

Abstract
A new lead class of antibacterial drug-like NAD synthetase (NADs) inhibitors was previously identified from a virtual screening study. Here a solution-phase synthetic library of 76 compounds, analogs of the urea-sulfonamide 5838, was synthesized in parallel to explore SAR on the sulfonamide aryl group. All library members were tested for enzyme inhibition against NADs and nicotinic acid mononucleotide adenylyltransferase (NaMNAT), the last two enzymes in the biosynthesis of NAD, and for growth inhibition in a B. anthracis antibacterial assay. Most compounds that inhibited bacterial growth also showed inhibition against one of the enzymes tested. While only modest enhancements in the enzyme inhibition potency against NADs were observed, of significance was the observation that the antibacterial urea-sulfonamides more consistently inhibited NaMNAT.
Introduction
As the threat of bioterrorism increases1,2 and the incidence of drug-resistant bacteria multiplies,3 the need for new antibiotics that act at novel targets becomes more pressing. Our earlier studies4–6 revealed that inhibition of one such target, the amidotransferase enzyme nicotinamide adenine dinucleotide (NAD) synthetase (NADs),7 which catalyzes the final step in the biosynthesis of NAD, could hinder both spore outgrowth and vegetative growth of Bacillus anthracis, which would provide antibacterial action at two different steps in this bacterium's life cycle.8,9 These were the first reported inhibitors of NADs and consisted of a general template of two aromatic end groups linked by a polymethylene tether 4,5 of 6–8 carbons. However, the permanent positive charge required on one end and the resulting detergent-like properties of this class of compounds were considered liabilities for further drug development, and we, therefore, sought to discover new, more drug-like lead classes of NADs inhibitors. A virtual screening study10,11 led to the identification of 4 new lead inhibitors with antibacterial activity, and their structures and activities are shown in Table 1.
Table 1.
| Cmpd ID | Structure | NADs IC50 (µM) | LBa B.a. MIC | MHbB.a. MIC |
|---|---|---|---|---|
| 5824 | 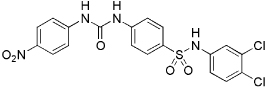 |
6.4 | 1.9 µM (0.9 µg/mL) | 2.8 µM (1.3 µg/mL) |
| 5599 | 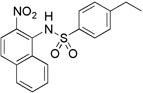 |
168 | 6.9 µM (2.4 µg/mL) | 3.8 µM (1.3 µg/mL) |
| 5617 | 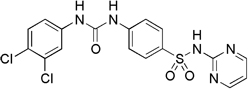 |
77.5 | 22.1 µM (9.7 µg/mL) | 3.8 µM (1.7 µg/mL) |
| 5833 | 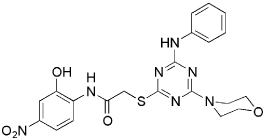 |
78.3 | 15 µM (7.2 µg/mL) | 7.5 µM (3.6 µg/mL) |
Luria-Bertani broth
Mueller-Hinton broth
In this work we noted that compounds closely related to the urea-sulfonamides 5617 and 5824, but containing a reverse sulfonamide, were not widely available commercially. We therefore sought to determine if the reverse sulfonamide analog of 5824 would maintain activity, and if the position of the nitro group was important for activity. Thus the synthesis of three 5824 analogs was proposed.
The syntheses of these parent compounds were performed in parallel according to Scheme 1. The ureas 3{1–3} were formed on a 100 mg scale according to a literature procedure,12 coupling p-phenylenediamine (1) to the appropriate 2-, 3-, and 4-nitrophenylisocyanates 2{1–3}. Reactions of these urea-anilines 3{1–3} with 3,4-dichlorobenzenesulfonyl chloride (4) gave chemset 5. Compounds 5{1–3} were characterized and were >90% pure by mass spectrometry, 1H and 13C NMR, and melting point determination.
Scheme 1.

Synthesis of parent compounds.
As shown in Table 2, the biological activities of 5{1} (4-nitro substituent) and 5824 were similar, while compounds 5{2} and 5{3} were less active. We next proposed SAR studies on the two most active leads, 5824 and 5. Here we describe an SAR study with 5, which we pursued first since we had on hand from another study a large group of substituted benzenesulfonyl chlorides suitable for analog synthesis.
Table 2.
The reverse sulfonamide analogs of 5824 and their biological activities.
| ID | NADs IC50 (µM) | LB B.a. MIC (µM[µg/mL]) | MH B.a. MIC (µM[µg/mL]) |
|---|---|---|---|
| 5824 | 6.4 | 1.9 [0.9] | 2.8 [1.3] |
| 5{1} | 32.7 | 2.8 [1.3] | 3.8 [1.8] |
| 5{2} | 95.5 | > 240 [>115] | – a |
| 5{3} | 443 | > 240 [>115] | – a |
Compounds which showed no inhibition in the LB MIC assay were not included in the MH assay.
Since many of the active NADs inhibitors from our previous virtual screening study10,11 contained nitro groups, as a first approach we decided to maintain the mononitro functionality on the urea N-aryl group in each of the 3 positions of 5, and to explore several different commercially available R-groups on the sulfonamide phenyl ring (Figure 2).
Figure 2.

SAR library proposed around parent compounds 5{1–3}.
