

Abstract
The budding yeast, Saccharomyces cerevisiae, accumulates the storage polysaccharide glycogen in response to nutrient limitation. Glycogen synthase, the major form of which is encoded by the GSY2 gene, catalyzes the key regulated step in glycogen storage. Here, we utilize Gsy2p fusions to green fluorescent protein (GFP) to determine where glycogen synthase is located within cells. We demonstrate that the localization pattern of Gsy2-GFP depends upon the glycogen content of the cell. When glycogen is abundant, Gsy2-GFP is found uniformly throughout the cytoplasm but under low glycogen conditions, Gsy2-GFP localizes to discrete spots within cells. Gsy2p is known to bind to glycogen and we propose that the subcellular distribution of Gsy2-GFP reflects the distribution of glycogen particles. In the absence of glycogen, Gsy2p translocates into the nucleus. We hypothesize that Gsy2p is normally retained in the cytoplasm through its interaction with glycogen particles. When glycogen levels are reduced, Gsy2p loses this anchor and can traffic into the nucleus.
Keywords: Glycogen synthase, Saccharomyces cerevisiae, localization, nucleus
Introduction
Glycogen is a branched polymer of glucose synthesized by a large variety of organisms as a storage form of both carbon and energy (Roach et al. 2001). Glycogen accumulation in yeast is initiated under conditions of nutrient limitation, such as the approach to stationary phase in liquid culture (Francois and Parrou 2001). Synthesis of glycogen requires the activities of glycogenin, a self-glucosylating initiator protein (Cheng et al. 1995), glycogen synthase (Farkas et al. 1991), which catalyses bulk synthesis, and the branching enzyme (Rowen et al. 1992). Glycogen breakdown requires glycogen phosphorylase and debranching enzyme (Hwang and Fletterick 1986; Teste et al. 2000) or, under certain conditions, glucoamylase (Yamashita and Fukui 1985). The main controls over glycogen storage are exerted at the levels of glycogen synthase and glycogen phosphorylase, both of which are subject to complex transcriptional and post-translational regulation (reviewed in Francois and Parrou 2001).
Yeast has two genes for glycogen synthase, GSY1 and GSY2. The GSY2 gene encodes the major isoform of glycogen synthase, accounting for 80 – 90% of enzyme activity in stationary phase (Farkas et al. 1991). Transcription of GSY2 is strongly induced towards the end of the logarithmic growth phase and this induction requires the cAMP-dependent protein kinase (Hardy et al. 1994; Ni and Laporte 1995; Smith et al. 1998). Post-translational control is also important in the regulation of Gsy2p and the enzyme can be inhibited by phosphorylation (Francois and Hers 1988; Francois et al. 1988; Rothman-Denes and Cabib 1970; Rothman-Denes and Cabib 1971), which occurs at three C-terminal sites, Ser650, Ser654 and Thr667 (Hardy and Roach 1993). The inhibition by phosphorylation is overcome by the presence of the allosteric activator glucose-6-phosphate (glucose-6-P) (Francois et al. 1988; Rothman-Denes and Cabib 1971). Mutations that affect the phosphorylation state of glycogen synthase, alter glucose-6-P levels, or influence glucose-6-P binding to glycogen synthase, all result in altered glycogen accumulation (Hardy and Roach 1993; Huang et al. 1997; Pederson et al. 2004)
Most work on the regulation of glycogen synthase has focused on the study of Gsy2p and kinases acting upon Gsy2p have been identified (Huang et al. 1996a). The cyclin-dependent protein kinase Pho85p, in complex with one of two related cyclins, Pcl8p or Pcl10p, can phosphorylate and inactivate Gsy2p (Huang et al. 1998; Wilson et al. 1999). Deletion of either PHO85 or both PCL8 and PCL10 results in hyperactivation of glycogen synthase and over-accumulation of glycogen (Huang et al. 1998; Timblin et al. 1996). Dephosphorylation and activation of Gsy2p is catalyzed by the Glc7p protein phosphatase in complex with the Gac1p targeting subunit (Feng et al. 1991; Francois et al. 1992; Stuart et al. 1994).
Work from mammalian systems has shown that phosphorylation controls not only the activity of glycogen synthase but also the subcellular localization of the enzyme, at least in some cell types (Ou et al. 2005; Prats et al. 2005). For example, Ou et al. reported that in 3T3-L1 adipocytes, glycogen synthase had a diffuse cytoplasmic distribution under basal conditions. Upon insulin treatment, dephosphorylation and activation of glycogen synthase occurred and the enzyme relocated to discrete punctate clusters (Ou et al. 2005). Additionally, Prats et al. reported phosphorylation-dependent translocation of the muscle isoform of glycogen synthase to spherical structures that contained actin using an electrically-stimulated rabbit muscle model (Prats et al. 2005). Guinovart's group has also investigated the translocation of muscle glycogen synthase, in this case using cultured human myotubes (Cid et al. 2005). Here, glycogen synthase was found to accumulate in the nucleus under low-glycogen conditions. Furthermore, this same group has reported translocation of the liver isoform of glycogen synthase towards the plasma membrane of cultured hepatocytes in response to glucose addition (Fernandez-Novell et al. 1992a; Fernandez-Novell et al. 1992b; Fernandez-Novell et al. 1996; Fernandez-Novell et al. 1997). Interestingly, changes in the phosphorylation state of glycogen synthase were found not to be important in the translocation observed in either the cultured myotubes or the cultured hepatocytes (Cid et al. 2005; Fernandez-Novell et al. 1992a; Fernandez-Novell et al. 1992b; Fernandez-Novell et al. 1996; Fernandez-Novell et al. 1997).
The subcellular localization of yeast glycogen synthase has not been addressed in any detail, being limited to data available from a high-throughput study (Huh et al. 2003). We addressed the localization of glycogen synthase in yeast cells through the expression of Gsy2p fused to either green fluorescent protein (GFP) or a cyan variant of GFP (CFP). We found that the distribution of Gsy2p was dependent upon glycogen content. In cells that contained high levels of glycogen, Gsy2p was distributed throughout the cytoplasm. In cells that had reduced glycogen content, Gsy2p localized to discrete regions of the cytoplasm. Lastly, in cells that contained no glycogen whatsoever, Gsy2p was concentrated within the nucleus. The significance of the variations in glycogen synthase distribution is discussed.
Materials and Methods
Materials
Standard laboratory reagents were obtained from Sigma-Aldrich, St. Louis, MO, USA and material for the preparation of yeast media were from BD, Franklin Lakes, NJ, USA. Restriction endonucleases and other molecular biology reagents were from New England Biolabs, Ipswich, MA, USA. The sources of other specialty chemicals are given in the text.
Yeast and bacterial strains and media
The yeast strains used are listed in Table 1. Standard techniques were used for manipulation (Guthrie and Fink 1991). A polymerase chain reaction strategy was used to generate the yellow fluorescent protein fusion to glycogen phosphorylase. Briefly, a cassette comprising 3' sequence from the GPH1 gene (with the stop codon removed), the yellow fluorescent protein coding sequence, and the KanMX6 selectable marker was amplified from the plasmid pKT140 (Sheff and Thorn 2004). The resulting PCR product was transformed into the yeast strain EG328-1A and transformants were selected on YPD medium (2% bacto peptone, 1% yeast extract, 2% glucose, 2% agar) supplemented with 200 μg ml-1 Geneticin (MP Biomedicals, Solon, OH, USA). Isolated colonies were streaked to fresh YPD+Geneticin plates. The insertion of the tag at the correct position in the chromosome was verified by a PCR-based test. Several clones having the correct insertion were identified and one, designated MB1-8, was chosen for further work. Mating and tetrad analysis were used to generate yeast strains carrying the GPH1-YFP fusion in conjunction with other mutations (Guthrie and Fink 1991).
Table 1.
