

Abstract
Drug-induced liver injury (DILI) causes significant patient morbidity and mortality, and is the most common reason for drug withdrawals. It is imperative to gain a thorough understanding of the underlying mechanisms of DILI to effectively predict and prevent these reactions. We have recently developed a murine model of halothane-induced liver injury (HILI). The aim of the present study was to investigate the role of hepatic natural killer T (NKT) cells in the pathogenesis of HILI. The degrees of HILI were compared between WT and CD1d−/−mice, which are deficient in NKT cells. The data revealed that CD1d−/−mice were resistant in developing HILI. This resistance appeared to be a direct result of NKT cell-depletion rather than an indirect one due to the absence of cross-talk between NKT cells and other hepatic innate immune cells. Compared with WT mice, CD1d−/− mice exhibited a significantly lower number of hepatic infiltrating neutrophils upon halothane challenge (470,000±100,000/liver in WT vs. 120,000±31,500/liver in CD1d−/− mice). This result in conjunction with our previous finding of an indispensable role of neutrophils in HILI strongly suggest that NKT cells play a critical role in regulating neutrophil recruitment, thereby contributing to the development of HILI. Collectively, the current study and published reports indicate that this murine model of HILI provides an experimental system for the investigation of the underlying mechanisms of DILI. In addition, this model may yield the discovery of susceptibility factors that may control the development of liver injury in patients treated with halothane and potentially other drugs.
Keywords: Drug, Hepatotoxicity, Neutrophils
1. Introduction
The liver is a primary target of adverse drug reactions because it is the predominant site for the biotransformation of drugs. The severity of drug-induced liver injury (DILI) can range from mild and transient increases in serum transaminase levels with no clinical consequences, to acute and chronic hepatitis, cholestatic disease, cirrhosis, fulminant hepatic failure (requiring liver transplantation) and even death. Compared with other types of liver disease, DILI has a greater risk of developing into fulminate hepatic failure. It is estimated that DILI accounts for over 50% of liver failure cases in the US.[1] DILI is also the most common reason for the withdrawal of FDA-approved drugs from the market.[2] In recent years, several drugs, most notably troglitazone and bromfenac have been withdrawn from the market during post-marketing surveillance due to severe hepatotoxicity. As such, it is imperative to gain a thorough understanding of the underlying mechanisms of DILI before strategies for developing diagnostic tests and designing enhanced clinical trials can be achieved. The enhanced usage and implementation of animal studies will provide insights into the complex molecular and cellular mechanisms of DILI.
We have previously reported the development of a murine model of liver injury caused by an inhalation anesthetic, halothane.[3] This model shares similar characteristics to those found in 20% of patients who develop acute halothane hepatotoxicity, which progresses into life-threatening halothane hepatitis in a small percentage of such patients. Based on our model, it was revealed that although the generation of a chemically reactive metabolite via halothane metabolism is necessary to initiate hepatocyte damage, various innate immune cells appear to play critical roles in the progression of tissue damage. The data showed that depletion of neutrophils markedly attenuated halothane-induced liver injury (HILI) in mice, indicating a pathological role for neutrophils. Natural killer T (NKT) cells, which express surface markers of both NK and T cells, represent another important type of hepatic innate immune cells. In mice, as a proportion of mature T cells, NKT cells are most frequent in the liver (30–50%) and bone marrow (20–30%), while they represent a smaller proportion of T cells in the spleen (3%) and lymph nodes (0.3%).[4] Numerous reports have described the important roles of murine hepatic NKT cells in inhibiting hepatitis B viral replication,[5] as well as suppressing cancer liver metastasis and the development of hepatocellular carcinoma.[6]
In contrast to their protective roles, NKT cells have also been demonstrated to exhibit pathological effects in animal models of liver injury. CD1d−/− mice, which lack NKT cells, were shown to be highly resistant to concanavalin A-induced hepatitis, a mouse model of human autoimmune and viral hepatitis.[7] Mechanistic studies have further revealed the link between the indispensable pathological role of NKT cells and their production of interleukin (IL)-4, interferon (IFN)-γ, tumor necrosis factor (TNF)-α, and FasL, all of which are critical in the development of concanavalin A-induced hepatitis.[7–10] In another murine model of hepatitis caused by the injection of the exogenous ligand of NKT cell receptor, alpha-galactosylceramide (α-GalCer), the production of TNF-α by NKT cells was found to be the key mediator of liver injury.[11] Furthermore, the prompt production of IFN-γ by NKT cells has been suggested to play a central role in endotoxin-induced liver injury.[12,13]
In spite of two studies investigating the role of NKT cells in a mouse model of acetaminophen overdose-induced liver injury,[14,15] the role of NKT cells in DILI remains largely unknown. The former of these studies demonstrated that depletion of both NK and NKT cells yielded a significant decrease in acetaminophen-induced liver injury and improved survival.[14] The data suggested that the secretion of IFN-γ by NK/NKT cells may induce inflammatory chemokines, enhance leukocyte recruitment and increase FasL expression, thereby contributing to liver injury. However, a more recent report revealed that the pathological role of NK/NKT cells in acetaminophen-induced liver injury was due to the presence of dimethyl sulfoxide used to aid in the dissolution of acetaminophen.[15] The latter study demonstrated that the low amounts of dimethyl sulfoxide caused a significant increase in both the number and activation of hepatic NK/NKT cells. These findings suggest that NKT cells, activated by either drug-dependent or independent stimuli, may play a critical role in the underlying mechanisms of DILI. Provided that NKT cells are readily activated by self-lipid antigens, bacterial and viral antigens, cytokines, as well as “danger signals” released from damaged cells, these factors may represent key risk factors that modulate an individual’s susceptibility to DILI.
