

Abstract
Due to limited penetration of the BBB, many therapeutic agents in clinical use require higher doses in order to reach effective concentrations in brain. In some instances, these high doses elicit severe side effects. In the case of erythropoietin (EPO), an established neuroprotectant against ischemic brain injury, its low BBB permeability requires such a high therapeutic dose that it can induce dangerous complications such as polycythmia and secondary stroke. The purpose of this study is to generate a modified EPO that has increased facility crossing the BBB without losing its neuroprotective element. We have engineered a fusion protein (EPO-TAT) by tagging a protein transduction domain derived from HIV TAT to the EPO protein. This sequence enhanced the capacity of EPO to cross the BBB in animals at least twofold when IP administered and up to five-fold when IV administered. In vitro experiments showed that this EPO fusion protein retained all its protective properties against neuronal death elicited by oxygen-glucose deprivation and NMDA insults. The needed therapeutic dose of the EPO-TAT was decreased by ~10-fold compared to that of regular EPO to achieve equivalent neuroprotection in terms of reducing volume of infarction induced by middle cerebral artery occlusion in mice. Our results support the approach of using a protein transduction domain coupled to therapeutic agents. In this way, not only can the therapeutic doses be lowered, but agents without BBB permeability may now be available for clinical applications.
Keywords: erythropoietin, drug delivery system, HIV TAT protein, blood-brain barrier, neuroprotection, ischemia/brain
INTRODUCTION
Erythropoietin (EPO) has recently emerged as a promising candidate for neuroprotection both in animal models of ischemia [1, 2] and in stroke patients [3]. This promising approach, especially for patients who are not suitable for tPA treatment, represents a new frontier in the treatment of stroke. However, a significant challenge for the future clinical use of EPO in CNS injury is how to deliver it more efficiently to the brain across the blood-brain barrier (BBB). As only a very small portion (0.5-1%) of systemically administered EPO crosses the BBB [4], large amounts and multiple doses of EPO have been required to achieve effective concentrations in the brain [1, 3]. Such administration regimens of EPO may potentially lead to an increase in hematocrit [5] and stimulate the production of platelets [6-8], increasing the likelihood of microinfarctions and macroinfarctions, thus severely limiting or even precluding the use of EPO as a therapeutic agent for stroke. Alternate strategies to facilitate delivery of EPO across the BBB will greatly broaden EPO’s clinical applications for the treatment of ischemic injury and other neurological diseases.
In our laboratory as well as others, it has been found that fusing a protein with a protein transduction domain (PTD) will enhance its capacity to cross the BBB. Fusion with the transacting activator of transcription (TAT) sequence derived from the HIV virus facilitated delivery of therapeutic agents across the BBB and reduced ischemic brain injury after systemic injection [9-11]. By the same reasoning, fusing the TAT sequence to EPO should enable a larger amount of EPO to cross the BBB, thereby reducing the therapeutic dose necessary to achieve the same level of neuroprotection afforded by higher doses.
In this study, we investigated the impact of fusing the TAT sequence to EPO on the biological activity of EPO and its capacity to protect against ischemic injury in in vitro and in vivo models. In addition, we demonstrated that inclusion of a PTD domain with EPO resulted in about ten-fold decrease in the therapeutic dose of EPO, indicating the efficacy of this approach in clinical applications of therapeutic agents that are hard to deliver across the BBB.
MATERIALS AND METHODS
Construction of the EPO fusion protein containing TAT
We constructed a plasmid that expresses human EPO containing TAT PTD and dihydrogen folate reductase (DHFR). In brief, a cDNA fragment encoding human EPO fused with HIV TAT (YGRKKRRQRRR) and his-6 tag in the stated order was amplified by the PCR method using human EPO cDNA (OriGene, Rockville, MD, USA) as a template. The resulting PCR product was inserted into the multiple cloning sites of plasmid pIRES-EYFP (Clontech, Mountain View, CA, USA) yielding a plasmid designated as pEPO-TAT. Then, mouse DHFR cDNA was PCR-amplified from a mouse cDNA library and inserted into the pEPO-TAT to substitute for EYFP. In the resulting plasmid, referred to as pEPO-TAT-DHFR, both genes were driven by the CMV promoter. Between the genes, the presence of an internal ribosome entry site (IRES) sequence enables their translation as separate genes. A control plasmid encoding EPO (without TAT) and DHFR was constructed in a similar manner.
To produce the EPO-TAT or EPO recombinant protein, plasmids were linearized with XhoI at the end of poly A signal, and then transfected into DHFR-/- Chinese hamster ovary cell line DG44 (a generous gift from Dr. Lawrence Chasin, Columbia University) using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Two days after transfection, the cells were split at a 1:10 ratio and maintained in α-MEM medium (Invitrogen, Carlsbad, CA, USA) until cell colonies were formed. Twenty-four colonies were picked and transferred into four six-well plates. Then, methotrexate was introduced at a gradually increasing concentration (5nM ~ 20μM) as selection pressure to perpetuate only cells that express the gene of interest. EPO expression in the supernatant was quantitatively monitored by the EPO immunoassay kit Quantikine IVD (R&D Systems, Minneapolis, MN, USA), and the clones with no or lower amplification of EPO were discarded. Finally, the clones with the highest EPO-TAT and EPO expression, respectively, were cultured on a large scale, and the medium was collected for the purification of the recombinant proteins.