Yeast strains used
| Strain* | Genotype | Source or Reference |
|---|---|---|
| EG328-1A | MATα leu2 trp1 ura3 | K. Tatchell |
| EG328-2D | MATα leu2 trp1 ura3 gac1::LEU2 | K. Tatchell |
| CC9 | MATα leu2 trp1 ura3 glg1-1::LEU2 glg2::URA3 | Cheng et al. 1995 |
| DH3 | MATα leu2 trp1 ura3 gsy1::LEU2 gsy2::URA3 | Hardy et al. 1994 |
| DH11-51 | MATα leu2 trp1 ura3 reg1::URA3 | Huang et al. 1996b |
| MB1-8 | MATα leu2 trp1 ura3 GPH1-yECitrine::KanMX6 | This study |
| WW10 | MATα leu2 trp1 ura3 pcl8::TRP1 pcl10::LEU2 | Wilson et al. 1999 |
| WW33-4C | MATa trp1 ura3 glg1-1::LEU2 glg2::URA3 GPH1-yECitrine::KanMX6 | This study |
| BY4743 |
MATa/MATα his3/his3 leu2/leu2 lys2/LYS2
MET15/met15 ura3/ura3 |
Open Biosystems |
| gsy2::KanMX |
MATa/MATα his3/his3 leu2/leu2 lys2/LYS2
MET15/met15 ura3/ura3 gsy2::KanMX4/gsy2::KanMX4 |
Open Biosystems |
Strains EG328-1A, EG328-2D, CC9, DH3, DH11-51, MB1-8, WW10, and WW33-4C are congenic. Strains BY4743 and gsy2::KanMX are isogenic.
Yeast strains were grown at 30 °C in synthetic complete (SC) medium from which tryptophan (SC-Trp), or uracil (SC-Ura) had been omitted as appropriate to allow for plasmid maintenance. SC contained 0.67 % (w/v) yeast nitrogen base without amino acids, 2% (w/v) glucose and the required Supplement Mix, either CSM-Trp or CSM-Ura as appropriate (MP Biomedicals, Solon, OH, USA). Agar was added at 2% w/v in solid media. Plasmids were maintained in E. coli strain DH5α or, in cases where the restriction endonuclease BclI was to be used, in the methylase-deficient strain ER2925. Standard molecular biology techniques were used for growth, transformation and plasmid isolation (Sambrook et al. 1989)
Plasmids
The vector encoding the C-terminal fusion between green fluorescent protein (GFP) and glycogen synthase, designated pJR1420-A, was a kind gift from Dr. Jared Rutter, University of Utah School of Medicine. Briefly, pJR1420-A comprises the promoter and coding region of the GSY2 gene, from which the stop codon had been deleted, cloned into pRS416 (Sikorski and Hieter 1989), containing the gene for enhanced GFP and a yeast terminator sequence from the UGP1 gene. Site directed mutagenesis to replace S650, S654, or T667 with alanine was performed using the QuickChange kit (Stratagene, La Jolla, CA, USA), generating the vectors pJR1420-B, pJR1420-C, and pWW213 respectively. To generate a TRP1-marked version of the Gsy2-GFP vector, the region containing the GSY2 promoter and coding sequence, the GFP coding sequence, and the terminator were transferred to pRS314 (Sikorski and Hieter 1989), producing pWW214. The S650A, S654A, and T667A mutants were similarly transferred to the pRS314 vector, generating pWW215, pWW217 and pWW218. A GFP-tagged GSY2 mutant in which all three phosphorylation sites were mutated to non-phosphorylatable residues was constructed by replacing a SacI/NdeI fragment of pJR1420-A, which spanned the phosphorylation sites, with a SacI/NdeI fragment from a phosphorylation site mutant form of GSY2 carried in the pET-28a vector (EMD Chemicals, Gibbstown, NJ, USA; Huang et al. 1998). The region of this modified pJR1420-A containing the GSY2 promoter and coding sequence, the GFP coding sequence, and the terminator was transferred to pRS314, producing pWW219. A PCR-based approach was used to generate pWW20, in which GFP was fused to a mutant form of GSY2 where R579, R580, and R582, which are involved in binding glucose-6-P, had been mutated to alanine (Pederson et al. 2000; Pederson et al. 2004). A region spanning two BclI sites was amplified from a pET-28a construct that carried GSY2 with the R579A/R580A/R582A mutations (Pederson et al. 2000). The resulting PCR product was sequenced and then digested with BclI and used to replace a BclI fragment of pJR1420-A. The region of this modified pJR1420-A containing the GSY2 promoter and coding sequence, the GFP coding sequence, and the terminator were transferred to pRS314, producing pWW220. To construct cyan fluorescent variants of Gsy2p, the yEmCFP cassette, encoding a yeast-enhanced version of blue-shifted GFP that does not form dimers, was amplified from vector pKT212 (Sheff and Thorn 2004) and sub-cloned to pRS314. The GSY2 promoter and coding sequences from pJR1420-A or pJR1420-B (S650A phosphorylation site mutant) were sub-cloned into this modified pRS314 vector, generating pWW216 or pWW221, respectively.
Microscopy
Aliquots of liquid growth medium (5 ml) were inoculated at a density of 4 × 105 to 6 × 105 cells/ml and incubated for 18 hours at 30 °C, with shaking. By the end of this incubation period, the cultures had reached a density of 1.0 × 108 to 2 × 108 cells/ml. Aliquots of yeast culture were transferred to microscope slides and covered with concanavalin A-coated coverslips. Images were then collected using a Zeiss Axioskop 2 Plus equipped with a 100x oil immersion lens and an AxioCam MRm camera. Axiovision 3.1 software was used for image analysis. The filter sets used for epifluorescence were Zeiss FITC (GFP), Chroma yellow GFP BP catalogue number 41028 (YFP), and Chroma Cyan GFP V2 catalogue number 31044v2 (CFP). The specimens were prepared immediately before use and the maximum time allowed to elapse between specimen preparation and the completion of data collection was 15 minutes.
Staining of yeast nuclei
The stain Hoechst 33342 (Invitrogen, Carlsbad, CA, USA) was used to visualize yeast nuclei. Hoechst 33342 was added to an aliquot of yeast culture (final concentration of 10 μg ml-1) from an aqueous stock solution. The cell aliquot was incubated in the dark at 30 °C and stained cells were then examined under the microscope using a Zeiss DAPI filter set and the conditions described above.
Enzyme assays
Glycogen synthase was assayed in extracts prepared from yeast cells by lysis with glass beads as described previously (Hardy et al. 1994; Thomas et al. 1968). Glycogen phosphorylase activity was measured in the direction of glycogen synthesis by monitoring the incorporation of [14C]glucose from [14C]glucose-1-phosphate into glycogen using a modification of published procedures (Gilboe et al. 1972)
Glycogen determination
Glycogen was determined enzymatically as described previously (Wang et al. 2001). Glycogen content was also assessed qualitatively by exposing yeast colonies growing on the surface of agar plates to iodine vapor. Yeast colonies stain brown in proportion to the amount of glycogen that they contain (Chester 1968).
Immunoblotting of GFP and YFP fusion proteins
Yeast cell extracts were prepared as described (Hardy et al. 1994) and resolved by polyacrylamide gel electrophoresis in the presence of sodium dodecyl sulphate (Laemmli 1970). The resolved proteins were transferred to a PVDF membrane, which was probed with Anti-GFP (Roche Applied Science, Indianapolis, IN, USA) or polyclonal antibodies raised to a peptide derived from Gsy2p (Hardy and Roach 1993). Enhanced chemiluminescence and a horse radish peroxidase-conjugated second antibody (Promega, Madison, WI, USA) were used for detection.
Protein determination
The protein content was estimated by the method of Bradford, using bovine serum albumin as a standard (Bradford 1976).