The aim of the present study was to investigate the role of NKT cells in HILI in mice. Susceptibility to HILI was compared between NKT cell-depleted CD1d−/− mice and their wild-type (WT) counterparts. Our data demonstrated that CD1d−/− mice developed minimal liver injury upon halothane challenge and exhibited a much lower number of infiltrating neutrophils in the liver compared with WT mice. These findings suggest that NKT cells are activated upon halothane exposure and that they subsequently play a critical role in the development and progression of HILI.
2. MATERIALS AND METHODS
2.1 Materials
All chemicals and reagents were obtained from Sigma (St. Louis, MO), unless stated otherwise. All antibodies used for flow cytometric analysis were purchased from eBioscience (San Diego, CA), except for phycoerythrin (PE)-conjugated α-GalCer-loaded mouse CD1d-tetramers (CD1d-tetramer, National Institutes of Health Tetramer Core Facility). Rabbit polyclonal anti-trifloroacetylchloride (TFA) antisera (anti-TFA antisera) was a kind gift from Dr. Lance Pohl (National Institutes of Health, Bethesda, MD).
2.2 Animal treatment
CD1d−/− mice (on Balb/cJ background) and WT Balb/cJ mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred in the Center for Laboratory Animal Care (CLAC) at the University of Colorado Denver (UC Denver). All animal experiments were performed in accordance with guidelines from the UC Denver Institutional Animal Care and Use Committee. For all experiments involving halothane treatment, female WT and CD1d−/− mice were injected intraperitoneally (i.p.) with 30 mmole/kg of halothane (Halocarbon Labs Inc., Hackensack, NJ), dissolved in 2 mL of olive oil. Control mice were treated with nothing (naïve) or vehicle (olive oil).
To deplete hepatic Kupffer cells (KCs), liposome-entrapped clodronate was prepared as previously described[16] and injected intravenously (i.v.) to mice 2 days prior to halothane treatment. Control mice were treated with empty liposomes. To deplete NK cells, mice were injected i.v. with anti-AsGM1 polyclonal antibody (100 μL; Wako Chemical USA, Richmond, VA) 2 days prior to halothane treatment. Control mice were injected with the same volume of normal rabbit serum.
2.3 Assessment of hepatotoxicity
At 15 h and 24 h after halothane treatment, mice were anesthetized and blood was collected by retro-orbital puncture. Serum alanine transaminase (ALT) levels were measured using a diagnostic assay kit (Teco Diagnostics, Anaheim, CA) following the manufacturer’s instruction. At 24 h after halothane administration, animals were sacrificed and the livers removed. Liver sections were fixed in 10% formaldehyde overnight before being transferred to a 70% ethanol solution. Paraffin-embedded liver sections were mounted onto glass slides and stained with hematoxylin and eosin (H/E; Department of Pathology, UC Denver).
2.4 Immunoblot analysis of TFA-protein adducts and CYP450 2E1 expression in the liver
WT and CD1d−/− mice were sacrificed 8 h after halothane treatment. Liver homogenate samples (30 μg/lane) were resolved on 12% polyacrylamide gels and transferred onto nitrocellulose membranes (Bio-Rad Laboratories Inc., Hercules, CA). Membranes were blocked with 5% (w/v) fat-free milk and probed with anti-TFA antisera (1:1000), or anti-cytochrome (CYP) 450 2E1 antibody (1:5000; Assay Designs Inc., Ann Arbor, MI) overnight at 4 °C. Membranes were then incubated with horseradish peroxidase-conjugated goat anti-rabbit IgG antibody (1:2000; Chemicon International Inc., Temcula, CA) for 1 h at room temperature. Protein signals were visualized using an ECL Plus Western Blotting Detection System (GE Healthcare Bio-Science Corp., Little Chalfont, UK), and the data were captured using a Storm 860 system (GE Healthcare Bio-Science Corp.).
2.5 Isolation of hepatic leukocytes and hepatocytes
Hepatic leukocytes were isolated following a previously described method with slight modification.[17] Mice were anesthetized and the liver perfused in situ with pre-warmed Hank’s balanced salt solution (HBSS) at 37°C for 5 min. Single cell suspensions were filtered through a 100 μm cell strainer (BD Falcon, Bedford, MA) and centrifuged at 300 × g for 5 min. The pellet was resuspended in 15 mL of 35% Percoll containing 50 U/mL of heparin (Baxter Healthcare Corporation, Deerfield, IL) and centrifuged at 500 × g for 15 min. The resulting pellet was collected and resuspended in 1.5 mL of red blood cell lysing buffer for 5 min. The cells were then washed in HBSS solution containing 0.6% acid citrate-dextrose (Acd-A) and 0.5% bovine serum albumin (BSA).