The fusion proteins were purified using a Ni-NTA superflow agarose column (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. The resulting fusion proteins were dialyzed four times against PBS using the 10K Dialysis Cassette (Pierce, Rockford, IL, USA). The purified proteins were verified by Coomassie blue staining and Western blot analysis, filtered through a 0.2-micron low-protein binding filter and stored at -70 °C. The concentrations of the purified recombinant proteins were determined by the EPO immunoassay kit Quantikine IVD.
Murine model of transient focal ischemia and determination of infarct volume
All animal experiments were approved by the University of Pittsburgh Institutional Animal Care and Use Committee and performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals. Focal cerebral ischemia was produced by intraluminal occlusion of the left middle cerebral artery (MCA) with a nylon monofilament suture as described [12]. Briefly, male 2- to 3-month-old C57BL/6 mice (25-30 g each; The Jackson Laboratory, Bar Harbor, ME, USA) were anesthetized with 1.5% isoflurane in a 30% O2/70% N2O mixture under spontaneous breathing. The rectal temperature was controlled at 37.0 ± 0.5°C via a temperature-regulated heating pad during surgery and MCA occlusion. Mean arterial blood pressure was monitored during MCA occlusion through a tail cuff, and arterial blood gas was analyzed at 15 min after the onset of ischemia. The animals underwent MCA occlusion for 60 min and then reperfusion for 72 hours. To determine the efficacy of EPO treatment, EPO-TAT or EPO at the indicated doses or PBS was administered into the animals through the tail vein at the onset of post-stroke reperfusion. At 72 hours after MCA occlusion, brains were removed and the forebrain was sliced into coronal sections 1 mm thick. Sections were stained with 2% 2,3,5-triphenyltetrazolium chloride (TTC). Infarct volume was determined using MCID image analysis [12]. In all experiments,the experimenter was blinded to the type and dose of injected EPO.
Quantitative analysis of EPO in the cerebrospinal fluid (CSF) and plasma
EPO-TAT and EPO were administered respectively into rats at a concentration of 5000 U/kg body weight (BW) via intraperitoneal injection. Another duplicate set of rats was treated with EPO-TAT and EPO by intravenous injection. Three hours post-injection, CSF samples were collected via cisternal puncture as described [13]. Briefly, rats were anesthetized with 1.5% isoflurane in a 30% O2/70% N2O mixture under spontaneous breathing, and each rat’s head was fixed at a specific forward angle. An incision was made in the skin over the occipital bone, and the first layer of the muscle was cut off. When the allanto-occipital membrane was exposed, the cisterna magna was cannulated by placing a 25-gauge needle there, and 200 μl of CSF was carefully withdrawn for ELISA analysis to determine the concentration of EPO. CSF samples contaminated with blood (>0.25% or 1.7×104 RBC/μl CSF) were discarded. EPO levels in the CSF or plasma were quantitatively determined by ELISA using a human EPO immunoassay kit Quantikine IVD (R&D systems, Minneapolis, MN, USA) as described by the manufacturer. Each sample was assayed in duplicate. PBS-injected animals were used as control.
In vitro models of ischemia-like injury
Primary cultures of cortical neurons were prepared from 17-d C57BL/6 mouse embryos as previously described [14]. Experiments were conducted at 12–14 d in vitro (DIV), when cultures consisted primarily of neurons (97%). Cortical neurons at 12 DIV were pretreated with 1 U/ml EPO-TAT or wild-type EPO for 24 hr, then subjected to either oxygen and glucose deprivation (OGD) or N-methyl D-aspartate (NMDA) neurotoxicity.
For the OGD model, cortical neurons treated with EPO-TAT were subjected to OGD for 60 min as described previously [14, 15], and then returned to normal culture condition and normal culture media supplied with the same concentration of EPO-TAT or EPO. Neurons without EPO-TAT or EPO treatment (OGD alone) and a TAT-tagged non-relevant fusion protein GFP-TAT served as controls. Twenty-four hours after OGD, cell death was quantitatively analyzed by Hoechst 33258 nuclear staining. The percentages of cells showing chromatin condensation or DNA damage were quantified under each experimental condition (three randomly selected fields per well, four to six wells per condition per experiment, and three independent experiments). In selected experiments, cell death was also evaluated after OGD by measuring lactate dehydrogenase (LDH) release from damaged cells into the culture medium, and expressed as percentage of LDH release compared with the OGD-alone group.
