~ 1.6-fold. In terms of total tolerance, the product of the fold increase and the fold decrease, the range varies from a low of ~ 2.7-fold variation to a high that is effectively infinite.
Table 2.
Global tolerances to large changes in parameter values for phenotypes 11 and 47 between the dashed lines in the design space of Fig. 3*. The parameters on the left are involved in the synthesis and loss of mRNA; those on the right are involved in the synthesis, loss, and inter-conversion of protein molecules.
| mRNA | Protein | ||
|---|---|---|---|
| Parameter | Tolerance | Parameter | Tolerance |
| γ M | [2.0, 4.2] † | δ C | [∞, 41] |
| γ M max | [2.4, 1.7] | δ C max | [1.7, 82] |
| KD | [2.8, 1.6] | KR | [82, ∞] |
| KI | [11, 2.4] | a | [5.6, ∞] |
| p | [3.0, 3.4] | γ C | [1.6, 1.7] |
| n | [∞,∞] | γ D | [1700, 2.8] |
| δ M | [1.7, 1.6] | β D | [3.4, 1900] |
| δ D | [11, 1.6] | ||
[fold decrease, fold increase]
Note that in Fig. 3 the upper boundaries of Regions 11 and 47 extend to the upper boundary of Region 7 and the c-d boundary of Region 43, respectively.
6. Discussion
Although it had long been thought that CRO and CI form a double-negative bistable switch that plays an essential role in initiating induction, there is now evidence suggesting that CRO is not required for the initiation process. This conclusion is based on experiments that attempt to eliminate repression of transcription from pRM by CRO using mutant strains in which transcription from pRM is otherwise unaltered. In none of the experiments [11 - 13] is this ideal achieved. The evidence suggests that induction can be initiated without the involvement of CRO, although CRO does affect the threshold level of RecA activity necessary for induction [12] and the subsequent development of the lytic program [13]. The results of the analysis in Section 5 support this revised view that CI itself is the core regulator governing initiation of the induction process. These results indicate that the cI gene circuit is robustly designed by natural selection when judged according to criteria for local performance (Table 1) and global performance (Table 2). Nevertheless, it is clear that factors outside the core cI gene circuit, such as the regulators CRO and CII, play roles in conjunction with CI. These roles can be interpreted in the context of the system design space in Fig. 3.
The regulator CRO influences the threshold level of DNA damage necessary to initiate induction. Atsumi and Little [12] have shown that without CRO, approximately 40% more DNA damage is required for induction. As damage and SOS activity increase, the level of CI is reduced. Transcription of cro then increases somewhat and the elevation in CRO leads to a partial repression of cI transcription that counteracts the derepression caused by the lowering of CI itself. The partial repression of cI transcription by CRO can be interpreted as an effective decrease in σ, the capacity for regulation of cI transcription, although this is a dynamic process and a more detailed analysis would be necessary to make quantitative predictions. When CRO is absent, this has the effect of shifting the d-e boundary in Fig. 3 to the right by the same amount as the decrease in σ. Thus, the 40% increase in the threshold level of r that is necessary for induction (or ~ 0.15 of a log unit in Fig. 3), represents approximately a 40% decrease in σ. Alternatively, when CRO is present, this has the effect of shifting the d-e boundary in Fig. 3 to the left by ~ 0.15 of a log unit, thus lowering the threshold level of r that is necessary for induction. Thus, induction and variations it its threshold are associated with movements in the horizontal direction between the dashed lines in Fig. 3.
The regulator CII assists in the establishment of lysogeny by providing an initial enhancement in the level of cI transcription from a separate promoter pRE that adds to the basal rate of transcription from the promoter pRM [41]. The transcription from pRE is shut off once the commitment to lysogeny is established. This enhancement corresponds to a transient elevation in σ, the capacity for regulation of cI transcription. This has the qualitative effect of shifting the boundaries in Fig. 3 upward (and the d-e boundary to the right), thus favoring lysogeny by enlarging the lysogenic regions at the expense of the lytic regions. Again, this is a dynamic process and a more detailed analysis would be necessary to make quantitative predictions.