Hepatocytes were isolated following a previously established method[18] with slight modifications. Briefly, mice were anesthetized with isoflurane and the livers perfused in situ with a Ca2+-free HBSS buffer for 5 min followed by a 0.05% collagenase solution for 25 min. After digestion, the liver was disrupted in Williams E media by gently shaking in a 50 mL Falcon tube. The single cell suspension was filtered through a 100 μm cell strainer and then centrifuged at 30 × g for 3 min to pellet hepatocytes. A density gradient centrifugation using 40% Percoll was subsequently employed to remove non-viable hepatocytes.
2.6 Flow cytometric analysis
Freshly isolated hepatic leukocytes were incubated with anti-FcγR II/III antibody (10 μg/mL, clone 93) plus rat serum (1:10 dilution) for 10 min on ice in order to prevent nonspecific binding. The cells were stained with various antibodies, including fluorescein isothiocyanate (FITC)-conjugated anti-Gr1 (clone RB6-8C5), PE-conjugated anti-CD11b (clone M1/70) in order to identify neutrophils (CD11b+Gr-1+). The NKT cell population (CD1d-Tetramer+CD3+) was recognized by staining the cells with PE-conjugated CD1d-tetramers and allophycocyanin (APC)-conjugated anti-CD3 (clone 145-2C11). In some experiments, cells were stained with PE-conjugated anti-CD1d antibody (clone 1B1) to examine the expression of CD1d on hepatocytes. After antibody staining, the cells were analyzed on a FACS Calibur using CellQuest software (BD Biosciences, San Jose, CA). The data were further analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
2.7 Statistical analysis
Data are presented as mean ± SEM. Two-tailed Student’s t-test was used to compare two groups. Comparison among multiple groups was performed using one-way analysis of variance (ANOVA) with a post-hoc test of significance between individual groups. Differences were considered significant when p < 0.05.
3. RESULTS
3.1 NKT cell-deficient CD1d−/− mice are resistant to HILI
NKT cell depletion in vivo has often been achieved by administering either an anti-NK1.1 antibody or α-GalCer to mice. However, the anti-NK1.1 antibody depletes not only NKT cells, but NK cells as well. α-GalCer-induced NKT cell depletion is preceded by activation of these cells, which may cause the further activation of additional cells, including NK cells. Together, these activated cells are capable of producing pro-inflammatory cytokines, such as IFN-γ, that can inhibit CYP450 activity.[19] The suppression of CYP450 activities inhibits halothane metabolism to TFA, thereby preventing the onset of HILI.[17] Due to these concerns associated with NKT cell depletion in vivo, we chose to use CD1d−/− mice, which lack NKT cells due to ablation of the CD1d gene that is critical for NKT cell development.
CD1d−/− mice on a Balb/cJ, rather than C57BL/6J, background were employed in the present study, as we have previously established the susceptibility of Balb/cJ WT mice to HILI.[3] Female mice were used as they were found to be more susceptible to HILI than their male counterparts.[3] Compared with WT mice, our data demonstrated that CD1d−/− mice were much less susceptible to HILI, evident from the nearly 20-fold lower ALT levels observed in CD1d−/− mice (Fig. 1A, 4797 ± 513 IU/L in WT mice vs 202 ± 114 IU/L in CD1d−/− mice 24 h post-halothane challenge). In line with the minimal ALT levels, histological evaluation of liver sections obtained at 24 h after halothane treatment revealed markedly reduced hepatocyte necrosis in CD1d−/− mice compared with WT mice (Fig. 1B).
Figure 1.

CD1d−/− mice were resistant to HILI. Female WT and CD1d−/− mice were treated with halothane. (A) Serum ALT levels were determined at 15 h and 24 h after halothane treatment. Results shown represent mean ± SEM of 10 mice per group, and individual samples were assayed in triplicate. (*, p <0.05 compared with WT mice). (B) Livers were excised at 24 h after halothane treatment and photomicrographs (200×, final magnification) of paraffin-embedded liver sections stained with H/E are shown (outlines indicate injured areas).
The metabolism of halothane to its reactive metabolite, TFA, and the subsequent formation of TFA-protein adducts are thought to be a prerequisite for the development of tissue damage. To investigate whether the resistance exhibited by CD1d−/− mice to HILI was due to inhibition of halothane metabolism, the levels of TFA-protein adducts were examined by immunoblot analysis using an anti-TFA antibody (kindly provided by Dr. Lance Pohl, NIH). Although a slight increase was evident, there was no dramatic decrease in the amount of TFA-protein adducts within the liver of CD1d−/− mice compared with those in WT mice at 8 h after halothane challenge (Fig. 2A). In addition, the levels of CYP450 2E1 expression were also similar between CD1d−/− and WT mice (Fig. 2B). These data clearly indicate that the resistance of CD1d−/− mice to HILI is not due to a compromise in either halothane metabolism or TFA generation.
Figure 2.