To induce NMDA neurotoxicity, EPO-TAT or EPO pretreated neurons were challenged with 200 μM of NMDA for 15 min and then returned to normal culture condition and normal culture media supplied with the same concentration of EPO-TAT or EPO. Cell death was analyzed at 24 hours after NMDA treatment using the methods described above.
To compare whether EPO-TAT and EPO has equal or similar dose-efficacy against ischemia-like injury, neurons were treated with EPO-TAT and EPO at concentrations of 0.1, 0.3, 1 and 10 U/ml, respectively, and then subjected to OGD insult using the same profile as above. Cell death was analyzed 24 hours after OGD.
Statistical analysis
Results are reported as mean ± SEM. The difference between means was assessed by the Student’s t test (single comparisons) or by ANOVA and post hoc Bonferroni/Dunn tests (for multiple comparisons), with p < 0.05 considered statistically significant.
RESULTS
1. Generation of EPO fusion protein containing TAT
In our previous study, we found that fusion of an HIV TAT sequence (YGRKKRRQRRR) to Bcl-xL facilitated its crossing of the BBB to confer neuroprotection [9]. In this study, we used the same TAT sequence and fused it to the EPO gene. We tagged the TAT sequence and his-6 tag at the C-terminal of EPO, since the N-terminal signal peptide of EPO is removed upon maturation. A mammalian expression system was used in lieu of a bacterial expression system because the function of EPO is dependent on being properly glycosylated. Moreover, to obtain EPO-TAT on a large scale, we insert a second gene, DHFR, which functions as a selection marker under the control of IRES. The resulting final plasmid pEPO-TAT-DHFR is a bicistronic construct that expresses both EPO-TAT and DHFR from single mRNA transcript (Fig. 1a). Hence, in the presence of the DHFR analogue methotrexate in DHFR-deficient Chinese hamster ovary cells DG44, we can selectively amplify and over-express the fusion protein EPO-TAT. Furthermore, we have performed clonal selections for 12 months to establish several clones that are stable high expressers of EPO-TAT, which is secreted into the culture medium (Fig. 1b). Typically, the concentrations of EPO or EPO-TAT in the culture medium can reach about 300 U/ml among the highest EPO-expressing clones, which is approximately 300-fold higher than those not selected by methotrexate. The molecular weight of the purified EPO-TAT fusion protein is approximately 36-40 kD, about 1 or 2 kD larger than EPO without TAT or commercial regular EPO (Fig. 1c), suggesting that EPO-TAT is likely to be properly glycosylated.
Figure 1. Generation of EPO fusion protein containing TAT.
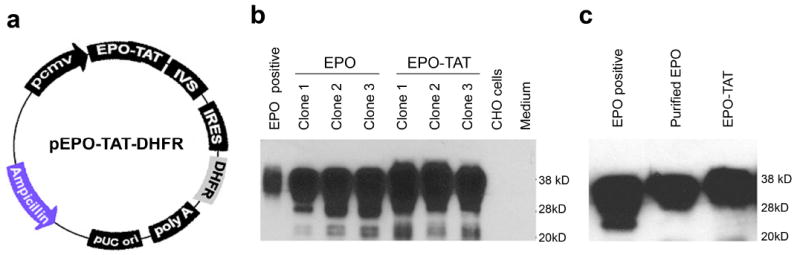
(a) A schematic map of plasmid pEPO-TAT-DHFR. TAT tag was PCR-added at the C-terminal of human EPO under the control of CMV promoter. Mouse DHFR was inserted downstream of IRES. A synthetic intron (IVS) was inserted between EPO-TAT and IRES to enhance the stability of the mRNA. (b) Representative graph of Western blot of stable cell lines expressing EPO-TAT. CHO DG44 cells (DHFR -/-) were transfected with pEPO-TAT-DHFR or p-EPO-DHFR. Stable cell lines were screened with methotrexate. EPO fusion protein in supernatant was analyzed by Western blot by loading 10 μl of medium from stable cell lines of EPO-TAT (lanes 5-7) and wild-type EPO without TAT (lanes 2-4). Commercial EPO (1 U) was used as control. (c) Western blot analysis of purified EPO-TAT and wild-type EPO recombinant protein.
2. EPO-TAT fusion protein is biologically active
To determine whether the purified EPO-TAT protein has biological activity, we tested its neuroprotective effects in primary cortical neuron cultures using in vitro models of ischemia-like insults: OGD and NMDA toxicity. Previous studies [2, 16, 17] have indicated that wild-type EPO is neuroprotective against either OGD or NMDA neurotoxicity at a dose of 1U/ml, therefore we used this dose in our experiments. In the first paradigm, OGD was induced for 60 min followed by 24 hr of normal oxygen and glucose concentrations, and cell death was assayed 24 hr later. As shown in Fig. 2a-e, OGD insult resulted in an increase in condensed nuclei in cultures (Fig. 2b). Pre-treatment with EPO (Fig. 2d) or EPO-TAT (Fig. 2e) significantly decreased the number of condensed nuclei. Quantitative analysis of Hoechst 33258 staining (Fig. 2f) demonstrated that treatment with EPO-TAT or EPO reduced cell death comparably by at least 60%. Similarly, using a non-biased LDH assay, we observed a decrease of LDH release by at least 70% upon treatments with EPO-TAT and EPO, respectively (Fig. 2g). Also, the presence of EPO-TAT or EPO significantly decreased cell death by 40% derived from NMDA toxicity (Fig. 2h). These results suggest that the inclusion of a TAT sequence did not alter the protective capacity of EPO against either OGD or NMDA toxicity.