An increased multiplicity of infection leads to early elevation of CII concentrations [42]. As just noted, this causes an increase in the parameter σ, which raises the boundary between the lytic region 1 and the lysogenic region 11. This implies an increased probability of lysogenic over lytic growth, which is consistent with the experimental observations of Kourilsky and Knapp [43]. On the other hand, when cells are growing exponentially in poor versus rich media, CI will have an increased half-life (since this is largely dictated by the dilution rate). This causes an increase in the value of the parameter α = δ Cmax / δ C, which raises the boundary between region 7 and the lysogenic region 11. For example, the lysogenic state for cells grown in glucose minimal media with a doubling time of 60 minutes will have a value of α that is 3-fold larger than that for cells grown in LB broth [27]. This corresponds to an upward shift of ~ 0.5 of a log unit in Fig. 3 and an increased tolerance for maintaining lysogeny, which is also consistent with the experimental observations of Kourilsky and Knapp [43]. This result highlights the critical role played by the half-life of CI, which is discussed further below. Thus, many of the pivotal events early in the lytic versus lysogenic decision appear to be associated with movements in the vertical direction across region 7 in Fig. 3.
The ratio of mRNA to CI half-lives has a dramatic influence on the global switching times. This ratio is ~ 0.3 for the lysogen in a host cell with a doubling time of 20 minutes. During induction, when the half-life of CI is sharply reduced, this ratio rises to ~ 65. This large change in ratio would be responsible for a marked difference in switching times (as seen in Section 5.2), if it were not for the early overriding effect of CII. The time for the core circuit to turn OFF lysogeny, ~ 8 minutes, should be shorter than the time that the earliest lytic functions appear, and this is consistent with the results of Osterhout et al. [44] showing that transcripts of early lytic functions are first detected at around 25 minutes following induction.
The results in Sections 4 and 5 suggest that the system design of the cI gene circuit is not only robust to local (small) changes in parameters, but it is also remarkably tolerant to global (large) changes in parameters and environmental stimuli. For example, the system is capable of tolerating an approximate 4-fold range in values of the activation constant for cI transcription (KD) without losing the ability to reciprocally switch from lysogenic to lytic growth and back. This is consistent with the work of Little and colleagues [39a, 45, 46] in which they have constructed variants with major changes in the binding constants, and indeed the identity of the circuit elements, and yet many of their constructs retain the essential qualitative features of the wild-type phage. It should be noted that large values of r (major DNA damage) combined with low values of κ (including the activation constant KD) will lower the concentration of CI dimer while maintaining a high rate of transcription from the promoter pRM, which we have defined qualitatively as a lysogen-like state. If the level of dimer were lowered sufficiently, then lysogeny could not be maintained and induction would ensue.
It must be emphasized that the boundaries, which are key to the construction of the system design space, are not arbitrarily defined or selected in an ad hoc way to provide the most accurate representation of the original system. Indeed, they represent a judicious approximation that illuminates the qualitative nature of the phenotypes. The boundaries are dictated by the form of the original equations and by the kinetic parameters found therein. It is difficult to imagine another method of defining these boundaries that is as tractable and revealing as the method we have described here.
7. Conclusions
A generic approach has been used to construct a system design spaces for the core cI gene circuit of phage lambda in which qualitatively distinct phenotypes can be identified and counted, their relative fitness analyzed and compared, and their tolerance to large changes measured quantitatively. This 2-D design space is a dimensional compression of the 15-D parameter space, yet it contains all of the original parameters and independent variables that characterize the original system. Moreover, the manner in which the original parameters are combined to define landmarks in the system design space reveals important relationships among the parameters that are not evident in the larger parameter space.