Results
Localization pattern observed for Gsy2-GFP
We expressed Gsy2p with a C-terminal fusion to GFP from a centromeric vector. The construct was under the control of the native GSY2 promoter. We transformed a gsy2 deletion strain with this plasmid and found that it restored glycogen synthase activity, yielding an activity only around 50% greater than that of the isogenic wild type strain (gsy2 mutant strain: 1.8 ± 0.13 nmol min-1 mg-1, isogenic wild type strain 18.7 ± 2.7 nmol min-1 mg-1, gsy2 mutant expressing Gsy2-GFP: 28.0 ± 1.0 nmol min-1 mg-1). In agreement with these activity measurements, Western blot analysis with antibody to Gsy2p indicated that the Gsy2-GFP construct was expressed at a level similar to that of wild type Gsy2p (not shown).
A large-scale screen of protein localization using GFP-fusions had indicated that Gsy2p was found both in the bulk cytoplasm and in punctate “speckles” (Huh et al. 2003). In addition, Gsy2p was said to be nuclear in a subpopulation of cells (Huh et al. 2003). We examined gsy2 mutant cells transformed with the Gsy2-GFP fusion using a microscope equipped for epifluorescence work.
We found that GFP-fluorescence was detectable in 58 ± 4% of cells and that, as reported by Huh et al., Gsy2-GFP was found in the bulk cytoplasm. However, careful examination of cells revealed three distinct localization patterns (Figure 1). The three patterns were one or a few well-defined, relatively large bright spots of fluorescence (spot), a spot accompanied by a haze that appeared to be due to the presence of out of focus fluorescence from smaller concentrations of Gsy2-GFP (spot plus haze), and a diffuse cytoplasmic staining, which remained diffuse upon focusing through the cell (diffuse). Roughly one third of the stained cells fell into each category in this particular genetic background (Figure 2). We could not detect Gsy2-GFP within the nucleus (not shown).
Figure1. The localization of Gsy2-GFP within wild type yeast cells.

The wild type yeast strain BY4743 was transformed with a centromeric plasmid encoding the GSY2 open reading frame fused at the C-terminus to GFP (pJR1420-A). After overnight growth in SC-Ura medium, aliquots of cells were examined by fluorescence microscopy. The left-hand series of images shows representative fields of cells observed using differential interference contrast. The right-hand series of panels shows fluorescence images of the same fields. Three distinct types of localization pattern could be distinguished within a single cell population; well defined bright spots (spot), spots accompanied with a haze (spot+haze), and diffuse cytoplasmic staining (diffuse). The scale bar represents 10 μm.
Figure 2. Mutation of the known phosphorylation sites in glycogen synthase causes a shift from more localized to more diffuse staining.

The wild type yeast strain BY4743 was transformed with centromeric plasmids encoding wild type Gsy2-GFP (pJR1420-A), S650A Gsy2-GFP (pJR1420-B), S654A Gsy2-GFP (pJR1420-C), or T667A Gsy2-GFP (pWW213). The localization of each Gsy2-GFP construct was then determined by fluorescence microscopy after overnight growth in SC-Ura medium. Panel A: In each case, the percentage of cells showing fluorescence characterized as well defined bright spots (spot), spots accompanied with a haze (spot+haze), and diffuse cytoplasmic staining (diffuse) was determined. A minimum of ten fields, containing a total of at least 250 cells were counted. The results show the mean ± standard error of the mean for three separate experiments. Panel B: The left-hand series of images shows representative fields of cells observed using differential interference contrast. The right-hand series of panels shows fluorescence images of the same fields. The three phosphorylation site mutants had a more diffuse localization pattern than did wild type glycogen synthase. The scale bar represents 10 μm.
The localization pattern is perturbed in phosphorylation site mutants of glycogen synthase
There is evidence from mammalian systems that the localization of muscle glycogen synthase is affected by its phosphorylation state, at least under some conditions (Ou et al. 2005; Prats et al. 2005). Therefore, we examined the localization patterns seen with modified forms of Gsy2-GFP in which the three known regulatory phosphorylation sites (S650, S654, and T667) had been mutated to alanine. These constructs were all fully functional, as judged by their ability to restore glycogen accumulation to a gsy2 mutant strain (not shown).
Mutation of any one of the three known phosphorylation sites in Gsy2p caused a shift in the distribution pattern of the GFP-fusion proteins away from the bright spot pattern towards the diffuse pattern (Figure 2).
The tendency of GFP to form dimers could confound our localization studies. To address this issue, we generated a cyan fluorescent protein (CFP) tagged version of Gsy2p using an optimized form of CFP that does not form dimers (Sheff and Thorn 2004). The results obtained were indistinguishable from those obtained with Gsy2-GFP (not shown). We chose to use GFP for most studies since it is brighter than CFP.
The localization of glycogen synthase is perturbed in mutant strains that fail to either phosphorylate or dephosphorylated glycogen synthase appropriately
We went on to analyze the distribution of the glycogen synthase-GFP fusion proteins in yeast strains that were defective in glycogen synthase kinase and glycogen synthase phosphatase activity. We analyzed Gsy2-GFP protein localization in yeast cells where both the PCL8 and PCL10 genes had been deleted (resulting in severely compromised ability to phosphorylate and inactivate glycogen synthase). In pcl8 pcl10 mutant yeast cells, glycogen synthase had a diffuse, cytoplasmic localization and the distinct spots and spot/haze structures were almost entirely absent (Figure 3).
Figure 3. Genetic manipulation of the phosphorylation state of glycogen synthase alters the intracellular distribution of the enzyme.

Wild type Gsy2-GFP (pJR1420-A) was expressed in cells that were defective in the phosphorylation of glycogen synthase (pcl8 pcl10 double mutant; WW10) and in cells that were unable to dephosphorylate glycogen synthase (gac1 mutant; EG328-2D). Cells were examined after overnight growth in SC-Ura medium. Panel A: The localization pattern of Gsy2-GFP was scored by fluorescence microscopy as described in the legend to Figure 2. The wild type cells were strain EG328-1A. Panel B: Representative images of pcl8 pcl10 mutant cells are shown (differential interference contrast on the left, fluorescence image of the same field on the right), where Gsy2-GFP showed a diffuse localization pattern. Panel C: Representative images of gac1 mutant cells are shown, where Gsy2-GFP tended to concentrate in particular areas within cells. Co-staining of DNA with Hoechst 33342 indicated that these spots of Gsy2-GFP fluorescence were not coincident with the nucleus. The scale bar represents 10 μm.
We also analyzed glycogen synthase localization in a strain in which the GAC1 gene had been deleted. Deletion of GAC1 prevented effective dephosphorylation and activation of Gsy2p, a process requiring the Glc7p phosphatase in complex with the Gac1p targeting subunit (Feng et al. 1991; Stuart et al. 1994). Deletion of the GAC1 gene resulted in the localization of Gsy2p to bright pinpoint spots (Figures 3). The spots were variable in size and typically only one or two were found per cell. Staining with Hoechst 33342 dye indicated that these spots of glycogen synthase fluorescence were not coincident with the nucleus (Figure 3). However, the bright spots of fluorescence were often found to coincide with the position of small, rounded, dark-appearing bodies in the differential interference contrast images (not shown).
Note that both the pcl8 pcl10 mutant and the gac1 mutant were constructed in the EG328-1A background, rather than the BY4743 background used in Figure 1 and 2. The distribution of Gsy2-GFP varies somewhat between the EG328-1A and BY4743 wild type backgrounds.