Figure 2. EPO-TAT fusion protein is neuroprotective against OGD and NMDA toxicity.
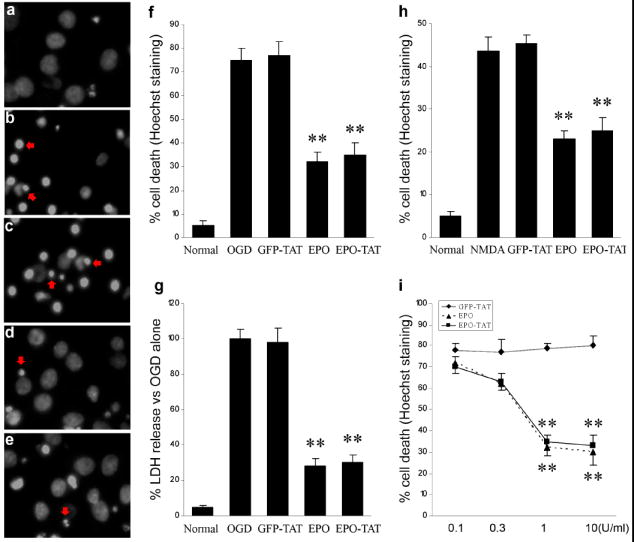
Cortical neurons at 13 DIV were pretreated for 24 hr with 1 U/ml wild-type EPO or EPO-TAT and then subjected to either OGD (a-g) or NMDA neurotoxicity (h). Representative graphs of Hoechst staining were shown (a-e) after OGD insults. Twenty-four hours of pre-treatment with either wild-type EPO (d) or EPO-TAT (e) reduced neuronal death, while the control protein GFP-TAT had no effect (c). Control neurons and OGD alone treated neuron are shown in panels a and b, respectively. Fragmented or condensed nuclei are indicated by red arrows. (f) Quantitative counting of cell death induced by OGD. (g) Relative LDH release compared with OGD alone. OGD resulted in increased LDH release, which was significantly prevented by EPO or EPO-TAT. (h) Neurons were challenged with 200 μM NMDA and cell death was quantitatively analyzed by Hoechst nucleus staining 24 hours after NMDA treatment. (i) Dose-response curve of EPO-TAT against OGD insults. Neurons were treated with a series concentration of EPO-TAT and EPO, and then subjected to OGD. Cell death was counted 24 hours after OGD. ** p<0.001 versus OGD control or GFP-TAT group or NMDA alone. Data are mean ± SEM, n=12 from three independent experiments.
To further examine whether the addition of TAT affects the dose-efficacy of EPO, neurons were treated with a series concentration of EPO-TAT, and then subjected to OGD. As shown in Fig. 2i, EPO-TAT inhibits cell death induced by OGD at a dose-dependent manner, with an optimal dose at 1 U/ml. Further increase of EPO-TAT concentration did not provide extra protection. Moreover, at the same dosage, EPO-TAT showed same or similar efficacy against OGD insult as compared with EPO in all groups. Finally EPO-TAT and EPO displayed a similar temporal profile of efficacy; there is no significant difference in the extent of protection between EPO-TAT and EPO when they were added at the same time point in their respective experiments. However, minimal protection was observed in both EPO-TAT and EPO treated groups if EPO-TAT and EPO were added after OGD or NMDA insults (data not shown). The distinction between these results and in vivo studies which reported that EPO conferred neuroprotection if administrated within 6 hours window may be due to the triggering of different EPO signaling pathways.
On the whole, these results demonstrate clearly that the inclusion of a TAT sequence did not alter the protective capacity, the dose response or the temporal profile of efficacy of EPO against either OGD or NMDA toxicity. In other words, the EPO-TAT fusion protein is an active therapeutic agent with a protective capacity comparable to that of EPO.
3. TAT fused to EPO enhances the delivery of EPO across the BBB
To examine the capacity of EPO-TAT to cross the BBB as compared to EPO after systemic administration, we injected intraperitoneally (IP) EPO-TAT and EPO into adult rats at the dose of 5000 U/kg BW. According to previous reports [18, 19], EPO injected via IP reaches peak concentration in the plasma 3-4 hours post-injection. Therefore, we collected plasma and CSF samples 3 hours post-injection for quantitative analysis of EPO via ELISA. At this time point, the concentration of EPO-TAT in the plasma was 1894 mU/ml, about 34% higher than that of EPO (Fig. 3a). However, the concentration of EPO-TAT in the CSF was 2.5 times higher than that of EPO (Fig. 3b), suggesting that EPO-TAT penetrates the BBB more efficiently than EPO.