The analysis within this framework reveals several interlocking principles that constitute the system design of phage lambda. The system design space provides a graphical means of representing these principles. One example is the size of the “hysteretic buffer” given by σ[(2p−1)/(2pa)]. Another example is the size of the switching zone across the hysteretic buffer given by α2θ−2ϕ−2σ(1−2p)/p. In addition to having a large size for this zone, it also is important for the normalized Michaelis constant for activation be located within this region, i.e., α−1σ2θϕ2 < κ <ασ1/p θ−1. Such relationships constitute key design principles governing the cI gene circuit. Others involving the half-lives of CI and its mRNA have been alluded to above.
While the specifics of the cI gene circuit may not be relevant to other gene circuits, the methods of constructing the system design space exhibited here are generic and might well lead to important insights when applied to the regulation of other systems. What will be needed for such an analysis is the formulation of a kinetic model that takes into account the essential interactions within the system and the appropriate experimental data that allows an estimation of the values for the model parameters.
Acknowledgments
We thank Pedro Coelho and Dean Tolla for fruitful discussions, and John Little for critical input in the construction of the model. This work was supported in part by a grant (RO1-GM30054) from the US Public Health Service to MAS, and by an Earl C. Anthony Fellowship to RAF.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Homer . The Odyssey. Vol. 4. 800 - 600 BC. Never, anywhere, have I seen so great a likeness in man or woman – but it is truly strange! This boy must be the son of Odysseus, Telémakhos, the child he left at home that year the Akhaian host made war on Troy; pp. 152–156. [Google Scholar]
- 2.Sturtevant AH. A History of Genetics. Harper & Row; New York: 1965. [Google Scholar]
- 3.Brenner S. Genomics: The end of the beginning. Science. 2000;287:2173–2174. doi: 10.1126/science.287.5461.2173. [DOI] [PubMed] [Google Scholar]
- 4.Hlavacek WS. How to deal with large models? Mol Sys Biol. 2009;5:240–242. doi: 10.1038/msb.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Daun S, Rubin J, Vodovotz Y, Roy A, Parker R, Clermont G. An ensemble of models of the acute inflammatory response to bacterial lipopolysaccharide in rats: Results from parameter space reduction. J Theoret Biol. 2008;253:843–853. doi: 10.1016/j.jtbi.2008.04.033. [DOI] [PubMed] [Google Scholar]
- 6.Gutenkunst RN, Waterfall JJ, Casey FP, Brown KS, Myers CR, et al. Universally sloppy parameter sensitivities in systems biology models. Plos Computational Biology. 2007;3:1871–1878. doi: 10.1371/journal.pcbi.0030189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Oppenheim AB, Kobiler O, Stavans J, Court DL, Adhya S. Switches in bacteriophage lambda development. Annu Rev Genet. 2005;39:409–429. doi: 10.1146/annurev.genet.39.073003.113656. [DOI] [PubMed] [Google Scholar]
- 8.Tian T, Burrage K. Bistability and switching in the lysis/lysogeny genetic regulatory network of bacteriophage lambda. J Theor Biol. 2004;227:229–237. doi: 10.1016/j.jtbi.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Court DL, Oppenheim AB, Adhya S. A new look at bacteriophage λ genetic networks. J Bacteriol. 2007;189:298–304. doi: 10.1128/JB.01215-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ptashne M. A Genetic Switch. Blackwell Scientific Publications; Cambridge, U.K: 1992. [Google Scholar]