Distribution of glycogen synthase in reg1 mutant cells
Perturbation of the phosphorylation state of glycogen synthase results in changes in the level of glycogen stored within the cell. The pcl8 pcl10 mutants (and cells expressing the Ser650Ala, Ser654Ala or Thr667Ala phosphorylation site mutants of glycogen synthase) store substantially more glycogen than do wild type cells. Conversely, the gac1 mutants show reduced levels of glycogen storage. In order to distinguish phosphorylation control of glycogen synthase localization from localization regulated in response to the abundance of glycogen, we examined the distribution of Gsy2-GFP within reg1 mutant yeast. REG1 encodes a subunit of the Glc7p protein phosphatase and deletion of REG1 results in increased glycogen storage without affecting the phosphorylation state of glycogen synthase (Huang et al. 1996b; Tu and Carlson 1995). We found that Gsy2-GFP had a more diffuse localization in reg1 mutant cells than in wild type yeast cells (Figure 4). The glycogen contents of the strains used were: wild type 20 ± 4.0 μg (107 cells)-1 and reg1 mutant 45 ± 4.5 μg (107 cells)-1.
Figure 4. Deletion of REG1 alters the subcellular distribution of glycogen synthase.

Deletion of the REG1 gene results in increased glycogen storage but does not alter the phosphorylation state of glycogen synthase. A strain in which REG1 had been deleted (DH11-51) was transformed with wild type Gsy2-GFP (pWW214) and grown overnight in SC-Trp medium. The localization pattern of Gsy2-GFP was scored by fluorescence microscopy as described in the legend to Figure 2 and the wild type cells were strain EG328-1A (Panel A). Representative images are shown (Panel B). The left-hand image shows a field of cells observed using differential interference contrast. The right-hand panel shows fluorescence images of the same fields. In reg1 mutant cells, Gsy2-GFP had a more diffuse distribution than in wild type cells. The scale bar represents 10 μm.
Glycogen degradation causes a shift in the localization of glycogen synthase from more diffuse to more compact patterns
To further investigate the relationship between glycogen content and the distribution of glycogen synthase within cells, we determined the effect of glucose removal upon Gsy2-GFP localization. Both wild type and pcl8 pcl10 double mutant cells were grown to saturation in YPD medium. Aliquots were taken and the Gsy2-GFP distribution patterns scored. Glycogen content was assessed in the same aliquot. The remainder of each culture was collected by centrifugation, rinsed with growth medium lacking glucose, and resuspended in this same medium. After 4 hr of incubation at 30 °C, aliquots were removed. The Gsy2-GFP localization was scored, and the glycogen content was measured.
We found that a four hour glucose starvation resulted in a significant decrease in glycogen content and a significant redistribution of Gsy2-GFP away from the more diffuse patterns and towards the more concentrated, bright spot pattern (Figure 5). We plotted glycogen content against the percentage of stained cells exhibiting the bright spot staining pattern, a striking negative correlation was observed where decreasing glycogen correlated with increasing bright spot staining and a concomitant decrease in the diffuse/spot plus haze staining patterns (Figure 6). Therefore, the subcellular distribution of glycogen synthase was sensitive to the glycogen content of the cell.
Figure 5. Removal of glucose from the growth medium results in a reduction in glycogen stores and a redistribution of glycogen synthase within the cell.

Wild type (EG328-1A) and pcl8 pcl10 mutant (WW10) cells were transformed with vector containing Gsy2-GFP (pJR1420-A). After overnight growth in SC-Ura medium, two aliquots of each culture were taken. Cells were collected from one aliquot by centrifugation, washed twice with sterile water, resuspended in fresh SC-Ura medium lacking glucose, and incubated at 30 °C (Starved). From the other aliquot, cells were collected by centrifugation and the growth medium was saved. The cells were washed twice with sterile water, resuspended in the saved growth medium, and incubated as above (Unstarved). After 4 hours, aliquots were taken for glycogen measurement (Panel A) and determination of the subcellular localization of glycogen synthase (Panel B). Four hours of starvation resulted in significant glycogen depletion and an increase in the number of cells exhibiting the bright spot staining pattern.
Figure 6. Reduced glycogen content correlates with increased localization of glycogen synthase.
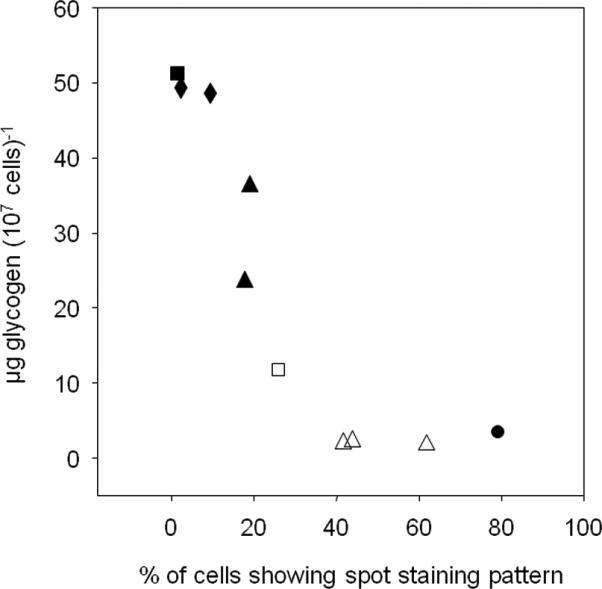
Glycogen content was plotted against the percentage of stained cells that showed the ‘spot’ pattern of tight glycogen synthase localization. Ten individual experiments were analyzed and both standard, overnight cultures (filled symbols) and cultures that had been starved of glucose for 4 hours (open symbols) were included. The yeast strains used were pcl8 pcl10 mutants (WW10; squares), reg1 mutants (DH11-51; diamonds), wild type cells (EG328-1A; triangles), and gac1 mutants (EG328-2D; circles).
Co-localization glycogen synthase and glycogen phosphorylase
Glycogen synthase is known to associate with glycogen particles (Meyer et al. 1970). This fact, coupled with the observed changes in the distribution pattern of Gsy2-GFP as glycogen content changed, led us to hypothesize that Gsy2-GFP fluorescence might act as a marker for glycogen particles within the cell. To further study the relationship between fluorescently-tagged Gsy2p and glycogen particles, we investigated the localization of another protein known to associate with glycogen, glycogen phosphorylase (Gph1p). We utilized a PCR-based approach to tag the GPH1 gene at the 3'-end with a yellow variant of GFP (yECitrine, referred to here as YFP; (Sheff and Thorn 2004) in our wild type background. We verified that the construct was functional by crossing the Gph1-YFP fusion construct into a strain in which the gph1 gene had been deleted and then measuring glycogen phosphorylase activity (gph1 mutant with Gph1-YFP: 120 ± 11 nmol min-1 mg-1, isogenic wild type cells: 100 ± 16 nmol min-1 mg-1). We also generated versions of GSY2 tagged at the 3'-end with a cyan variant of GFP that does not form dimers (yEmCFP, referred to here as CFP; Sheff and Thorn 2004) by swapping the GFP cassette in our expression vectors with a CFP cassette. We transformed the Gph1-YFP strain with vector encoding Gsy2-CFP and determined the localization patterns of both glycogen phosphorylase and glycogen synthase. We found that Gph1-YFP and Gsy2-CFP co-localized (Figure 7A). The distribution of Gsy2-GFP (and therefore Gph1-YFP) between the bright spot, spot plus haze, and diffuse staining patterns was similar to that observed in Figure 3. We also expressed the S650A mutant of Gsy2-CFP in our Gph1-YFP background. Both proteins were found distributed diffusely throughout the cytoplasm (Figure 7B). Therefore, the subcellular distribution of both glycogen phosphorylase and glycogen synthase is influenced in the same way by the glycogen content of the cell. Since both proteins are known to interact physically with glycogen particles but not to interact directly with each other, the distribution of these proteins within the yeast cell might be indicative of the distribution of glycogen particles (Meyer et al. 1970; Ito et al. 2001; Ho et al. 2002).
Figure 7. Co-localization of glycogen synthase and glycogen phosphorylase.