To this end, larger quantities of compounds 5 were prepared for use in the sulfonamide library synthesis. Though the literature procedure used for the small scale urea reactions called for refluxing conditions, at that temperature we observed a diurea byproduct resulting from the reaction of each of the desired products 3{1–3} with another molecule of 2{1–3}. Thus, subsequent reactions were performed at 0 ºC; these byproducts could not be completely avoided, however, resulting in improved, yet moderate to poor yields, especially in the case of 5{2} (see experimental section).
Next, the parallel sulfonamide-forming reactions were carried out in 10-mL reaction vials with pyridine as solvent at 0 ºC according to a literature procedure (Scheme 2).13 After work-up and solvent removal, unreacted starting materials were removed by partially dissolving the crude reaction mixtures in 6% i-PrOH in CHCl3 and filtering off the nearly pure product 7{1–3, 1–22}. In a few instances, parallel column chromatography carried out in silica-packed 10-mL syringes was required to obtain sufficiently pure products. No product was obtained for chemset members 7{1, 15} and 7{2, 15}. Compound identities were supported by mass spectroscopy and the purity of all but two library members was determined to be at least 80% by HPLC/MS. Purities were confirmed for 20 library members (see Table 3) using 1H NMR with an internal standard (hexamethyldisiloxane). Two final compounds (7{2,4} and 7{2,17}) were found to be less than 80% pure by LC/MS, but were tested in that form with the intention of purifying further those which showed significant biological activity.
Scheme 2.

Parallel sulfonamide library synthesis.
Table 3.
Yield and purity data for chemset 7.
| Chemset 7 | %Yield | LCMS purity | NMR purity |
|---|---|---|---|
| {1,1} | 36 | 87 | 93 |
| {2,1} | 54 | 82 | –a |
| {3,1} | 70 | 82 | –a |
| {1,2} | 45 | 90 | 94 |
| {2,2} | 38 | 82 | –a |
| {3,2} | 66 | 89 | –a |
| {1,3} | 70 | 87 | –a |
| {2,3} | 58 | 86 | –a |
| {3,3} | 61 | 88 | 96 |
| {1,4} | 50 | 80 | –a |
| {2,4} | 49 | 69 | –a |
| {3,4} | 57 | 88 | –a |
| {1,5} | 40 | 93 | 99 |
| {2,5} | 50 | 100 | 94 |
| {3,5} | 26 | 88 | –a |
| {1,6} | 29 | 86 | 91 |
| {2,6} | 60 | 83 | –a |
| {3,6} | 75 | 88 | –a |
| {1,7} | 33 | 85 | –a |
| {2,7} | 65 | 100 | 98 |
| {3,7} | 61 | 90 | –a |
| {1,8} | 63 | 85 | –a |
| {2,8} | 45 | 98 | 96 |
| {3,8} | 56 | 94 | –a |
| {1,9} | 68 | 91 | –a |
| {2,9} | 57 | 85 | –a |
| {3,9} | 66 | 98 | 96 |
| {1,10} | 70 | 94 | –a |
| {2,10} | 26 | 84 | –a |
| {3,10} | 65 | 80 | 96 |
| {1,11} | 61 | 90 | 91 |
| {2,11} | 47 | 84 | –a |
| {3,11} | 54 | 86 | –a |
| {1,12} | 46 | 79 | –a |
| {2,12} | 55 | 87 | –a |
| {3,12} | 56 | 88 | 91 |
| {1,13} | 51 | 84 | –a |
| {2,13} | 58 | 81 | –a |
| {3,3} | 75 | 90 | 99 |
| {114} | 60 | 84 | - |
| {2,14} | 63 | 78 | - |
| {3,14} | 74 | 88 | 96 |
| {1,15} | –b | - | - |
| {2,15} | –b | - | - |
| {3,15} | 29 | 87 | –a |
| {1,16} | 79 | 83 | –a |
| {2,16} | 60 | 100 | 94 |
| {3,16} | 51 | 85 | –a |
| {1,17} | 63 | 100 | 96 |
| {2,17} | 56 | 59 | –a |
| {3,17} | 76 | 88 | –a |
| {1,18} | 17 | 100 | –a |
| {2,18} | 71 | 93 | –a |
| {3,18} | 45 | 94 | –a |
| {1,19} | 70 | 92 | –a |
| {2,19} | 49 | 100 | 93 |
| {3,19} | 70 | 89 | –a |
| {1,20} | 66 | 88 | –a |
| {2,20} | 60 | 100 | 97 |
| {3,20} | 36 | 89 | –a |
| {1,21} | 41 | 88 | –a |
| {2,21} | 48 | 81 | –a |
| {3,21} | 13 | 95 | –a |
| {1,22} | 69 | 92 | 94 |
| {2,22} | 52 | 84 | 90 |
| {3,22} | 21 | 93 | –a |
Data not obtained
No product obtained
While most library products could be synthesized in one step from compounds 3{1–3}, a few required additional steps. After the nitrile products 7{1–3,18–20}were obtained, small amounts of each were reduced with 1M BH3-THF according to a literature procedure14 to obtain the corresponding aminomethyl compounds 8{1–3,18–20}, as shown is Scheme 3. Nitrile 7{1,18} was not obtained in sufficient quantity to carry forward to the reduction step. The other nitriles provided reduction products in at least 80% purity (as confirmed by HPLC/MS) after parallel column chromatography using silica-packed 10-mL syringes.