- 11.Svenningsen SL, Costantino N, Court DL, Adhya S. On the role of Cro in lambda prophage induction. Proc Natl Acad Sci USA. 2005;102:4465–4469. doi: 10.1073/pnas.0409839102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atsumi S, Little JW. Role of the lytic repressor in prophage induction of phage λ as analyzed by a module-replacement approach. Proc Natl Acad Sci USA. 2006;103:4558–4563. doi: 10.1073/pnas.0511117103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schubert RA, Dodd IB, Egan JB, Shearwin KE. CRO’s role in the CI-CRO bistable switch is critical for λ’s transition from lysogeny to lytic development. Genes Dev. 2009;21:2461–2472. doi: 10.1101/gad.1584907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Santillan M, Mackey MC. Why the lysogenic state of phage lambda is so stable: a mathematical modeling approach. Biophys J. 2004;86:75–84. doi: 10.1016/S0006-3495(04)74085-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vilar JM, Saiz L. DNA looping in gene regulation: from the assembly of macromolecular complexes to the control of transcriptional noise. Curr Opin Genet Dev. 2005;15:136–144. doi: 10.1016/j.gde.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 16.Lou C, Yang X, Liu X, He B, Ouyang Q. A quantitative study of λ-phage switch and its components. Biophys J. 2007;92:2685–2693. doi: 10.1529/biophysj.106.097089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Anderson LM, Yang H. DNA looping can enhance lysogenic CI transcription in phage lambda. Proc Natl Acad Sci USA. 2008;105:5827–5832. doi: 10.1073/pnas.0705570105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson LM, Yang H. A simplified model for lysogenic regulation Through DNA looping. 30th Annual International IEEE EMBS Conference; Vancouver, British Columbia, Canada. August 20-24; 2008. pp. 607–610. [DOI] [PubMed] [Google Scholar]
- 19.Aurell E, Brown S, Johanson J, Sneppen K. Stability puzzles in phage lambda. Phys Rev E. 2002;65:051914–051919. doi: 10.1103/PhysRevE.65.051914. [DOI] [PubMed] [Google Scholar]
- 20.Aurell E, Sneppen K. Epigenetics as a first exit problem. Phys Rev Lett. 2002;88:048101. doi: 10.1103/PhysRevLett.88.048101. [DOI] [PubMed] [Google Scholar]
- 21.Zhu XM, Yin L, Hood L, Ao P. Calculating biological behaviors of epigenetic states in the phage lambda life cycle. Funct Integr Genomics. 2004;4:188–195. doi: 10.1007/s10142-003-0095-5. [DOI] [PubMed] [Google Scholar]
- 22.Dodd IB, Perkins AJ, Tsemitsidis D, Egan JB. Octamerization of λ CI repressor is needed for effective repression of pRM and efficient switching from lysogeny. Genes Dev. 2001;15:3013–3022. doi: 10.1101/gad.937301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Little JW. The SOS regulatory system: control of its state by the level of RecA protease. J Mol Biol. 1983;167:791–808. doi: 10.1016/s0022-2836(83)80111-9. [DOI] [PubMed] [Google Scholar]
- 24.Powell MJD. A Fast Algorithm for Nonlinearly Constrained Optimization Calculations. In: Watson GA, editor. Numerical Analysis. Vol. 630. Lecture Notes in Mathematics, Springer Verlag; 1978. pp. 144–157. [Google Scholar]
- 25.Han SP. A Globally Convergent Method for Nonlinear Programming. J Optimization Theory and Applications. 1977;22:297–309. [Google Scholar]
- 26.Burz D, Becket D, Benson N, Ackers GK. Self-assembly of bacteriophage CI repressor: effects of single-site mutations on the monomer-dimer equilibrium. Biochemistry. 1994;33:8399–8405. doi: 10.1021/bi00194a003. [DOI] [PubMed] [Google Scholar]
- 27.Court DL, Crombrugghe B, Adhya S, Gottesman M. Bacteriophage lambda Hin function II. Enhanced stability of lambda messenger RNA. J Mol Biol. 1980;138:731–743. doi: 10.1016/0022-2836(80)90062-5. [DOI] [PubMed] [Google Scholar]