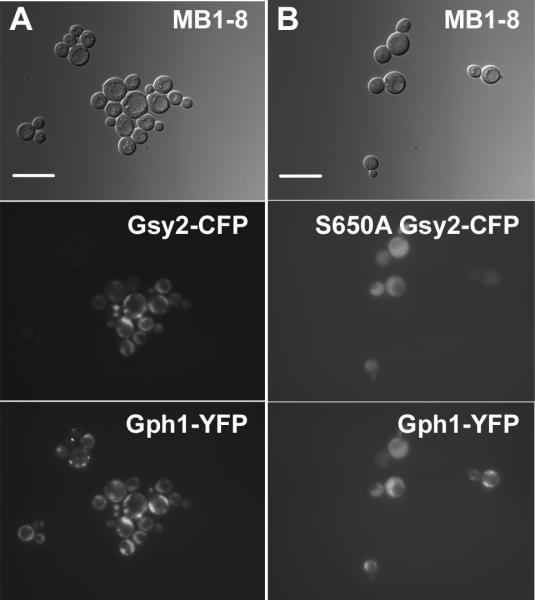
Strain MB1-8, which contains a fusion between GPH1 and a yellow-fluorescent protein cassette at the GPH1 locus, was transformed with centromeric plasmids encoding wild type Gsy2-CFP (pWW216; Panel A) or S650A Gsy2-CFP (pWW221; Panel B). The subcellular localization of both glycogen synthase and glycogen phosphorylase was determined after overnight growth in SC-Trp. Glycogen synthase and glycogen phosphorylase were found to co-localize. The scale bar represents 10 μm.
Distribution of glycogen synthase and glycogen phosphorylase in cells that lack glycogen
If the subcellular distribution of glycogen synthase and glycogen phosphorylase was indeed dependent upon binding to glycogen, then what would the localization of these proteins be in the absence of glycogen? Our initial hypothesis was that Gsy2-CFP and Gph1-YFP, having nothing to bind to, would be distributed uniformly throughout the cytoplasm. To investigate, we crossed our Gph1-YFP strain with a strain that lacked both isoforms of glycogenin (glg1 glg2 double mutant). In the absence of glycogenin, yeast cannot synthesize the primer oligosaccharide that initiates glycogen chains. Cells that lack glycogenin are therefore glycogen-deficient. We transformed the resulting glg1 glg2 strain bearing Gph1-YFP with a vector expressing the Gsy2-CFP fusion. We then determined the subcellular localization of both Gph1-YFP and Gsy2-CFP (Figure 8). Whilst glycogen phosphorylase was distributed throughout the cytoplasm, glycogen synthase tended to concentrate in one specific region of the cell. Staining of DNA with Hoechst 33342 dye indicated that the region in question was the nucleus. Glycogen synthase tended to accumulate within the nucleus of the great majority of cells whereas Gph1p was found in the nucleus of <3% of cells.
Figure 8. In the absence of glycogen, glycogen synthase but not glycogen phosphorylase localizes to the nucleus.

Gsy2-CFP (pWW216) was expressed in strain WW33-4C, in which both glycogenin genes were deleted and the GPH1-YFP fusion described in the legend to Figure 7 was present at the GPH1 locus. The subcellular distributions of glycogen synthase and glycogen phosphorylase were determined after overnight growth in SC-Trp medium (Panel A). Glycogen synthase was found to accumulate in the nucleus of the majority of cells (indicated by the arrows in the center panel) but glycogen phosphorylase did not (note absence of Gph1-YFP fluorescence at positions indicated by arrows in the lower panel). Panel B: Nuclear localization of glycogen synthase was confirmed by co-staining with Hoechst 33342 dye. The scale bar represents 10 μm.
Translocation of mammalian muscle glycogen synthase into the nucleus has been reported to occur in the absence of glycogen (Cid et al. 2005). Guinovart and colleagues demonstrated that an arginine-rich region at the C-terminus of the protein was required for nuclear uptake (Cid et al. 2005). This region is conserved in Gsy2p and had previously been shown to be required for glucose-6-P binding in both the yeast and mammalian enzymes (Hanashiro and Roach 2002; Pederson et al. 2000). We expressed a mutant form of Gsy2-GFP in which three arginine residues implicated in both glucose-6-P binding and nuclear uptake of glycogen synthase were mutated to alanine (R579A/R580A/R582A) in the glg1 glg2 double mutant yeast strain CC9. Nuclear uptake of Gsy2-GFP was still observed, indicating that the arginine-rich sequence in question was not involved in the uptake process (Figure 9).
Figure 9. Nuclear localization of glycogen synthase does not require the presence of a basic region at the C-terminus, implicated in glucose-6-P binding.

The yeast strain CC9, which lacks both isoforms of glycogenin, was transformed with a construct encoding a R579A/R580A/R582A mutant of Gsy2-GFP (pWW220). This region had been implicated in binding glucose-6-P and also in the nuclear uptake of mammalian muscle glycogen synthase. The subcellular distribution of the R579A/R580A/R582A mutant was determined after overnight growth in SC-Trp medium. DNA was stained using Hoechst 33342 dye. The R579A/R580A/R582A mutant was found in the nucleus. The scale bar represents 10 μm.
Transport into and out of the nucleus is frequently sensitive to phosphorylation. In order to investigate any potential role for phosphorylation of glycogen synthase on the nuclear uptake of the protein seen in glg1 gl2 mutants, we transformed strain CC9 with a variety of different phosphorylation site mutants of glycogen synthase. However, the introduction of each construct resulted in the generation of a yeast strain that was capable of synthesizing significant quantities of glycogen (Figure 10). The amount of glycogen synthesized ranged from 30% to 120% of wild type levels, making interpretation of the importance of phosphorylation in the control of Gsy2-GFP localization hard to determine. Note that despite encoding a de-regulated form of glycogen synthase (Pederson et al. 2000), the R579A/R580A/R582A mutant was incapable of restoring glycogen storage to a glg1 glg2 mutant (Figure 10).
Figure 10. Expression of non-phosphorylatable glycogen synthase mutants restores glycogen storage to yeast strain lacking glycogenin.

The yeast strain CC9, which lacks both isoforms of glycogenin, was transformed with constructs encoding the S650A (pWW215), S654A (pWW217), T667A (pWW218), S650D/S654D/T667D (pWW219), R579A/R580A/R582A (pWW220), mutant forms of Gsy2-GFP, wild type Gsy2-GFP (pWW214), or empty vector (pRS314). The strain EG328-1A, from which CC9 was derived, was transformed with empty vector (pRS314). Glycogen content was assessed by staining by exposing patches of cells to iodine vapor. Expression of phosphorylation site mutants of glycogen synthase, but not expression of wild type glycogen synthase or the R579A/R580A/R582A mutant, restored glycogen accumulation to glycogenin-deficient yeast.
Discussion
We have determined that the subcellular localization of the glycogen metabolizing enzymes glycogen synthase (Gsy2p) and glycogen phosphorylase (Gph1p) is dependent upon the glycogen content of cells. In cells that contain high levels of glycogen, Gsy2p and Gph1p take up a diffuse localization, being distributed throughout the cytoplasm but excluded from the vacuole and nucleus. In cells depleted for glycogen, either by removal of glucose from the growth medium or by ‘forcing’ phosphorylation of glycogen synthase via deletion of GAC1, glycogen synthase and glycogen phosphorylase localized to intensely-fluorescent spots. Gsy2p and Gph1p do not interact directly with each other yet both bind to glycogen particles. The co-localization of Gsy2p and Gph1p that we observed, coupled with the changes in localization pattern seen in response to changes in the level of glycogen, led us to propose that our tagged proteins were acting as markers of glycogen particle distribution.