Figure 3. TAT fused to EPO enhances the delivery of EPO across the BBB.

EPO-TAT or EPO (5000 U/kg BW) was injected either IP (a, b) or IV (c); plasma and CSF were collected 3 hr post-injection. Total EPO protein in the plasma (a) or CSF (b, c) was measured by ELISA. Data are mean ± SEM (n=4 per group). * p<0.05, ** p<0.01 versus PBS controls; ## p<0.01 versus EPO.
We also performed the same analysis under a different injection paradigm. EPO-TAT and EPO were administered intravenously (IV) and the CSF samples were collected 3 hours post-injection. In this case, the concentration of EPO in the CSF was 14.4±3.7 mU/ml whereas the corresponding concentration of EPO-TAT in the CSF was 67±22.4 mU/ml (Fig. 3c), a fivefold difference between the two in this paradigm. Between the two paradigms, the concentrations of EPO and EPO-TAT in the CSF derived from IV injection were six times higher than those derived from IP injection, suggesting that IV injection is a more effective means of therapeutic delivery of this neuroprotective agent and hence of greater clinical relevance. Indeed, this observation is consistent with another reported study [20].
Since EPO-TAT was more efficient in crossing the BBB than was EPO, a dose of EPO-TAT significantly lower than that of EPO may yield the same EPO concentration in the CSF. As expected, EPO-TAT at 1000 U/kg yielded a concentration in the CSF comparable to that of EPO at 5000U/kg (Fig. 3c). This result indeed supports our notion that the protein transduction domain TAT is able to facilitate EPO’s crossing the BBB.
4. EPO-TAT reduces infarct volume more efficiently than EPO in an MCAO model
To examine whether the enhanced capacity of EPO-TAT to cross the BBB would translate into a lower effective dose, the neuroprotective effects of EPO-TAT were determined in an MCA occlusion model in mice. MCA occlusion for 60 min produced ipsilateral cerebral infarction averaging ~32 mm3 in volume, as determined at 72 hr based on the loss of TTC staining (Fig. 4a). Intravenous administration of EPO at 1000 U/kg at the onset of post-ischemic reperfusion appeared to reduce infarct volume. However, this reduction is not statistically significant when compared with control group administrated with PBS. On the other hand, EPO-TAT injection at 1000 U/kg resulted in a significant reduction of the infarct volume averaging ~53% (p<0.05 versus PBS), with an efficacy similar to that of EPO at 5000 U/kg, but more efficient than EPO at the same dose (Fig. 4a and b, p<0.05). Further dose increase of EPO-TAT at 5000U/kg did not exhibit a significantly higher protection than either EPO-TAT at 1000U/kg or EPO at 5000U/kg, suggesting that the protective capacity of EPO-TAT reached a plateau at 1000U/kg. Interestingly, EPO-TAT at a lower dose 400U/kg is still capable of significant protection, reducing the infarct volume by 38%. In all three groups (PBS, EPO and EPO-TAT), physiological parameters (blood pressure, blood gases, and blood glucose) were measured with no significant changes among groups. In addition, cortical blood flow as determined using laser Doppler flowmetry showed no significant differences between groups during and after MCAO (data not shown). These results, in concert, support the notion that increasing the capacity of a therapeutic agent to cross the BBB will enable the use of a lower dose to achieve the same degree of protection.
Figure 4. EPO-TAT protects against focal ischemic infarction.
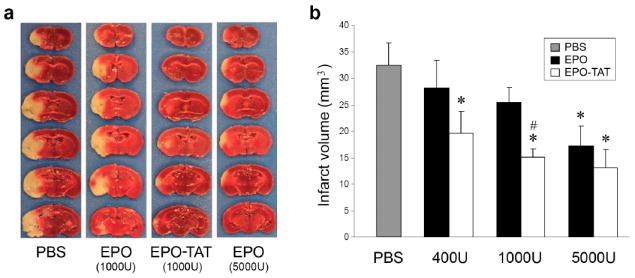
Mice were subjected to 60 min of MCAO followed by 72 hr of reperfusion. EPO-TAT or EPO was injected intravenously at the onset of reperfusion. TTC staining was assessed 72 hr after MCAO. (a) Representative photographs of TTC-stained coronal sections of mouse brains subjected to MCAO followed by administration of PBS, EPO-TAT or EPO. (b) Quantitative measurement of infarct volume. Data are presented as mean ± SEM, n=9 per group. * p<0.05 versus PBS; # p<0.05 versus EPO at 1000 U/kg.