The lack of NKT cells in CD1d−/− mice did not inhibit halothane metabolism. Female WT and CD1d−/− mice (3 mice/group in panel A and 4 mice/group in panel B) were treated with halothane, and after 8 h, liver homogenates were prepared and used (30 μg/lane) for immunoblot detection of (A) TFA-protein adducts with anti-TFA polyclonal antibodies (1:1000 dilution) and (B) CYPP450 2E1 with anti-2E1 polyclonal antibodies (1:5000 dilution). Molecular mass markers (kDa) are indicated on the right. β-actin served as a loading control (indicated by arrow).
3.2 KCs and NK cells do not play a role in HILI
The resistance of CD1d−/− mice against the development of HILI may be either a direct result of the lack of NKT cells or an indirect effect due to the absence of cross-talk between NKT cells and other hepatic innate immune cells, such as KCs and NK cells. To determine whether KCs contribute to the pathogenesis of HILI, these cells were depleted prior to halothane treatment by using liposome-entrapped clodronate. The data demonstrated that KC depletion did not affect degree of liver injury caused by halothane (Fig. 3A, 5403 ± 1029 IU/L in KC-intact mice vs 4385 ± 783 IU/L in KC-depleted mice). Activated NKT cells often induce NK cell activation, resulting in the production of pro-inflammatory cytokines, including IFN-γ. Therefore, it is highly likely that rather than their direct involvement, NKT cells indirectly contribute to HILI via activation of NK cells. In this case, depletion of NK cells should result in the inhibition of HILI. However, our data revealed that the administration of an anti-AsGM-1 antibody to specifically deplete NK cells did not affect HILI (Fig. 3B, 5601 ± 754 IU/L in NK-intact mice vs 4675 ± 756 IU/L in NK-depleted mice). These findings indicate the dispensable role of KCs and NK cells in the development of HILI, and further demonstrate that these cells are not essential in either activating NKT cells or mediating the pathological effects of NKT cells.
Figure 3.

KC and NK cell depletion did not affect hepatic HILI. (A) KCs were depleted in female WT mice by i.v. injection with liposome/clodronate (Lipo-Cld). Control mice were treated with empty liposomes (Lipo-PBS). (B) NK cells were depleted in female WT mice by i.v. injection of anti-AsGM-1 antibody. Control mice were treated with normal rabbit serum. Two days after liposome/clodronate or anti-AsGM-1 antibody treatment, mice were treated with halothane, and serum ALT levels were determined at 15 h after halothane treatment. Results shown represent mean ± SEM of 10 mice per group, and individual samples were assayed in triplicate.
3.3 Halothane treatment induces NKT cell activation
The fact that CD1d−/− mice are resistant to HILI strongly suggests that NKT cells are activated upon halothane treatment, and subsequently contribute to the development of HILI. To examine this hypothesis, hepatic leukocytes were isolated from WT mice at 4 h after halothane challenge. The cells were then stained with anti-CD3 antibody and CD1d-tetramer to identify NKT cells by flow cytometry. As shown in Fig 4, the percentage of NKT cells (CD3+CD1d-tetramer+) among total hepatic leukocytes increased following halothane treatment, indicating the activation of such cells.
Figure 4.
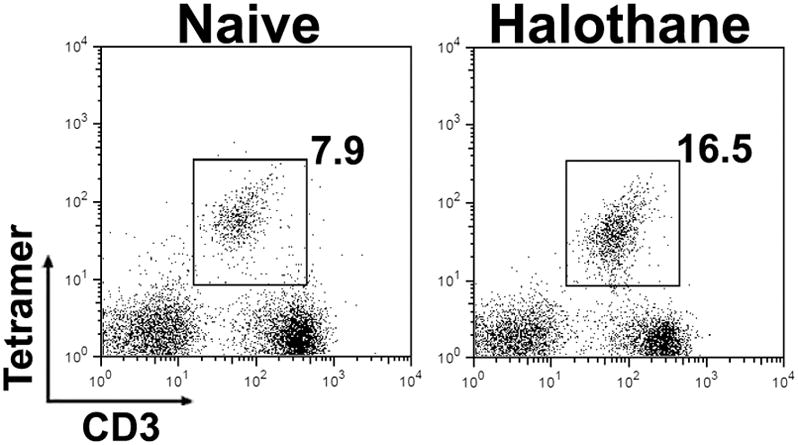
Halothane treatment of WT mice led to an increase in the number of hepatic NKT cells. Female WT mice were treated with nothing (naive) or halothane, and hepatic leukocytes were isolated 4 h later. The cells were stained with PE-conjugated CD1d-tetramer and APC-conjugated anit-CD3, and analyzed by flow cytometry. The percentage of NKT cells (CD1d-tetramer+ CD3+) is presented in the upper right quadrant of each graph. Data shown are representative of 4 mice per group.
NKT cell activation occurs via their T cell receptor recognition of glycolipids presented in complex with the MHC class I-like molecule, CD1d.[20] It is possible that halothane challenge may increase the amount of, or induce the generation of new glycolipids, thereby stimulating the activation of NKT cells. In addition, to confirm the ability of hepatocytes to present endogenous lipid antigens to NKT cells, the expression of CD1d on the surface of hepatocytes was examined by flow cytometry. The data revealed that the majority of hepatocytes isolated from both naïve and halothane-treated mice expressed CD1d on their surface (Fig. 5).