Scheme 3.

Reduction of nitriles 7 to the corresponding aminomethyls.
We were also interested in preparing the aniline derivatives of the N-acetyl sulfonamide products; however, only the 2-nitro derivative 7{3,15} was obtained successfully from the parallel sulfonamide reactions. Therefore, a small amount of 7{3,15} was hydrolyzed with hydrochloric acid in methanol according to a literature procedure15 to give the unprotected aniline 9(Scheme 4). Isolation of the crude reaction product gave a pale yellow oil, which was confirmed to be greater than 80% pure by HPLC/MS and was, therefore, not purified further.
Scheme 4.

Hydrolysis of N-acetyl 7{3,15} to the corresponding aniline.
All of the 75 final products, including the three parents, were first tested as inhibitors of B. anthracis NAD synthetase in an HPLC assay and were evaluated for antibacterial activity against B. a. according to our previously reported procedure.11 Only compounds which showed some activity in the Luria-Bertani (LB) broth antibacterial assay were assayed again using Mueller Hinton (MH) broth as specified by the Clinical and Laboratory Standards Institute MIC broth microdilution protocol.16 Subsequently, these same compounds were evaluated as inhibitors of NaMNAT (see below). The structural and biological data are shown in Table 5.
Table 5.
Structures and biological activities of final library members.
| Cmpd ID | Nitro position | R | NADs IC50(µM) | NaMNAT50(µM) | LB MIC (µM [µg/mL]) | MH MIC (µM [µg/mL]) |
|---|---|---|---|---|---|---|
| 5824 | 10 | 2 | 1.9 [0.9] | 2.8 [1.3] | ||
| 5{1} | 4 | 3,4,-Cl2 | 32.7 | 7.3 | 2.8 [1.3] | 3.8 [1.8] |
| 5{2} | 3 | 3,4,-Cl2 | 95.5 | 382 | > 240 [>115] | –a |
| 5{3} | 2 | 3,4,-Cl2 | 443 | > 600 | > 240 [>115] | – |
| 7{1,1} | 4 | 2-Me | 600 | 16.3 | 15 [6.4] | 7.5 [3.2] |
| 7{2,1} | 3 | 2-Me | 513 | >150 | > 240 [>102] | – |
| 7{3,1} | 2 | 2-Me | 538 | >150 | > 240 [>102] | – |
| 7{1,2 } | 4 | 3-Me | 321 | 19.9 | 15 [6.4] | 7.5 [3.2] |
| 7{2,2} | 3 | 3-Me | >600 | >150 | > 240 [>102] | – |
| 7{3,2} | 2 | 3-Me | 569 | >150 | > 240 [>102] | – |
| 7{1,3} | 4 | 4-Et | 492 | >150 | > 240 [>106] | – |
| 7{2,3} | 3 | 4-Et | 330 | 12 | 7.5 [3.3] | < 3.8[<1.7] |
| 7{3,3} | 2 | 4-Et | >600 | >150 | > 240 [>106] | – |
| 7{1,4} | 4 | 4-Ph | 427 | >150 | > 240 [>117] | – |
| 7{2,4} | 3 | 4-Ph | 480 | >150 | > 240 [>117] | – |
| 7{3,4} | 2 | 4-Ph | >600 | >150 | > 240 [>117] | – |
| 7{1,5} | 4 | 2-F | 422 | 22.2 | 30 [12.9] | 15 [6.5] |
| 7{2,5} | 3 | 2-F | 494 | >150 | > 240 [>103] | – |
| 7{3,5} | 2 | 2-F | >600 | >150 | 240 [103] | 30 [12.9] |
| 7{1,6} | 4 | 3-F | >600 | 35.8 | 15 [6.5] | 7.5 [3.2] |
| 7{2,6} | 3 | 3-F | 535 | >150 | 30 [12.9] | 15 [6.5] |
| 7{3,6} | 2 | 3-F | >600 | 36.1 | 30 [12.9] | 15 [6.5] |
| 7{1,7} | 4 | 4-F | 353 | 370 | 15 [6.5] | 15 [6.5] |
| 7{2,7} | 3 | 4-F | 463 | >150 | > 240 [>103] | – |
| 7{3,7} | 2 | 4-F | >600 | >150 | > 240 [>103] | – |
| 7{1,8} | 4 | 2-Cl | 423 | 17.3 | 30 [13.4] | 7.5 [3.3] |
| 7{2,8} | 3 | 2-Cl | >600 | 20.4 | 30 [13.4] | 7.5 [3.3] |
| 7{3,8} | 2 | 2-Cl | >600 | >150 | > 240 [>107] | – |
| 7{1,9} | 4 | 3-Cl | >600 | 19.3 | 7.5 [3.3] | 7.5 [3.3] |
| 7{2,9 } | 3 | 3-Cl | 414 | 17.5 | 7.5 [3.3] | 7.5 [3.3] |
| 7{3,9} | 2 | 3-Cl | >600 | >150 | > 240 [>107] | – |
| 7{1,10 } | 4 | 4-Cl | 336 | >150 | > 240 [>107] | – |
| 7{2,10 } | 3 | 4-Cl | 287 | >150 | > 240 [>107] | – |
| 7{3,10 } | 2 | 4-Cl | >600 | >150 | > 240 [>107] | – |
| 7{1,11 } | 4 | 2-CF3 | 505 | 13.3 | 15 [7.2] | 15 [7.2] |
| 7{2,11 } | 3 | 2-CF3 | >600 | >150 | > 240 [>115] | – |