DISCUSSION
In the evaluation of therapeutic agents, many of the candidates with high promise in cell culture models are deemed unsuitable for use in animals because of their inability to cross the BBB. Even for those with limited capacity to cross the BBB, the therapeutic doses must be set to high enough levels to ensure sufficient amounts of them will reach the injury site. Typically, treatments using such high doses inevitably give rise to side effects. For example, EPO has a limited capacity to cross the BBB even under compromised vascular integrity following ischemia [20, 21], and therefore large doses or multiple injections of EPO have been required for the treatment of stroke. Such an administration regime may cause a series of severe blood-stimulating side effects, hence limiting EPO’s clinical application. To resolve the EPO delivery problem, in this study, we have generated a brain-penetrating EPO by fusing EPO with protein transduction domain derived from HIV TAT. Our study indicates that the additon of TAT does not affect the biological activity of EPO, but significantly increases its delivery across the BBB. The increased delivery is translated into a lower effective therapeutic dose against ischemic brain injury. HIV TAT-tagged EPO therefore may be a promising approach for EPO delivery into the CNS.
One of the approaches to effectively deliver therapeutic proteins into the brain is to fuse the protein of interest with protein transduction domain. So far, the HIV TAT is the most widely used protein transduction domain for protein delivery. Our own studies [9, 11] and others [10, 22, 23] have consistently demonstrated the effectiveness of HIV TAT in delivering a variety of proteins across the BBB into the brain and offering neuroprotection against ischemic brain injury as well as other neurological diseases. In this study, we found that HIV TAT can significantly increase EPO delivery across the BBB, and the therapeutic dose of EPO thereby is reduced tenfold. Interestingly, EPO has a higher innate capacity to cross the BBB in humans than in rodents. EPO injection at 1500 U/kg under physiological condition [24] or 550 U/kg under ischemic condition [3] can reach similar therapeutic concentrations in the human CSF (~17 mU/ml according to [3]) as compared with 5000 U/kg in rodents. Assuming that EPO-TAT has similar efficacy in crossing the human BBB and reduce the therapeutic dose at least ten-fold (5~10 fold according to figure 4b) in rodent MCAO models, it is highly probable that EPO-TAT can be used as a therapetuic agent for stroke at a clinically safe range. Furthermore, other delivery approaches such as poly-arginine, nanoparticle and receptor-mediated transcytosis have been used for protein delivery into the CNS. Comparison of the delivery capacity of HIV TAT with these approaches will warrant further research to determine the optimal system for EPO delivery into the CNS.
Thus far, another approach to overcome caveats related to EPO therapy in the CNS lies in the development of EPO derivatives such as asialo-erythropoietin [25], carbamylated EPO [17] and EPO mimetic peptide [26] to lessen the side effects. Indeed, these derivatives showed neuroprotective effects against ischemic brain injury and other CNS diseases with none of the aforementioned side effects, even when administered at high doses. Nevertheless, large doses and multiple EPO administrations could have other side effects unrelated to EPO’s blood-stimulating side effect. For instance, a recent clinical trial indicated that EPO treatment significantly increased the death of stroke patients due to increased incidence of intracranial hemorrhage [27]. Thus, reducing the therapeutic dose of wild-type EPO as well as EPO derivatives by increasing their brain-penetrating ability might greatly improve EPO’s clinical application in the CNS.
In our experiments, the EPO concentration in the CSF was ~14.4 mU/ml 3 hours after IV injection of EPO at 5000 U/kg. This concentration was compatible to the previously reported rat study by Leist et al. [17, 28], but substantially lower than the concentration of ~200 mU/ml reported in another study [17, 28]. The reason for this discrepancy is unknown; however, different CSF sampling procedures could account for the fluctuation in EPO concentrations found in different studies. We noticed that CSF sampling in small animals could easily be affected by blood contamination. Since EPO concentration in plasma is at least 100 times higher than that in the CSF, a minute cross-contamination of plasma in the CSF could result in substantial elevation of the EPO value. In our study, we monitored blood contamination by counting red blood cells (RBC) in all CSF samples using a hemacytometer under microscopy. Samples with RBC over 1.7×104 RBC/μl CSF (equivalent to 0.25% blood contamination) were not subjected to further EPO concentration determinations.
Finally, our study essentially focused on the delivery efficacy crossing the BBB and the impact of EPO fused with a TAT on infarction size. Whether EPO-TAT at a lower therapeutic dose has the same effect on long-term functional recovery warrants further investigation. Furthermore, since TAT increases the delivery efficiency of EPO across the BBB, EPO-TAT may reach effective therapeutic concentrations in the brain more rapidly than wild-type EPO, and therefore it could be very interesting to examine whether rapid delivery of EPO-TAT can be translated into a wider therapeutic window. Our results, in concert, suggest that PTD can mitigate the dose-dependent side effects and also open an avenue for evaluating the efficacy of reagents that are otherwise unable to transverse the BBB without PTD in rodent ischemic models.