Figure 5.

Cell surface expression of CD1d by hepatocytes. Female WT mice were treated with nothing (naïve) or halothane, and hepatocytes were isolated at 4 h later. The cells were stained with PE-conjugated anti-CD1d antibody and analyzed by flow cytometry. Data are displayed as histograms depicting fluorescence intensity (x-axis) versus cell number (y-axis). Solid black lines represent isotype control antibodies and shaded histograms represent specific CD1d staining. The percentage of hepatocytes that express CD1d is presented in each graph. Data shown are representative of 3 mice per group.
3.4 Hepatic neutrophil infiltration is inhibited in CD1d−/− mice
We have reported that aside from the formation of TFA and TFA-protein adducts, activation of the innate immune system contributes to the overall extent of HILI.[3] Our previous data have demonstrated that halothane treatment induced neutrophil recruitment into the liver and that HILI was significantly attenuated upon depletion of neutrophils.[3] These data indicate a crucial role for neutrophils in the progression of HILI. To investigate whether neutrophil recruitment into the liver upon halothane challenge was suppressed in the absence of NKT cells, hepatic leukocytes were isolated from individual WT and CD1d−/− mice. The cells were stained with antibodies against Gr-1 and CD11b and analyzed by flow cytometry. The data revealed a significant reduction in the number of neutrophils that infiltrated into the liver of CD1d−/− mice, compared with WT mice, at 6 h (data not shown) and 12 h (Fig. 6A and B) following halothane treatment (470,000±100,000 neutrophils/liver in WT mice vs. 120,000±31,500 neutrophils/liver in CD1d−/− mice). This finding suggests a critical role for NKT cells in the pathogenesis of HILI through promoting neutrophil infiltration into the liver. In contrast, depletion of KCs (Fig. 6C) or NK cells (data not shown) did not affect hepatic neutrophil infiltration.
Figure 6.

Halothane-induced hepatic recruitment of neutrophils was not affected by KC depletion, but was inhibited in CD1d−/− mice compared with WT mice. (A and B) Female WT and CD1d−/− mice were treated with halothane. Hepatic leukocytes were isolated 12 h later from individual mice and stained with FITC-conjugated anti-Gr-1 and PE-conjugated anti-CD11b antibodies and analyzed by flow cytometry. (A) Data shown are representative dot plots of neutrophil infiltration into the liver of halothane-treated WT and CD1d−/− mice. The percentage of neutrophils (Gr-1+CD11b+) is presented in the upper right quadrant of each graph. (B) The absolute number of infiltrated neutrophils was calculated as the product of the total number of hepatic leukocytes isolated and the percentage of neutrophils. The absolute number of hepatic neutrophils per liver from halothane-treated WT and CD1d−/− mice (10 mice per group) are plotted as mean ± SEM. (*, p <0.05 compared with WT mice). (C) KCs were depleted as described in Fig. 3, and treated with halothane 2 days later. Hepatic leukocytes were isolated 12 h after halothane treatment, and the cells were stained and analyzed by flow cytometry as described above. The absolute number of infiltrated neutrophils per liver were calculated and plotted as mean ± SEM.
4. DISCUSSION
DILI is a significant cause of patient morbidity and mortality. The idiosyncratic nature and poor diagnosis of such reactions represent a major safety issue in drug therapy and new drug development. Research aimed at understanding the pathogenesis of DILI has been hampered by the lack of animal models. Our previous development of a murine model of HILI demonstrated a strain-dependent susceptibility that was not due to variations in the metabolism of halothane to TFA but rather the extent of inflammatory response.[3] Moreover, our studies revealed that co-treatment of mice with a viral RNA mimetic, polyinosinic-polycytidylic acid (polyI:C), caused an exacerbation of HILI by stimulating hepatic macrophages and NK cells to produce multiple pro-apoptotic factors.[17] Collectively, these findings suggest that in addition to halothane-induced direct hepatotoxicity, the activation of various innate immune cells within the liver plays an important role in the pathogenesis of HILI.
The current study examined the involvement of NKT cells in HILI and demonstrated that NKT cell-depleted CD1d−/− mice were resistant to the development of HILI (Fig. 1). This resistance was not due to a compromise in halothane metabolism or the subsequent formation of TFA-protein adducts, which are critical events in the initiation of tissue damage. In addition, the data revealed similar levels of hepatic CYP450 2E1 expression between CD1d−/− and WT mice (Fig. 2). While the levels of TFA-protein adducts were slightly increased, they were certainly not decreased in CD1d−/− mice compared with WT mice (Fig. 2). This result, in conjunction with our previous finding that the strain-dependent susceptibility to HILI was not due to variations in halothane metabolism,[3] suggest that TFA-protein adduct formation is a prerequisite for the development of HILI. Nonetheless, the degree of tissue damage does not closely correlate with the extent the adduct formation.