| 7{3,11 } | 2 | 2-CF3 | >600 | >150 | > 240 [>115] | – |
| 7{1,12 } | 4 | 3-CF3 | >600 | 26.7 | 7.5 [3.6] | 7.5 [3.6] |
| 7{2,12 } | 3 | 3-CF3 | 293 | >150 | > 240 [>115] | – |
| 7{3,12} | 2 | 3-CF3 | 298 | >150 | > 240 [>115] | – |
| 7{1,13} | 4 | 4-CF3 | 168 | >150 | > 240 [>115] | – |
| 7{2,13} | 3 | 4-CF3 | 143 | >150 | > 240 [>115] | – |
| 7{3,13} | 2 | 4-CF3 | 560 | 150 | > 240 [>115] | – |
| 7{1,14} | 4 | 4-OPh | 327 | >150 | > 240 [>121] | – |
| 7{2,14} | 3 | 4-OPh | 362 | 225 | 30 [15] | > 60 [30] |
| 7{3,14 } | 2 | 4-OPh | >600 | >150 | > 240 [>121] | – |
| 7{3,15} | 2 | 4-NHAc | >600 | >150 | > 240 [>113] | – |
| 7{1,16} | 4 | 3-OMe | 370 | >150 | 15 [6.6] | 15 [6.6] |
| 7{2,16} | 3 | 3-OMe | 600 | 25.0 | 60 [26] | 15 [6.6] |
| 7{3,16} | 2 | 3-OMe | >600 | >150 | > 240 [>106] | – |
| 7{1,17} | 4 | 4-OMe | >600 | >150 | 30 [13] | 7.5 [3.3] |
| 7{2,17} | 3 | 4-OMe | >600 | 10.1 | > 240 [>106] | – |
| 7{3,17} | 2 | 4-OMe | >600 | >150 | > 240 [>106] | – |
| 7{1,18} | 4 | 2-CN | 526 | >150 | > 240 [>105] | – |
| 7{2,18} | 3 | 2-CN | >600 | >150 | > 240 [>105] | – |
| 7{3,18} | 2 | 2-CN | 590 | >150 | > 240 [>105] | – |
| 7{1,19} | 4 | 3-CN | 546 | >150 | > 240 [>105] | – |
| 7{2,19} | 3 | 3-CN | >600 | >150 | > 240 [>105] | – |
| 7{3,19} | 2 | 3-CN | >600 | >150 | > 240 [>105] | – |
| 7{1,20} | 4 | 4-CN | >600 | >150 | 60 [26] | 15 [6.6] |
| 7{2,20} | 3 | 4-CN | >600 | >150 | > 240 [>105] | – |
| 7{3,20} | 2 | 4-CN | >600 | >150 | > 240 [>105] | – |
| 7{1,21} | 4 | 3-CO2H | 537 | >150 | > 240 [>109] | – |
| 7{2,21} | 3 | 3-CO2H | 274 | >150 | > 240 [>109] | – |
| 7{3,21} | 2 | 3-CO2H | 456 | 150 | > 240 [>109] | – |
| 7{1,22} | 4 | 4-CO2H | 498 | >150 | > 240 [>109] | – |
| 7{2,22 } | 3 | 4-CO2H | >600 | >150 | > 240 [>109] | – |
| 7{3,22} | 2 | 4-CO2H | 432 | 150 | > 240 [>109] | – |
| 8{2,18} | 3 | 2-CH2NH2 | >600 | >150 | > 240 [>106] | – |
| 8{3,18} | 2 | 2-CH2NH2 | >600 | >150 | > 240 [>106] | – |
| 8{1,19} | 4 | 3-CH2NH2 | 33.2 | 74.6 | 60 [26] | – |
| 8{2,19} | 3 | 3-CH2NH2 | 30 | >200 | 120 [53] | – |
| 8{3,19} | 2 | 3-CH2NH2 | 245 | >150 | > 240 [>106] | – |
| 8{1–20} | 4 | 4-CH2NH2 | 26 | 12.1 | > 240 [>106] | – |
| 8{2,20} | 3 | 4-CH2NH2 | 15.3 | 51.2 | > 240 [>106] | – |
| 8{3,20} | 2 | 4-CH2NH2 | 24.5 | >150 | > 240 [>106] | – |
| 9 | 2 | 4-NH2 | 17.1 | >150 | 120 [53] | – |
Data not obtained due to poor LB MIC
For NAD synthetase, the enzyme inhibition data in Table 5 suggests a few SAR trends. For example, as with the parent compounds, the 2-nitro is the least active of the three nitro positions, with few exceptions (9). Interestingly, most changes made on the sulfonamide end of the library members resulted in much poorer NADs enzyme activity. Only 13 compounds exhibited NADs inhibition at or below 300 µM, and none of those significantly inhibited bacterial growth. While the 4-CF3 group (library members 7{1–3,13}) gave moderate activity, the only substituents with activity comparable to the parent 5{1} were the aminomethyl and aniline derivatives. At physiological pH the aminomethyl compounds (though not the aniline) should be protonated, bearing a striking resemblance to our earlier class of tethered dimer inhibitors that require a terminal permanent positive charge for enzyme inhibition.4,6 These results suggest that additional examples of the urea-sulfonamides containing basic groups should be explored, as well as additional H-bond donors and acceptors. The aminomethyl derivatives, unlike 5{1}, were not antibacterial, possibly due to much poorer entry of the protonated amine into the bacterial cell.