Acknowledgments
The project was supported by National Institutes of Health Grants NS053473 (to G. C.), NS43802, NS45048, NS36736, and NS 56118 (to J.C.) and the American Heart Association Scientist Development Grant 06300064N (to G.C.). Y.L. is a recipient of the Chinese Natural Science Foundation Grant 30670725.
References
- 1.Brines ML, Ghezzi P, Keenan S, Agnello D, de Lanerolle NC, Cerami C, et al. Erythropoietin crosses the blood-brain barrier to protect against experimental brain injury. Proc Natl Acad Sci U S A. 2000 Sep 12;97(19):10526–31. doi: 10.1073/pnas.97.19.10526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sakanaka M, Wen TC, Matsuda S, Masuda S, Morishita E, Nagao M, et al. In vivo evidence that erythropoietin protects neurons from ischemic damage. Proc Natl Acad Sci U S A. 1998 Apr 14;95(8):4635–40. doi: 10.1073/pnas.95.8.4635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ehrenreich H, Hasselblatt M, Dembowski C, Cepek L, Lewczuk P, Stiefel M, et al. Erythropoietin therapy for acute stroke is both safe and beneficial. Mol Med. 2002 Aug;8(8):495–505. [PMC free article] [PubMed] [Google Scholar]
- 4.Brines M, Cerami A. Emerging biological roles for erythropoietin in the nervous system. Nat Rev Neurosci. 2005 Jun;6(6):484–94. doi: 10.1038/nrn1687. [DOI] [PubMed] [Google Scholar]
- 5.Wiessner C, Allegrini PR, Ekatodramis D, Jewell UR, Stallmach T, Gassmann M. Increased cerebral infarct volumes in polyglobulic mice overexpressing erythropoietin. J Cereb Blood Flow Metab. 2001 Jul;21(7):857–64. doi: 10.1097/00004647-200107000-00011. [DOI] [PubMed] [Google Scholar]
- 6.Leyland-Jones B. Breast cancer trial with erythropoietin terminated unexpectedly. Lancet Oncol. 2003 Aug;4(8):459–60. doi: 10.1016/s1470-2045(03)01163-x. [DOI] [PubMed] [Google Scholar]
- 7.Wolf RF, Peng J, Friese P, Gilmore LS, Burstein SA, Dale GL. Erythropoietin administration increases production and reactivity of platelets in dogs. Thromb Haemost. 1997 Dec;78(6):1505–9. [PubMed] [Google Scholar]
- 8.Wun T, Law L, Harvey D, Sieracki B, Scudder SA, Ryu JK. Increased incidence of symptomatic venous thrombosis in patients with cervical carcinoma treated with concurrent chemotherapy, radiation, and erythropoietin. Cancer. 2003 Oct 1;98(7):1514–20. doi: 10.1002/cncr.11700. [DOI] [PubMed] [Google Scholar]
- 9.Cao G, Pei W, Ge H, Liang Q, Luo Y, Sharp FR, et al. In Vivo Delivery of a Bcl-xL Fusion Protein Containing the TAT Protein Transduction Domain Protects against Ischemic Brain Injury and Neuronal Apoptosis. J Neurosci. 2002 Jul 1;22(13):5423–31. doi: 10.1523/JNEUROSCI.22-13-05423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kilic U, Kilic E, Dietz GP, Bahr M. Intravenous TAT-GDNF is protective after focal cerebral ischemia in mice. Stroke. 2003 May;34(5):1304–10. doi: 10.1161/01.STR.0000066869.45310.50. [DOI] [PubMed] [Google Scholar]
- 11.Yin W, Cao G, Johnnides MJ, Signore AP, Luo Y, Hickey RW, et al. TAT-mediated delivery of Bcl-xL protein is neuroprotective against neonatal hypoxic-ischemic brain injury via inhibition of caspases and AIF. Neurobiol Dis. 2006 Feb;21(2):358–71. doi: 10.1016/j.nbd.2005.07.015. [DOI] [PubMed] [Google Scholar]
- 12.Chen J, Nagayama T, Jin K, Stetler RA, Zhu RL, Graham SH, et al. Induction of caspase-3-like protease may mediate delayed neuronal death in the hippocampus after transient cerebral ischemia. J Neurosci. 1998 Jul 1;18(13):4914–28. doi: 10.1523/JNEUROSCI.18-13-04914.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frankmann SP. A technique for repeated sampling of CSF from the anesthetized rat. Physiol Behav. 1986;37(3):489–93. doi: 10.1016/0031-9384(86)90211-8. [DOI] [PubMed] [Google Scholar]
- 14.Cao G, Pei W, Lan J, Stetler RA, Luo Y, Nagayama T, et al. Caspase-activated DNase/DNA fragmentation factor 40 mediates apoptotic DNA fragmentation in transient cerebral ischemia and in neuronal cultures. J Neurosci. 2001 Jul 1;21(13):4678–90. doi: 10.1523/JNEUROSCI.21-13-04678.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cao G, Clark RS, Pei W, Yin W, Zhang F, Sun FY, et al. Translocation of apoptosis-inducing factor in vulnerable neurons after transient cerebral ischemia and in neuronal cultures after oxygen-glucose deprivation. J Cereb Blood Flow Metab. 2003 Oct;23(10):1137–50. doi: 10.1097/01.WCB.0000087090.01171.E7. [DOI] [PubMed] [Google Scholar]