The only other study addressing the localization of glycogen synthase and glycogen phosphorylase in yeast was the high-throughput study of Huh et al., which reported a fairly diffuse cytoplasmic distribution for both enzymes (Huh et al. 2003). In the case of Gsy2p, some cells were reported to show aggregates of staining in spot or speckle patterns, and nuclear localization was apparently seen in others. Examination of the images available on the website that accompanies the study of Huh et al. indicates that the majority of cells could be fitted into our categories of either ‘diffuse’ or ‘spot plus haze’, although the background was quite pronounced.
Cells showing the speckle-like pattern observed by Huh et al. were not seen in our study, nor did we observe nuclear localization of Gsy2p unless we took steps to prevent glycogen storage (discussed below). We believe that these discrepancies likely arose due to differences in the growth phase of the cells being examined. We utilized saturated cultures that had been grown for sixteen hours, by which point we know glycogen synthase expression and glycogen storage to be substantial. In contrast, the study of Huh et al. utilized cells in the mid-logarithmic phase of growth, which is before the onset of appreciable glycogen accumulation and a point at which GSY2 expression is far from maximal.
When considering the localization of glycogen phosphorylase reported by Huh et al. (diffuse, cytoplasmic), the same argument relating to the use of cultures very early in growth can be made. Furthermore, it is important to recognize that the scale of the study conducted by Huh et al. precluded rigorous functional testing of fusion proteins. Therefore, it is possible that full length, functional glycogen phosphorylase was not expressed and that the diffuse cytoplasmic localization pattern seen was an artifact of this.
It is perhaps worth commenting here on the heterogeneity of staining patterns observed for Gsy2-GFP and Gph1-YFP in our saturated cultures of wild type cells (spot, spot plus haze, and diffuse). One might have expected that, at the end of growth, most cells would be in a similar metabolic state, would have acquired a similar glycogen content and therefore would exhibit a similar subcellular distribution of glycogen synthase/phosphorylase. However, Cahill et al. have examined the glycogen content of yeast populations at the individual cell level (Cahill et al. 2000). Even after 48 hr of growth in batch culture, these workers found substantial variation in glycogen content between cells (Cahill et al. 2000). The distribution of different Gsy2-GFP and Gph1-YFP staining patterns that we observed, presumably corresponding to differences in glycogen content, is entirely consistent with these earlier results.
In the absence of glycogen, achieved by deletion of the GLG1 and GLG2 genes that encode glycogenin (Cheng et al. 1995), Gsy2p localized to the nucleus. We propose that, as glycogen storage begins in wild type cells, glycogen synthase binds to glycogen particles and is therefore retained in the cytoplasm. The absence of glycogen in the glycogenin mutant allows nuclear uptake. Notably, we found that Gph1p did not localize to the nucleus in glycogen-deficient cells and instead took up a diffuse, cytoplasmic distribution. This was the only instance where we could drive glycogen synthase and glycogen phosphorylase apart from each other.
A high-throughput study examining protein-protein interactions in yeast found a specific association between Gsy2p and the nuclear importin Pse1p (Krogan et al. 2006). This immediately raised the possibility that specific nuclear uptake of Gsy2p might be driven by Pse1p. However, a novel nuclear localization sequence recognized by Pse1p and four other importins (Kap95p, Kap104p, Kap114p and Kap123p) has been defined (Fries et al. 2007). This sequence is R/KxxL(x)nV/YxxV/IxK/RxxxK/R and a match is not found in the sequence of Gsy2p. Although not conclusive, this observation, coupled with results from mammalian systems, hints that an alternative mechanism of nuclear uptake might be operating.
Guinovart's group described translocation of the muscle isoform of glycogen synthase between the cytoplasm and nucleus (Cid et al. 2005; Ferrer et al. 1997). They went on to demonstrate that the nuclear uptake process required a sequence of basic residues found towards the carboxyl-terminus of glycogen synthase, which previous work had shown to be required for activation of glycogen synthase in response to glucose-6-P (Cid et al. 2005; Hanashiro and Roach 2002). These residues are conserved in yeast glycogen synthase and, again, are implicated in binding of glucose-6-P (Pederson et al. 2000; Pederson et al. 2004). However, we found that mutation of the conserved basic residues to alanine did not prevent nuclear uptake of Gsy2-GFP in our strain that did not express glycogenin.
Transport of many proteins into and out of the nucleus is regulated by phosphorylation. However, it proved hard to ascertain what role, if any, phosphorylation of glycogen synthase played in the nuclear uptake process. Expression of mutant forms of glycogen synthase lacking regulatory phosphorylation sites in the glycogenin-deficient strains resulted in significant glycogen accumulation. Previously, it had been shown that expression of a C-terminally truncated glycogen synthase, which lacked all known regulatory phosphorylation sites, could restore glycogen storage to a glycogenin-deficient yeast strain (Torija et al. 2005). We had hoped that expression of single phosphorylation site mutants might not allow glycogen storage to occur in the absence of glycogenin, but this proved not to be the case. The accumulation of glycogen in glycogenin mutants expressing Gsy2-GFP phosphorylation site mutants confounded our localization studies. It was difficult to determine if phosphorylation per se influenced nuclear uptake of glycogen synthase or if the increase in glycogen stores produced by expression of the mutant glycogen synthase acted to exclude the enzyme from the nucleus. Thus, the mechanism by which Gsy2p enters the yeast nucleus remains enigmatic and is a focus of our current work.
Glycogen serves as a store of both carbon and energy and the nucleocytoplasmic distribution of glycogen synthase is regulated by the cellular glycogen content in both yeast and muscle cells. When glycogen levels are greatly reduced, glycogen synthase is no longer anchored in the cytoplasm but can traffic into the nucleus. This is similar to the situation observed with mammalian muscle glycogen synthase in cultured cells (Cid et al. 2005). Here, a model has been developed where translocation of the enzyme from the cytoplasm to nucleus is proposed to act as a form of cellular ‘fuel gauge’, nuclear entry of glycogen synthase indicating that energy reserves were low (Cid et al. 2005). Might a similar model hold for yeast too and what could glycogen synthase do within the nucleus?
The glycolytic enzyme, glyceraldehyde-3-phosphate dehydrogenase has been shown to play a role in a variety of nuclear events, including telomere maintenance, transcriptional control, and nuclear membrane fusion (Sirover 2005). Therefore, precedents exist for the involvement of metabolic enzymes in the regulation of nuclear processes. It is possible that glycogen synthase could regulate transcription in response to energy availability by some as yet undetermined means.
Acknowledgements
This work was supported by grants (to WAW) from the National Institutes of Health (GM081810) and the Iowa Osteopathic Educational and Research Fund. Thanks to Drs. Bart Pederson and Anna DePaoli-Roach for many helpful discussions and to Dr. Jared Rutter, for the gift of the pJR1420-A, -B, and -C vectors, which spurred this investigation.