The resistance of CD1d−/− mice against the development of HILI may be either a direct consequence of NKT cell-deficiency or an indirect result due to the absence of cross-talk between NKT cells and other hepatic innate immune cells. NKT cell stimulation may be dependent upon the prior activation of KCs by either halothane itself or halothane-challenged hepatocytes. On the other hand, NKT cells may cause the activation of hepatic KCs and NK cells, resulting in their further production of pro-inflammatory cytokines.[21,22] It has been demonstrated that KCs play an important role in murine models of T cell- and TNF-α-dependent liver injury.[23] Furthermore, we have reported that KCs and NK cells play a key part in mediating polyI:C-induced exacerbation of HILI in mice.[17] However, our present study demonstrated that the specific depletion of either KCs or NK cells did not modulate the extent of HILI in WT mice (Fig. 3), thereby negating the involvement of these cells in the pathogenesis of HILI. These data further suggest that the resistance of CD1d−/− mice in developing HILI cannot be explained by the lack of interaction between NKT cells and KCs or NK cells.
We found that CD1d−/− mice, which were resistant to HILI, exhibited significantly decreased numbers of infiltrating neutrophils within the liver upon halothane challenge, compared with that in WT mice (Fig. 6A and B). The pathological role of neutrophils in HILI has been established in our previous studies, as depletion of neutrophils resulted in a marked attenuation of liver injury.[3] Collectively, these findings suggest that hepatic NKT cells are an important factor in the regulation of neutrophil recruitment into the liver, thereby contributing to HILI. In support of this hypothesis, a report has described that hepatic NKT cells, through secretion of osteopontin, mediated the degree of neutrophil infiltration and activation within the liver, thereby modulating the severity of concanavalin A-induced liver injury.[24] Another possible pathway by which NKT cells facilitate neutrophil recruitment is through the production of IL-17. IL-17, a major pro-inflammatory cytokine produced by activated T helper cells (Th17 cells), has been shown to cause neutrophil infiltration during tissue inflammation.[25,26] A number of recent studies have revealed that, compared with Th17 cells, NKT cells produce IL-17 much more rapidly upon activation, [27–29] and that this NKT cell-derived IL-17 mediates neutrophil infiltration.[28,29] Furthermore, it was recently reported that IL-17 plasma level was increased in WT Balb/c mice treated with halothane. The same study also found that neutralization of IL-17 attenuated HILI, and that administration of rIL-17 exacerbated it.[30] Based on these findings, our future study will focus on investigating the role of osteopontin and IL-17 in mediating NKT cell-regulated neutrophil recruitment and contributing to HILI.
Our data revealed that hepatic NKT cells were activated in mice upon halothane treatment (Fig. 4). NKT cells can be activated in many ways, for which pro-inflammatory cytokines, including IL-12, IL-15, and IL-18 have been shown to be one such mechanism.[31] It is therefore possible that hepatic KCs, which represent a major cellular source of these cytokines, are involved in activating NKT cells. However, our data demonstrating that depletion of KCs does not affect HILI (Fig. 3) negate such a role for KCs in NKT cell activation and thereby mediating NKT cell-induced liver injury. It is also known that NKT cells can be activated via T cell receptor recognition of glycolipids presented in complex with CD1d. An exogenous glycosphingolipid (GSL), α-GalCer, has been shown to cause a robust activation of NKT cells.[32] The physiological ligands recognized by NKT cells remain unidentified, although β-D-glycosylceramides (β-D-GlcCer), as self-GSL antigens, have gained experimental support.[20,33] Our data demonstrated that the majority of hepatocytes isolated from both naïve and halothane-treated WT mice express CD1d on their surface (Fig. 5). Furthermore, intravital microscopy studies demonstrating direct contact between hepatocytes and NKT cells (4 cells/h) support the notion that NKT cells regularly survey CD1d-bound lipid antigens presented by hepatocytes.[34] As such, it is likely that halothane treatment may induce the generation of new self-GSL antigens or increase their amount in hepatocytes, thereby resulting in direct NKT cell activation.
In summary, our studies demonstrated that CD1d−/−mice deficient in NKT cells were resistant to the development of HILI. This resistance appeared to be a direct result of NKT cell depletion rather than an indirect consequence due to the loss of cross-talk between NKT cells and other hepatic innate immune cells. Compared with WT mice, CD1d−/− mice exhibited significantly lower number of infiltrating neutrophils in the liver upon halothane challenge. This finding, together with an existing wealth of literature reports describing the role of NKT cells in regulating neutrophil recruitment, strongly suggests that NKT cells play a critical role in stimulating neutrophil infiltration and activation, thereby contributing to the development and progression of HILI. Collectively, the current study and our previous reports indicate that this murine model of HILI provides an experimental system for the investigation of the underlying mechanisms of DILI. Furthermore, this model may yield the identification of susceptibility factors that contribute to the development of liver injury in patients treated with halothane and potentially other drugs.
Acknowledgments
The authors wish to thank Dr. Lance Pohl (NIH, Bethesda, MD) for the generous gift of anti-TFA antisera. This work was supported by U.S. National Institutes of Health grant R01ES012914 (to Cynthia Ju).