A striking observation about the biological activity of this library is the lack of correlation between NADs inhibition and antibacterial activity. We also observed this trend in previous virtual screening studies,10,11 although our tethered dimer NADs inhibitors exhibited a good correlation between NADs inhibition and antibacterial action.6 Several possibilities may reasonably explain the lack of antibacterial actions for some NADs inhibitors (e.g., may not permeate into the bacterial cell; may be removed by efflux pumps; may undergo metabolism by bacteria). On the other hand, there are several compounds (7{1,1}, 7 {1,6}, 7 {1,9}, 7 {1,12}, 7 {1,16}, etc.) that are antibacterial, but which do not inhibit NAD synthetase – a behavior also exhibited by select compounds in previous studies. These compounds must be inhibiting bacterial growth by some mechanism other than NADs inhibition.
In an attempt to explore the latter, all library members were assayed against the enzyme which immediately precedes NADs in the NAD biosynthetic pathway, nicotinic acid mononucleotide adenylyltransferase (NaMNAT).17 While NaMNAT contains a smaller catalytic site than NADs, both enzymes share ATP as substrate and bind to an N-ribosylated nicotinic acid. Thus some small molecule inhibitors designed for NADs might reasonably inhibit NaMNAT. The NaMNAT inhibition data given in Table 5 confirms this supposition.
As with the NADs SAR, there are thus far no clear structural trends among the NaMNAT enzyme inhibitors. In general, the 4-NO2 position is the most favorable for NaMNAT inhibition (as seen with compounds 7{1–3,11}). The remaining SAR trends cannot be explained solely by hydrophobicity or electron-donating/withdrawing properties of the substituents on the sulfonamide terminus. The 4 most active NaMNAT inhibitors contain R groups that vary from methoxy, to ethyl, to aminomethyl, to trifluoromethyl, representing 4 very different substituent types. The only group which is not tolerated in the NaMNAT series is the nitrile. Of the 17 NaMNAT inhibitors (IC50 ≤ 100 µM), the position for the R group consistently giving the best activity is the 3-position, with 8 inhibitors (almost half) containing this substitution pattern. Unlike the NADs inhibition data, a number of different substituents give good NaMNAT inhibition, and there is a relationship between NaMNAT inhibition and antibacterial activity. Of the 20 antibacterial library compounds (MH MIC ≤ 30 µM), 15 (75%) have B.a. NaMNAT IC50 ≤ 50 µM. Likewise, among the 19 NaMNAT inhibitors with IC50 < 100 µM (including two parent compounds), 16 (or 84%) also inhibited bacterial growth below 30 µM, though the direct correlation between IC50 and MIC is modest (R2 = 0.29).
This SAR study provided very few low µM inhibitors of NADs, making it difficult at this time to draw conclusions related to the antibacterial efficacy of good inhibitors from this structural class, although our earlier work strongly supported the utility of this target. The current study does, however, support NaMNAT as a promising target for the urea-sulfonamides.
Conclusions
We have utilized parallel solution-phase synthetic chemistry to begin exploring the SAR of a new class of drug-like NAD synthetase inhibitors. 75 new compounds were synthesized and tested in our NADs and NaMNAT enzyme inhibition and B. anthracis antibacterial assays. Though we have found no direct correlation between either NADs or NaMNAT IC50 and MIC, all but 3 antibacterial compounds from this compound library inhibited at least one of the enzymes. Thus far the hydrophobicity and electronic properties of aryl substituents do not appear to predict the enzyme activity for this class of compounds. Further SAR data appears warranted and is being pursued.
Experimental Section
LC/MS Purity Assessment
HPLC analysis was performed using an HP1100 series system with diode array detection coupled with a MicroMass® Platform LCZ mass spectrometer. A Phenomenex® Luna 5µm, C18, 100Å, 100 × 4.60mm column was used for separations. The mobile phase was A: H2O (0.05% formic acid) and B: acetonitrile (0.05% formic acid). The gradient is listed in Table 6. The flow rate was 0.7 mL/min and diode array detection from 190 – 600 nm was used for each 10 µL injection. The mass spectrometer was equipped with an electrospray ionization (ESI) probe and was operated in both the ESI(+) and ESI(-) mode. Peak height estimation for each analyte was based on baseline integration of peaks observed by the diode array detector.
Table 6.
Linear gradient used for purity estimation of library members by LC/MS.
| Time (minutes) | %A | %B |
|---|---|---|
| 0.00 | 80.0 | 20.0 |
| 10.00 | 10.0 | 90.0 |
| 11.00 | 10.0 | 90.0 |
| 11.50 | 80.0 | 20.0 |
NMR Internal Standard Purity Assessment
A selection of 20 library members was also examined for purity via an internal standard NMR purity assessment. The stock NMR solution was created by combining CDCl3 and MeOH-d4 in a 1:1 ratio; 10% DMSO-d6 was added to aid in solubility, and hexamethyldisiloxane (HMDSO; NMR grade, Aldrich) was added to yield a final HMDSO concentration of 12 µM. A known amount (between 5 and 10 mg) of compound was dissolved into 0.5 mL of the NMR solvent, and the 1H NMR spectrum was recorded using a 400 MHz Bruker spectrometer. Peaks were integrated and calibrated according to a known peak area (methyl, when available; otherwise, a urea NH). Compound purity was determined by comparing the calculated weight based on HMDSO peak integration to the actual weight measured upon sample preparation.