- 16.Chong ZZ, Maiese K. Erythropoietin involves the phosphatidylinositol 3-kinase pathway, 14-3-3 protein and FOXO3a nuclear trafficking to preserve endothelial cell integrity. Br J Pharmacol. 2007 Apr;150(7):839–50. doi: 10.1038/sj.bjp.0707161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leist M, Ghezzi P, Grasso G, Bianchi R, Villa P, Fratelli M, et al. Derivatives of erythropoietin that are tissue protective but not erythropoietic. Science. 2004 Jul 9;305(5681):239–42. doi: 10.1126/science.1098313. [DOI] [PubMed] [Google Scholar]
- 18.Ranchon Cole I, Bonhomme B, Doly M. Pre-treatment of adult rats with high doses of erythropoietin induces caspase-9 but prevents light-induced retinal injury. Exp Eye Res. 2007 Dec;85(6):782–9. doi: 10.1016/j.exer.2007.08.013. [DOI] [PubMed] [Google Scholar]
- 19.Statler PA, McPherson RJ, Bauer LA, Kellert BA, Juul SE. Pharmacokinetics of high-dose recombinant erythropoietin in plasma and brain of neonatal rats. Pediatr Res. 2007 Jun;61(6):671–5. doi: 10.1203/pdr.0b013e31805341dc. [DOI] [PubMed] [Google Scholar]
- 20.Juul SE, McPherson RJ, Farrell FX, Jolliffe L, Ness DJ, Gleason CA. Erytropoietin concentrations in cerebrospinal fluid of nonhuman primates and fetal sheep following high-dose recombinant erythropoietin. Biol Neonate. 2004;85(2):138–44. doi: 10.1159/000074970. [DOI] [PubMed] [Google Scholar]
- 21.Grasso G, Buemi M, Alafaci C, Sfacteria A, Passalacqua M, Sturiale A, et al. Beneficial effects of systemic administration of recombinant human erythropoietin in rabbits subjected to subarachnoid hemorrhage. Proc Natl Acad Sci U S A. 2002 Apr 16;99(8):5627–31. doi: 10.1073/pnas.082097299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doeppner TR, Nagel F, Dietz GP, Weise J, Tonges L, Schwarting S, et al. TAT-Hsp70-mediated neuroprotection and increased survival of neuronal precursor cells after focal cerebral ischemia in mice. J Cereb Blood Flow Metab. 2009 Jun;29(6):1187–96. doi: 10.1038/jcbfm.2009.44. [DOI] [PubMed] [Google Scholar]
- 23.Zhou M, Xu W, Liao G, Bi X, Baudry M. Neuroprotection against neonatal hypoxia/ischemia-induced cerebral cell death by prevention of calpain-mediated mGluR1alpha truncation. Exp Neurol. 2009 Jul;218(1):75–82. doi: 10.1016/j.expneurol.2009.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xenocostas A, Cheung WK, Farrell F, Zakszewski C, Kelley M, Lutynski A, et al. The pharmacokinetics of erythropoietin in the cerebrospinal fluid after intravenous administration of recombinant human erythropoietin. Eur J Clin Pharmacol. 2005 May;61(3):189–95. doi: 10.1007/s00228-005-0896-7. [DOI] [PubMed] [Google Scholar]
- 25.Erbayraktar S, Grasso G, Sfacteria A, Xie QW, Coleman T, Kreilgaard M, et al. Asialoerythropoietin is a nonerythropoietic cytokine with broad neuroprotective activity in vivo. Proc Natl Acad Sci U S A. 2003 May 27;100(11):6741–6. doi: 10.1073/pnas.1031753100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brines M, Patel NS, Villa P, Brines C, Mennini T, De Paola M, et al. Nonerythropoietic, tissue-protective peptides derived from the tertiary structure of erythropoietin. Proc Natl Acad Sci U S A. 2008 Aug 5;105(31):10925–30. doi: 10.1073/pnas.0805594105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Early Communication about an Ongoing Safety Review Epoetin Alpha. US Food and Drug Administration. 2008 [Google Scholar]
- 28.Lieutaud T, Andrews PJ, Rhodes JK, Williamson R. Characterization of the pharmacokinetics of human recombinant erythropoietin in blood and brain when administered immediately after lateral fluid percussion brain injury and its pharmacodynamic effects on IL-1beta and MIP-2 in rats. J Neurotrauma. 2008 Oct;25(10):1179–85. doi: 10.1089/neu.2008.0591. [DOI] [PubMed] [Google Scholar]