Abbreviations
- DILI
drug-induced liver injury
- HILI
halothane-induced liver injury
- NKT
natural killer T
- IL
interleukin
- IFN
interferon
- TNF
tumor necrosis factor
- α-GalCer
alpha-galactosylceramide
- i.p
intraperitoneally
- KCs
Kupffer cells
- i.v
intravenously
- ALT
alanine transaminase
- H/E
hematoxylin and eosin
- CYP450 2E1
cytochrome P450 2E1
- TFA
trifloroacetylchloride
- FITC
fluoresceinisothiocyanate
- PE
phycoerythrin
- APC
allophycocyanin
- polyI
C, polyinosinic-polycytidylic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ostapowicz G, Fontana RJ, Schiodt FV, et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann Intern Med. 2002;137:947–54. doi: 10.7326/0003-4819-137-12-200212170-00007. [DOI] [PubMed] [Google Scholar]
- 2.Temple RJ, Himmel MH. Safety of newly approved drugs: implications for prescribing. JAMA. 2002;287:2273–5. doi: 10.1001/jama.287.17.2273. [DOI] [PubMed] [Google Scholar]
- 3.You Q, Cheng L, Reilly TP, Wegmann D, Ju C. Role of neutrophils in a mouse model of halothane-induced liver injury. Hepatology. 2006;44:1421–31. doi: 10.1002/hep.21425. [DOI] [PubMed] [Google Scholar]
- 4.Godfrey DI, Hammond KJ, Poulton LD, Smyth MJ, Baxter AG. NKT cells: facts, functions and fallacies. Immunol Today. 2000;21(11):573–83. doi: 10.1016/s0167-5699(00)01735-7. [DOI] [PubMed] [Google Scholar]
- 5.Kakimi K, Guidotti LG, Koezuka Y, Chisari FV. Natural killer T cell activation inhibits hepatitis B virus replication in vivo. J Exp Med. 2000;192(7):921–30. doi: 10.1084/jem.192.7.921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Margalit M, Shibolet O, Klein A, et al. Suppression of hepatocellular carcinoma by transplantation of ex-vivo immune-modulated NKT lymphocytes. Int J Cancer. 2005;115(3):443–9. doi: 10.1002/ijc.20889. [DOI] [PubMed] [Google Scholar]
- 7.Takeda K, Hayakawa Y, Van Kaer L, et al. Critical contribution of liver natural killer T cells to a murine model of hepatitis. Proc Natl Acad Sci U S A. 2000;97:5498–503. doi: 10.1073/pnas.040566697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kusters S, Gantner F, Kunstle G, Tiegs G. Interferon gamma plays a critical role in T cell-dependent liver injury in mice initiated by concanavalin A. Gastroenterology. 1996;111:462–71. doi: 10.1053/gast.1996.v111.pm8690213. [DOI] [PubMed] [Google Scholar]
- 9.Mizuhara H, O’Neill E, Seki N, et al. T cell activation-associated hepatic injury: mediation by tumor necrosis factors and protection by interleukin 6. J Exp Med. 1994;179:1529–37. doi: 10.1084/jem.179.5.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toyabe S, Seki S, Iiai T, et al. Requirement of IL-4 and liver NK1+ T cells for concanavalin A-induced hepatic injury in mice. J Immunol. 1997;159:1537–42. [PubMed] [Google Scholar]
- 11.Biburger M, Tiegs G. Alpha-galactosylceramide-induced liver injury in mice is mediated by TNF-alpha but independent of Kupffer cells. J Immunol. 2005;175(3):1540–50. doi: 10.4049/jimmunol.175.3.1540. [DOI] [PubMed] [Google Scholar]
- 12.Dieli F, Sireci G, Russo D, et al. Resistance of natural killer T cell-deficient mice to systemic Shwartzman reaction. J Exp Med. 2000;192(11):1645–52. doi: 10.1084/jem.192.11.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ogasawara K, Takeda K, Hashimoto W, et al. Involvement of NK1+ T cells and their IFN-gamma production in the generalized Shwartzman reaction. J Immunol. 1998;160(7):3522–7. [PubMed] [Google Scholar]
- 14.Liu ZX, Govindarajan S, Kaplowitz N. Innate immune system plays a critical role in determining the progression and severity of acetaminophen hepatotoxicity. Gastroenterology. 2004;127:1760–74. doi: 10.1053/j.gastro.2004.08.053. [DOI] [PubMed] [Google Scholar]
- 15.Masson MJ, Carpenter LD, Graf ML, Pohl LR. Pathogenic role of natural killer T and natural killer cells in acetaminophen-induced liver injury in mice is dependent on the presence of dimethyl sulfoxide. Hepatology. 2008;48(3):889–97. doi: 10.1002/hep.22400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ju C, Pohl LR. Immunohistochemical detection of protein adducts of 2,4-dinitrochlorobenzene in antigen presenting cells and lymphocytes after oral administration to mice: lack of a role of Kupffer cells in oral tolerance. Chem Res Toxicol. 2001;14:1209–17. doi: 10.1021/tx0100587. [DOI] [PubMed] [Google Scholar]