NADs HPLC Enzyme Assay
Library compounds were tested for activity against the enzyme using a HPLC assay that we developed and described previously.10,11 Briefly, the assay was carried out in two steps: sample preparation and sample analysis. The preparation of sample plates was performed using a BioMek® FX liquid handling system. The standard reaction volume was 200 µL. The reaction mixture contained 60 mM HEPPS, pH 8.5, 0.5 mM NH4Cl, 20 mM KCl, 10 mM MgCl2, 0.1 mM NaAD, 0.2 mM ATP, 6 µg/mL purified B. anthracis NADs, 2.5% (v/v) DMSO, 0.3% BOG, and inhibitors at various concentrations. Compounds were assayed beginning at 600 µM and at doubling dilutions down to 0.6 µM. The reaction was initiated by adding 0.2 mM ATP, and quenched after 10 minutes by adding 50 µL of 6 M guanidine-HCl. The plates were sealed by aluminum tape, and centrifuged at 2500 rpm for 10 minutes in order to pellet any precipitation that may have been caused by the inhibitors. Plates were stored at 4 oC prior to the HPLC analysis.
The HPLC procedure utilized a Gilson® 215 Liquid Handler, two Gilson® 306 pumps, and a Gilson® 170 diode array detector. A Phenomenex® Luna 5µm, C5, 100Å, 100 × 4.60 mm column was used for separations. The mobile phase was A: 20 mM NaH2PO4 pH 6.90 and B: acetonitrile. The gradient was 100% A from 0 – 3 minutes, to 5% A / 95% B from 3 – 4 minutes for each 20 µL injection. The flow rate was 1.0 mL/min and diode array detection was from 190 – 400 nm. Peak height estimation for NAD was based on baseline integration. The % inhibition at each inhibitor concentration was calculated by the difference in peak height of NAD compared to reactions without inhibitor. The IC50 was determined from the plot of NAD peak height vs. inhibitor concentration, and is defined as the concentration of inhibitor required to produce NAD peak height at 50% of the uninhibited reaction. In developing this assay, peak areas were also used to calculate the IC50 for selected active compounds, and similar results were obtained. Each compound was tested in duplicate, and the IC50 is reported as the average of duplicate runs.
NaMNAT HPLC Enzyme Assay
This assay monitors the production of NaAD in the enzymatic reaction by separating the reactants and products on an HPLC system. The assay system at pH 7.5 contained 50 mM HEPES, 10 mM MgCl2, 25 µM nicotinic acid mononucleotide (NaMN), 44 µM ATP, 0.3% BOG, 0.25 µg/ml B.a. NaMNAT, and inhibitors at eleven different concentrations (with 2.5% v/v final DMSO concentration). Under these conditions, the NaMN and ATP concentrations were the same as their Michaelis-Menton constants, which we reported previously17 . The enzymatic inhibition assay was carried out in 96-well microtiter plates with a total reaction volume of 200 µL.
In each well, 5 µL of DMSO with variable amount of compounds and 170 µL assay buffer containing everything except ATP were first incubated at room temperature for 10 min. The reaction was then initiated by adding 25 µL of ATP solution, and allowed to proceed for 10 min. Addition of 50 µL of 6M guanidine-HCl stopped the reaction. The reaction mixture was next separated on a 4.6mm × 100mm Synergi® Polar-RP column, using a Shimadzu (Columbia, MD) liquid chromatography system consisting of two pumps, a temperature controlled autosampler with a 12-plate rack changer, a column oven and a photo diode assay (PDA) detector. Separation of NaAD from the other component was achieved in less than 5 min by isocratic elution using 50 mM sodium phosphate as the running buffer at a flow rate of 1.0 mL/min. The peak area at 260 nm was used to quantify NaAD. Percent inhibition was calculated based on the difference in NaAD production between controls (DMSO only) and samples containing the compounds. The IC50 value was determined by plotting % inhibition vs. compound concentrations and is reported as the average of duplicate runs.
Antibacterial Assay
All library members were also screened against Bacillus anthracis Sterne in an antibacterial assay, which we also previously reported.10,11 Briefly, B. a. Sterne spores were subcultured from stock cultures into Luria-Bertani (LB) broth and incubated for 2–3 hours at 37 °C in ambient air until the OD600 measurement reached 0.5 to 0.6 when the bacteria are in mid-log phase. The cultures were diluted 1:1 into LB Broth with an absorbance at 600 nm measuring 0.25 to 0.3, then were added to plates containing 240 µM samples of the compounds to be tested. Compounds were tested at a final DMSO concentration of 1%. The plates were incubated at 37 ºC, and absorbance at 600 nm was read at 0 h and every hour for 5 hours. Any compounds which inhibited growth of the vegetative cell (as compared to the control containing only DMSO) were screened in the full MIC determination starting at 240 µM and creating doubling dilutions down to 7.5 µM in quadruplicate wells. A plot of cell density vs. time yields inhibition of growth results, and the MIC is defined as the lowest concentration of compound required to completely inhibit growth (100% inhibition). MIC is reported as the average of the four data points acquired for each compound. Controls for each assay measured sterility, B. anthracis Sterne viability, and MIC100 for the clinical antibiotic ciprofloxacin hydrochloride (from MP Biomedicals).