- 28.Sezonov G, Joseleau-Petit D, D’Ari R. Escherichia coli Physiology in Luria-Bertani Broth. J Bacteriol. 2007;189:8746–8749. doi: 10.1128/JB.01368-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Savageau MA. Design principles for elementary gene circuits: Elements, methods, and examples. Chaos. 2001;11:142–159. doi: 10.1063/1.1349892. [DOI] [PubMed] [Google Scholar]
- 30.Savageau MA, Coelho PMBM, Fasani RA, Tolla DA, Salvador A. Phenotypes and tolerances in the design space of biochemical systems. Proc Natl Acad Sci USA. 2009;106:6435–6440. doi: 10.1073/pnas.0809869106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coelho PMBM, Salvador A, Savageau MA. Global tolerance of biochemical systems and the design of moiety-transfer cycles. PLOS Comp Biol. 2009;5(3):e1000319. doi: 10.1371/journal.pcbi.1000319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Savageau MA, Voit EO. Recasting nonlinear differential equations as S-systems: a canonical nonlinear form. Math Biosci. 1987;87:83–115. [Google Scholar]
- 33.Savageau MA. Biochemical systems analysis II. The steady state solutions for an n-pool system using a power-law approximation. J Theoretical Biol. 1969;25:370–379. doi: 10.1016/s0022-5193(69)80027-5. [DOI] [PubMed] [Google Scholar]
- 34.Savageau MA. Biochemical Systems Analysis: A Study of Function and Design in Molecular Biology. Addison-Wesley; Reading, Mass: 1976. [Google Scholar]
- 35.Savageau MA. Concepts relating the behavior of biochemical systems to their underlying molecular properties. Arch Biochem Biophys. 1971;145:612–621. doi: 10.1016/s0003-9861(71)80021-8. [DOI] [PubMed] [Google Scholar]
- 36.Savageau MA. Parameter sensitivity as a criterion for evaluating and comparing the performance of biochemical systems. Nature. 1991;229:542–544. doi: 10.1038/229542a0. [DOI] [PubMed] [Google Scholar]
- 37.Savageau MA. Optimal design of feedback control by inhibition: dynamic considerations. J Mol Evolution. 1975;5:199–222. doi: 10.1007/BF01741242. [DOI] [PubMed] [Google Scholar]
- 38.Salvador A. Synergism analysis of metabolic processes: II. Tensor formulation and treatment of stoichiometric constraints. Mathematical Biosciences. 2000;163:131–158. doi: 10.1016/s0025-5564(99)00057-7. [DOI] [PubMed] [Google Scholar]
- 39.Little JW, Shepley DP, Wert DW. Robustness of a gene regulatory circuit. EMBO J. 1999;18:4299–4307. doi: 10.1093/emboj/18.15.4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goebel R, Sanfelice RG, Teel AR. Hybrid dynamical systems. IEEE Control Systems Magazine. 2009;29:28–93. [Google Scholar]
- 41.Reichardt L, Kaiser AD. Control of lambda repressor synthesis. Proc Natl Acad Sci USA. 1971;68:2185–2189. doi: 10.1073/pnas.68.9.2185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kobiler O, Rokney A, Friedman N, Court DL, Stavans J, Oppenheim AB. Quantitative kinetic analysis of the bacteriophage λ genetic network. Proc Natl Acad Sci USA. 2005;102:5310–5311. doi: 10.1073/pnas.0500670102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kourilsky P, Knapp A. Lysogenization by bacteriophage λ. III. Multiplicity dependent phenomena occurring upon infection by λ. Biochimie. 1974;56:1517–1523. [PubMed] [Google Scholar]
- 44.Osterhout RE, Figueroa IA, Keasling JD, Arkin AP. Global analysis of host response to induction of a latent bacteriophage. BMC Microbiol. 2007;7:82–93. doi: 10.1186/1471-2180-7-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Michalowski CB, Short MD, Little JW. Sequence tolerance of the phage lambda PRM promoter: implications for evolution of gene regulatory circuitry. J Bacteriol. 2004;186:7988–7999. doi: 10.1128/JB.186.23.7988-7999.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Atsumi S, Little JW. A synthetic phage λ regulatory circuit. Proc Natl Acad Sci USA. 2006;103:19045–19050. doi: 10.1073/pnas.0603052103. [DOI] [PMC free article] [PubMed] [Google Scholar]