References
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Cahill G, Walsh PK, Donnelly D. Determination of yeast glycogen content by individual cell spectroscopy using image analysis. Biotechnol. Bioeng. 2000;69(3):312–322. doi: 10.1002/1097-0290(20000805)69:3<312::aid-bit9>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- Cheng C, Mu J, Farkas I, Huang D, Goebl MG, Roach PJ. Requirement of the self-glucosylating initiator proteins Glg1p and Glg2p for glycogen accumulation in Saccharomyces cerevisiae. Mol. Cell. Biol. 1995;15(12):6632–6640. doi: 10.1128/mcb.15.12.6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chester VE. Heritable glycogen-storage deficiency in yeast and its induction by ultra-violet light. J. of Gen. Microbiol. 1968;51(1):49–56. doi: 10.1099/00221287-51-1-49. [DOI] [PubMed] [Google Scholar]
- Cid E, Cifuentes D, Baque S, Ferrer JC, Guinovart JJ. Determinants of the nucleocytoplasmic shuttling of muscle glycogen synthase. FEBS J. 2005;272(12):3197–3213. doi: 10.1111/j.1742-4658.2005.04738.x. [DOI] [PubMed] [Google Scholar]
- Farkas I, Hardy TA, Goebl MG, Roach PJ. Two glycogen synthase isoforms in Saccharomyces cerevisiae are coded by distinct genes that are differentially controlled. J. Biol. Chem. 1991;266(24):15602–15607. [PubMed] [Google Scholar]
- Feng Z, Wilson SE, Peng ZY, Schlender KK, Reiman EM, Trumbly RJ. The yeast GLC7 gene required for glycogen accumulation encodes a type 1 protein phosphatase. J. Biol. Chem. 1991;266(35):23796–23801. [PubMed] [Google Scholar]
- Fernandez-Novell JM, Bellido D, Vilaro S, Guinovart JJ. Glucose induces the translocation of glycogen synthase to the cell cortex in rat hepatocytes. Biochem. J. 1997;321(Pt1):227–231. doi: 10.1042/bj3210227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Novell JM, Arino J, Vilaro S, Guinovart JJ. Glucose induces the translocation and the aggregation of glycogen synthase in rat hepatocytes. Biochem. J. 1992a;281(Pt2):443–448. doi: 10.1042/bj2810443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Novell JM, Roca A, Bellido D, Vilaro S, Guinovart JJ. Translocation and aggregation of hepatic glycogen synthase during the fasted-to-refed transition in rats. Eur. J. Biochem. 1996;238(2):570–575. doi: 10.1111/j.1432-1033.1996.0570z.x. [DOI] [PubMed] [Google Scholar]
- Fernandez-Novell JM, Arino J, Vilaro S, Bellido D, Guinovart JJ. Role of glucose 6-phosphate in the translocation of glycogen synthase in rat hepatocytes. Biochem. J. 1992b;288(Pt2):497–501. doi: 10.1042/bj2880497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer JC, Baque S, Guinovart JJ. Muscle glycogen synthase translocates from the cell nucleus to the cytosol in response to glucose. FEBS Lett. 1997;415(3):249–252. doi: 10.1016/s0014-5793(97)01136-8. [DOI] [PubMed] [Google Scholar]
- Francois J, Parrou JL. Reserve carbohydrates metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Rev. 2001;25(1):125–45. doi: 10.1111/j.1574-6976.2001.tb00574.x. [DOI] [PubMed] [Google Scholar]
- Francois J, Hers HG. The control of glycogen metabolism in yeast. 2. A kinetic study of the two forms of glycogen synthase and of glycogen phosphorylase and an investigation of their interconversion in a cell-free system. Eur. J. Biochem. 1988;174(3):561–567. doi: 10.1111/j.1432-1033.1988.tb14135.x. [DOI] [PubMed] [Google Scholar]
- Francois J, Villanueva ME, Hers HG. The control of glycogen metabolism in yeast. 1. Interconversion in vivo of glycogen synthase and glycogen phosphorylase induced by glucose, a nitrogen source or uncouplers. Eur. J. Biochem. 1988;174(3):551–559. doi: 10.1111/j.1432-1033.1988.tb14134.x. [DOI] [PubMed] [Google Scholar]
- Francois JM, Thompson-Jaeger S, Skroch J, Zellenka U, Spevak W, Tatchell K. GAC1 may encode a regulatory subunit for protein phosphatase type 1 in Saccharomyces cerevisiae. EMBO J. 1992;11(1):87–96. doi: 10.1002/j.1460-2075.1992.tb05031.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fries T, Betz C, Sohn K, Caesar S, Schlenstedt G, Bailer SM. A novel conserved nuclear localization signal is recognized by a group of yeast importins. J. Biol. Chem. 2007;282(27):19292–19301. doi: 10.1074/jbc.M700217200. [DOI] [PubMed] [Google Scholar]
- Gilboe DP, Larson KL, Nuttall FQ. Radioactive method for the assay of glycogen phosphorylases. Anal. Biochem. 1972;47(1):20–7. doi: 10.1016/0003-2697(72)90274-6. [DOI] [PubMed] [Google Scholar]
- Guthrie C, Fink GR. Guide to yeast genetics and molecular biology. Methods Enzymol. 1991;194 [PubMed] [Google Scholar]
- Hanashiro I, Roach PJ. Mutations of muscle glycogen synthase that disable activation by glucose 6-phosphate. Arch. Biochem. Biophys. 2002;397(2):286–92. doi: 10.1006/abbi.2001.2623. [DOI] [PubMed] [Google Scholar]
- Hardy TA, Roach PJ. Control of yeast glycogen synthase-2 by COOH-terminal phosphorylation. J. Biol. Chem. 1993;268(32):23799–23805. [PubMed] [Google Scholar]
- Hardy TA, Huang D, Roach PJ. Interactions between cAMP-dependent and SNF1 protein kinases in the control of glycogen accumulation in saccharomyces cerevisiae. J. Biol. Chem. 1994;269(45):27907–27913. [PubMed] [Google Scholar]
- Ho Y, Gruhler A, Heilbut A, Bader GD, Moore L, Adams SL, Millar A, Taylor P, Bennett K, Boutilier K, Yang L, Wolting C, Donaldson I, Schandorff S, Shewnarane J, Vo M, Taggart J, Goudreault M, Muskat B, Alfarano C, Dewar D, Lin Z, Michalickova K, Willems AR, Sassi H, Nielsen PA, Rasmussen KJ, Andersen JR, Johansen LE, Hansen LH, Jespersen H, Podtelejnikov A, Nielsen E, Crawford J, Poulsen V, Sorensen BD, Matthiesen J, Hendrickson RC, Gleeson F, Pawson T, Moran MF, Durocher D, Mann M, Hogue CW, Figeys D, Tyers M. Systematic identification of protein complexes in Saccharomyces cerevisiae by mass spectrometry. Nature. 2002;415(6868):180–183. doi: 10.1038/415180a. [DOI] [PubMed] [Google Scholar]
- Huang D, Wilson WA, Roach PJ. Glucose-6-P control of glycogen synthase phosphorylation in yeast. J. Biol. Chem. 1997;272(36):22495–22501. doi: 10.1074/jbc.272.36.22495. [DOI] [PubMed] [Google Scholar]
- Huang D, Farkas I, Roach PJ. Pho85p, a cyclin-dependent protein kinase, and the Snf1p protein kinase act antagonistically to control glycogen accumulation in Saccharomyces cerevisiae. Mol. Cell. Biol. 1996a;16(8):4357–4365. doi: 10.1128/mcb.16.8.4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Chun KT, Goebl MG, Roach PJ. Genetic interactions between REG1/HEX2 and GLC7, the gene encoding the protein phosphatase type 1 catalytic subunit in Saccharomyces cerevisiae. Genetics. 1996b;143(1):119–127. doi: 10.1093/genetics/143.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Moffat J, Wilson WA, Moore L, Cheng C, Roach PJ, Andrews B. Cyclin partners determine Pho85 protein kinase substrate specificity in vitro and in vivo: Control of glycogen biosynthesis by Pcl8 and Pcl10. Mol. Cell. Biol. 1998;18(6):3289–3299. doi: 10.1128/mcb.18.6.3289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh WK, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O'Shea EK. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- Hwang PK, Fletterick RJ. Convergent and divergent evolution of regulatory sites in eukaryotic phosphorylases. Nature. 1986;324:80–84. doi: 10.1038/324080a0. [DOI] [PubMed] [Google Scholar]