All compounds which showed antibacterial action in the LB assay were then assayed according to the Clinical and Laboratory Standards Institute MIC broth microdilution protocol,16 which standardizes the number of bacteria used in the inoculum as 5 × 105 cfu/mL, using cation-adjusted Mueller-Hinton (MH) broth, except that measurements were taken at 5 hours, as opposed to 20 hours.
Synthetic Chemistry. General
Melting points were determined using a Mel-Temp Electrothermal 1201-D apparatus and are uncorrected. All 1H and 13C NMR spectra were recorded on a Bruker 400 MHz (1H) spectrometer using tetramethylsilane (TMS) as internal standard. Reactions were monitored by TLC (Whatmann silica gel, UV254, 25 µm plates), and flash column chromatography utilized Baker silica gel (40 µm) in the solvent system indicated. Anhydrous solvents used for reactions were purchased in SureSeal bottles from Aldrich Chemical Co. Other reagents were purchased from Aldrich, Alfa Aesar or Acros chemical companies and used as received. Parallel reactions were carried out in 10 mL screw-cap vials and were agitated by hand. Parallel work-ups were carried out in 50 mL conical Falcon tubes, were concentrated in 15 mL glass vials using a Savant SpeedVac Plus SC210A, and, where indicated, were purified by parallel silica gel chromatography (gravity) in 10 mL disposable syringes.
Preparation of Lead Compounds 5{1–3} (Scheme 1)
N-(4-Aminophenyl)-N’-(4-nitrophenyl)urea (3{1})
p-Phenylenediamine (1) (12 g, 0.11 mol) was partially dissolved in anhydrous CH2Cl2 (60 mL) under a nitrogen atmosphere, and the reaction vessel was submerged in an ice bath. A solution of 4-nitrophenylisocyanate 2{1} (22 g, 0.13 mol) in anhydrous CH2Cl2 (60 mL) was added slowly to the cooled reaction vessel via an addition funnel over a course of 20 minutes with vigorous mechanical stirring, resulting in immediate precipitation of product. Once the addition was complete, the ice bath was removed, and the reaction continued with stirring at room temperature for an additional 20 minutes. TLC (15% i-PrOH in CHCl3) showed that the diamine and isocyanate starting materials were gone; there was one new product spot (reaction with ninhydrin confirmed the presence of an amine), and one base-line spot corresponding to the diurea byproduct. Solvent was removed under vacuum to obtain a mixture of the two products (crude weight 29 g, 95% yield), which were then stirred in hot acetone (2 L). The diurea byproduct remained insoluble and was filtered off. Solvent was removed under vacuum, and the pure product 3{1} was obtained as a dense yellow powder (21 g, 71%): mp 221–223 ºC (decomposed). 1H NMR (DMSO-d6) δ 9.26 (s, 1H, NH), 8.39 (s, 1H, NH), 8.16 (dd, 2H, J = 9.33, 3.06 Hz), 7.66 (dd, 2H, J = 9.39, 3.06 Hz), 7.09 (dd, 2H, J=8.79, 3.06 Hz), 6.52 (dd, 2H, J = 8.76, 3.09 Hz), 4.85 (s, 2H, NH2).13C NMR (DMSO-d6) δ 152.18, 146.88, 144.66, 140.59, 127.66, 125.15, 121.18, 117.10, 114.08. MS (ES+): m/z 273 (M + H); MS (ES−): m/z 271 (M - H).
By this method were also prepared the following:
N-(4-Aminophenyl)-N’-(3-nitrophenyl)urea (3{2})
From 2{2} (12 g, 0.11 mol) was obtained 3{2} (8.2 g, 27 %) as a pure yellow powder: mp 212–214 ºC (decomposed). 1H NMR (DMSO-d6) δ9.04 (s, 1H, NH), 8.55 (t, 1H, J = 2.21), 8.31 (s, 1H, NH), 7.78 (m, 1H), 7.67, (m, 1H), 7.53 (t, 1H, J = 8.15 Hz), 7.09 (dd, 2H, J = 8.57, 3.06 Hz), 6.52 (dd, 2H, J = 8.56, 3.03 Hz), 4.84 (s, 2H, NH2). 13C NMR (DMSO-d6) δ 152.76, 148.16, 144.53, 141.55, 129.97, 127.90, 124.01, 121.26, 115.78, 114.08, 111.81. MS (ES+): m/z 273 (M + H); MS (ES-): m/z 271 (M - H).