- 17.Cheng L, You Q, Yin H, et al. Effect of polyI:C cotreatment on halothane-induced liver injury in mice. Hepatology. 2009;49(1):215–26. doi: 10.1002/hep.22585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rafeiro E, Barr SG, Harrison JJ, Racz WJ. Effects of N-acetylcysteine and dithiothreitol on glutathione and protein thiol replenishment during acetaminophen-induced toxicity in isolated mouse hepatocytes. Toxicology. 1994;93(2–3):209–24. doi: 10.1016/0300-483x(94)90079-5. [DOI] [PubMed] [Google Scholar]
- 19.Sakai H, Okamoto T, Kikkawa Y. Suppression of hepatic drug metabolism by the interferon inducer, polyriboinosinic acid:polyribocitidylic acid. J Pharmacol Exp Ther. 1992;263:381–6. [PubMed] [Google Scholar]
- 20.Stanic AK, De Silva AD, Park JJ, et al. Defective presentation of the CD1d1-restricted natural Va14Ja18 NKT lymphocyte antigen caused by beta-D-glucosylceramide synthase deficiency. Proc Natl Acad Sci U S A. 2003;100(4):1849–54. doi: 10.1073/pnas.0430327100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tacke F, Luedde T, Trautwein C. Inflammatory pathways in liver homeostasis and liver injury. Clin Rev Allergy Immunol. 2009;36:4–12. doi: 10.1007/s12016-008-8091-0. [DOI] [PubMed] [Google Scholar]
- 22.Tiegs G. Cellular and cytokine-mediated mechanisms of inflammation and its modulation in immune-mediated liver injury. Z Gastroenterol. 2007;45:63–70. doi: 10.1055/s-2006-927397. [DOI] [PubMed] [Google Scholar]
- 23.Schumann J, Wolf D, Pahl A, et al. Importance of Kupffer cells for T-cell-dependent liver injury in mice. Am J Pathol. 2000;157:1671–83. doi: 10.1016/S0002-9440(10)64804-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Diao H, Kon S, Iwabuchi K, et al. Osteopontin as a mediator of NKT cell function in T cell-mediated liver diseases. Immunity. 2004;21(4):539–50. doi: 10.1016/j.immuni.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 25.Laan M, Cui ZH, Hoshino H, et al. Neutrophil recruitment by human IL-17 via C-X-C chemokine release in the airways. J Immunol. 1999;162(4):2347–52. [PubMed] [Google Scholar]
- 26.Ruddy MJ, Shen F, Smith JB, Sharma A, Gaffen SL. Interleukin-17 regulates expression of the CXC chemokine LIX/CXCL5 in osteoblasts: implications for inflammation and neutrophil recruitment. J Leukoc Biol. 2004;76(1):135–44. doi: 10.1189/jlb.0204065. [DOI] [PubMed] [Google Scholar]
- 27.Coquet JM, Chakravarti S, Kyparissoudis K, et al. Diverse cytokine production by NKT cell subsets and identification of an IL-17-producing CD4-NK1.1- NKT cell population. Proc Natl Acad Sci U S A. 2008;105(32):11287–92. doi: 10.1073/pnas.0801631105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lee KA, Kang MH, Lee YS, et al. A distinct subset of natural killer T cells produces IL-17, contributing to airway infiltration of neutrophils but not to airway hyperreactivity. Cell Immunol. 2008;251(1):50–5. doi: 10.1016/j.cellimm.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 29.Michel ML, Keller AC, Paget C, et al. Identification of an IL-17-producing NK1.1(neg) iNKT cell population involved in airway neutrophilia. J Exp Med. 2007;204(5):995–1001. doi: 10.1084/jem.20061551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kobayashi E, Kobayashi M, Tsuneyama K, et al. Halothane-induced liver injury is mediated by interleukin-17 in mice. Toxicol Sci. 2009;111:302–10. doi: 10.1093/toxsci/kfp165. [DOI] [PubMed] [Google Scholar]
- 31.Hao Q, Liu J, Pappu R, et al. Contribution of bone marrow-derived cells associated with brain angiogenesis is primarily through leukocytes and macrophages. Arterioscler Thromb Vasc Biol. 2008;28(12):2151–7. doi: 10.1161/ATVBAHA.108.176297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kawano T, Cui J, Koezuka Y, et al. CD1d-restricted and TCR-mediated activation of valpha14 NKT cells by glycosylceramides. Science. 1997;278(5343):1626–9. doi: 10.1126/science.278.5343.1626. [DOI] [PubMed] [Google Scholar]
- 33.Paget C, Mallevaey T, Speak AO, et al. Activation of invariant NKT cells by toll-like receptor 9-stimulated dendritic cells requires type I interferon and charged glycosphingolipids. Immunity. 2007;27(4):597–609. doi: 10.1016/j.immuni.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 34.Geissmann F, Cameron TO, Sidobre S, et al. Intravascular immune surveillance by CXCR6+ NKT cells patrolling liver sinusoids. PLoS Biol. 2005;3(4):e113. doi: 10.1371/journal.pbio.0030113. [DOI] [PMC free article] [PubMed] [Google Scholar]