- Ito T, Chiba T, Ozawa R, Yoshida M, Hattori M, Sakaki Y. A comprehensive two-hybrid analysis to explore the yeast protein interactome. Proc. Natl. Acad. Sci. U S A. 2001;98:4569–4574. doi: 10.1073/pnas.061034498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogan N, Cagney G, Yu H, Zhong G, Guo X, Ignatchenko A, Li J, Pu S, Datta N, Tikuisis A, Punna T, Peregrin-Alvarez J, Shales M, Zhang X, Davey M, Robinson M, Paccanaro A, Bray J, Sheung A, Beattie B, Richards D, V C, Lalev A, Mena F, Wong P, Starostine A, Canete M, Vlasblom J, Wu S, Orsi C, Collins S, Chandran S, Haw R, Rilstone J, Gandi K, Thompson N, Musso G, St Onge P, Ghanny S, Lam M, Butland G, Altaf-Ul A, Kanaya S, Shilatifard A, O'Shea E, Weissman J, Ingles C, Hughes T, Parkinson J, Gerstein M, Wodak S, Emili A, Greenblatt J. Global landscape of protein complexes in the yeast Saccharomyces cerevisiae. Nature. 2006;440:637–643. doi: 10.1038/nature04670. [DOI] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of sructural head proteins during the assembly of the ead of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Meyer F, Heilmeyer LM, Jr, Haschke RH, Fischer EH. Control of phosphorylase activity in a muscle glycogen particle. I. Isolation and characterization of the protein-glycogen complex. J. Biol. Chem. 1970;245(24):6642–6648. PMID:4320610. [PubMed] [Google Scholar]
- Ni HT, Laporte DC. Response of a yeast glycogen synthase gene to stress. Mol. Microbiol. 1995;16(6):1197–1205. doi: 10.1111/j.1365-2958.1995.tb02342.x. [DOI] [PubMed] [Google Scholar]
- Ou H, Yan L, Osmanovic S, Greenberg CC, Brady MJ. Spatial reorganization of glycogen synthase upon activation in 3T3-L1 adipocytes. Endocrinology. 2005;146(1):494–502. doi: 10.1210/en.2004-1022. [DOI] [PubMed] [Google Scholar]
- Pederson BA, Wilson WA, Roach PJ. Glycogen synthase sensitivity to glucose-6-P is important for controlling glycogen accumulation in Saccharomyces cerevisiae. J. Biol. Chem. 2004;279(14):13764–8. doi: 10.1074/jbc.M312335200. [DOI] [PubMed] [Google Scholar]
- Pederson BA, Cheng C, Wilson WA, Roach PJ. Regulation of glycogen synthase. Identification of residues involved in regulation by the allosteric ligand glucose-6-P and by phosphorylation. J. Biol. Chem. 2000;275(36):27753–61. doi: 10.1074/jbc.M003342200. [DOI] [PubMed] [Google Scholar]
- Prats C, Cadefau JA, Cusso R, Qvortrup K, Nielsen JN, Wojtaszewki JF, Hardie DG, Stewart G, Hansen BF, Ploug T. Phosphorylation-dependent translocation of glycogen synthase to a novel structure during glycogen resynthesis. J. Biol. Chem. 2005;280(24):23165–23172. doi: 10.1074/jbc.M502713200. [DOI] [PubMed] [Google Scholar]
- Roach PJ, Skurat AV, Harris RA. Regulation of glycogen metabolism. In: Jefferson LS, Cherrington AD, editors. Handbook of Physiology Section 7, Volume II The Endocrine pancreas and regulation of metabolism. Lippincott Company; Philadelphia: 2001. pp. 609–647. [Google Scholar]
- Rothman-Denes LB, Cabib E. Glucose 6-phosphate dependent and independent forms of yeast glycogen synthetase. Their properties and interconversions. Biochemistry. 1971;10(7):1236–42. doi: 10.1021/bi00783a021. [DOI] [PubMed] [Google Scholar]
- Rothman-Denes LB, Cabib E. Two forms of yeast glycogen synthetase and their role in glycogen accumulation. Proc. Natl. Acad. Sci. U.S.A. 1970;66(3):967–74. doi: 10.1073/pnas.66.3.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowen DW, Meinke M, LaPorte DC. GLC3 and GHA1 of Saccharomyces cerevisiae are allelic and encode the glycogen branching enzyme. Mol. Cell. Biol. 1992;12(1):22–29. doi: 10.1128/mcb.12.1.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A laboratory manual. Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sheff MA, Thorn KS. Optimized cassettes for fluorescent protein tagging in Saccharomyces cerevisiae. Yeast. 2004;21(8):661–670. doi: 10.1002/yea.1130. [DOI] [PubMed] [Google Scholar]
- Sikorski RS, Hieter P. A system of shuttle vectors and yeast host strains designed for efficient manipulation of DNA in Saccharomyces cerevisiae. Genetics. 1989;122(1):19–27. doi: 10.1093/genetics/122.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirover MA. New nuclear functions of the glycolytic protein, glyceraldehyde-3-phosphate dehydrogenase, in mammalian cells. J. Cell. Biochem. 2005;95(1):45–52. doi: 10.1002/jcb.20399. [DOI] [PubMed] [Google Scholar]
- Smith A, Ward MP, Garrett S. Yeast PKA represses Msn2p/Msn4p-dependent gene expression to regulate growth, stress response and glycogen accumulation. EMBO J. 1998;17(13):3556–3564. doi: 10.1093/emboj/17.13.3556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart JS, Frederick DL, Varner CM, Tatchell K. The mutant type 1 protein phosphatase encoded by glc7-1 from saccharomyces cerevisiae fails to interact productively with the GAC1-encoded regulatory subunit. Mol. Cell. Biol. 1994;14(2):896–905. doi: 10.1128/mcb.14.2.896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teste MA, Enjalbert B, Parrou JL, Francois JM. The Saccharomyces cerevisiae YPR184w gene encodes the glycogen debranching enzyme. FEMS Microbiol. Lett. 2000;193(1):105–10. doi: 10.1111/j.1574-6968.2000.tb09410.x. [DOI] [PubMed] [Google Scholar]
- Thomas JA, Schlender KK, Larner J. A rapid filter paper assay for UDPglucose-glycogen glucosyltransferase, including an improved biosynthesis of UDP-14C-glucose. Anal. Biochem. 1968;25(1):486–99. doi: 10.1016/0003-2697(68)90127-9. [DOI] [PubMed] [Google Scholar]
- Timblin BK, Tatchell K, Bergman LW. Deletion of the gene encoding the cyclin-dependent protein kinase Pho85 alters glycogen metabolism in Saccharomyces cerevisiae. Genetics. 1996;143(1):57–66. doi: 10.1093/genetics/143.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torija MJ, Novo M, Lemassu A, Wilson W, Roach PJ, Francois J, Parrou JL. Glycogen synthesis in the absence of glycogenin in the yeast Saccharomyces cerevisiae. FEBS Lett. 2005;579(18):3999–4004. doi: 10.1016/j.febslet.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Tu JL, Carlson M. REG1 binds to protein phosphatase type 1 and regulates glucose repression in Saccharomyces cerevisiae. EMBO J. 1995;14(23):5939–5946. doi: 10.1002/j.1460-2075.1995.tb00282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Wilson WA, Fujino MA, Roach PJ. Antagonistic controls of autophagy and glycogen accumulation by Snf1p, the yeast homolog of the AMP-activated protein kinase, and the cyclin-dependent kinase Pho85p. Mol. Cell. Biol. 2001;21(17):5742–5752. doi: 10.1128/MCB.21.17.5742-5752.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WA, Mahrenholz AM, Roach PJ. Substrate targeting of the yeast cyclin-dependent kinase Pho85p by the cyclin Pcl10p. Mol Cell Biol. 1999;19(10):7020–30. doi: 10.1128/mcb.19.10.7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita I, Fukui S. Transcriptional control of the sporulation-specific glucoamylase gene in the yeast Saccharomyces cerevisiae. Mol. Cell. Biol. 1985;5(11):3069–3073. doi: 10.1128/mcb.5.11.3069. [DOI] [PMC free article] [PubMed] [Google Scholar]