N-(4-Aminophenyl)-N’-(2-nitrophenyl)urea (3{3})
From 2{3} (12 g, 0.11 mol) was obtained 3{3} (14 g, 48 %) as a bright orange powder: mp 192–194 ºC (decomposed). 1H NMR (DMSO-d6) δ 9.51 (s, 1H, NH), 9.36 (s, 1H, NH), 8.34 (d, 1H, J = 8.49), 8.08 (dd, 1H, J = 8.37, 1.42 Hz), 7.67 (td, 1H, J = 7.83, 1.48 Hz), 7.15 (td, 1H, J = 7.81, 1.21 Hz), 7.11 (d, 2H, J = 8.57 Hz), 6.53 (d, 2H, J = 8.55 Hz), 4.88 (s, 2H, NH2). 13C NMR (DMSO-d6) δ 151.98, 144.75, 136.98, 135.66, 135.04, 127.75, 125.40, 122.14, 121.62, 121.25, 114.11. MS (ES+): m/z 273 (M + H); MS (ES−): m/z 271 (M - H).
3,4-Dichloro-(N-(4-(((4-nitrophenyl)amino)carbonyl)aminophenyl))benzenesulfonamide (5{1})
To a solution of N-(4-aminophenyl)-N ’-(4-nitrophenyl) urea 3{1} (1.5 g, 5.5 mmol) in anhydrous pyridine (15 mL) at 0 ºC was slowly added 3,4-dichlorobenzenesulfonyl chloride (4) (1.0 mL, 1.6 g, 6.6 mmol). The reaction was stirred under a nitrogen atmosphere for 40 minutes and was diluted with EtOAc (100 mL). The reaction was quenched by adding 2 N HCl (50 mL) and the layers separated; the organic layer was washed further with 2 N HCl (2 × 50 mL), water (100 mL) and brine (75 mL), and was dried over anhydrous Na2SO4. The drying agent was filtered, and the solvent was removed under reduced pressure. The residue (2.1 g, 84%) was taken up in hot methanol (300 mL) and was decolorized with activated charcoal, boiling for 30 minutes. The decolorizing agent was removed by gravity filtration, the filtrate was reduced to 150 mL, and the pure product crystallized to give 5{1} as an off-white solid (1.2 g, 47 %): mp 207–209 ºC. 1H NMR (DMSO-d6) δ 10.25 (s, 1H, NH), 9.41 (s, 1H, NH), 8.91 (s, 1H, NH), 8.18 (dd, 2H, J = 9.29, 3.03 Hz), 7.89 (d, 1H, J = 2.10 Hz), 7.85 (d, 1H, J = 8.45 Hz), 7.66 (dd, 2H, J = 9.38, 3.11 Hz), 7.63 (dd, 1H, J = 8.45, 2.16 Hz), 7.38 (dd, 2H, J = 8.96, 2.96 Hz), 7.02 (dd, 2H, J = 8.95, 2.97 Hz). 13C NMR (DMSO-d6) δ 151.99, 146.40, 141.09, 139.77, 136.45, 136.04, 132.20, 131.78, 131.36, 128.49, 126.93, 125.25, 122.73, 119.60, 117.56. MS (ES-): m/z 479 (M - H).
By this method were prepared the following, with minor changes in purification as noted:
3,4-Dichloro-(N-(4-(((3-nitrophenyl)amino)carbonyl)aminophenyl))benzenesulfonamide (5{2})
From 3{2} (30 mg, 0.11 mmol) was obtained 5{2} (26 mg, 49%): mp 210.5–212 ºC (MeOH). Pure product was obtained by recrystallization from the decolorization solvent MeOH. 1H NMR (DMSO-d6) δ 10.22 (s, 1H, NH), 9.18 (s, 1H, NH), 8.81 (s, 1H), 8.53 (s, 1H, NH), 7.88 (s, 1H), 7.82 (m, 2H), 7.65 (m, 2H), 7.54 (t, 1H, J = 8.12 Hz), 7.37 (d, 2H, J = 8.60), 7.01 (d, 2H, J = 8.58 Hz). MS (ES-): m/z 479 (M - H).
3,4-Dichloro-(N-(4-(((2-nitrophenyl)amino)carbonyl)aminophenyl))benzenesulfonamide (5{3})
From 3{3} (50 mg, 0.18 mmol) was obtained 5{3} (32 mg, 37%): mp 206.5–208 ºC (MeOH). Pure product was obtained by recrystallization from the decolorization solvent MeOH. 1H NMR (DMSO-d6) δ 10.22 (s, 1H, NH), 9.83 (s, 1H, NH), 9.56 (s, 1H, NH), 8.26 (d, 1H, J = 8.48 Hz), 8.09 (dd, 1H, J = 8.32, 1.25 Hz), 7.89 (d, 1H, J = 2.13 Hz), 7.85 (d, 1H, J = 8.45 Hz), 7.69 (t, 1H, J = 7.86 Hz), 7.62 (dd, 1H, J = 8.44, 2.12 Hz), 7.39 (d, 2H, J = 8.80 Hz), 7.20 (td, 1H, J = 7.80, 1.14 Hz), 7.02 (d, 2H, J = 8.84 Hz). MS (ES-): m/z 479 (M - H).
Preparation of Library Members 7 (Scheme 2)
Procedure for Parallel Sulfonamide Synthesis
The starting urea-amines (3) (0.55 mmol) were partially dissolved in pyridine (1.5 mL) in 10-mL, screw-cap vials, and the reaction vials were placed in a rack and submerged in an ice bath. The appropriate sulfonyl chlorides (1.2 equiv) were added to each vial; the vials were capped and the entire apparatus was shaken manually at 0 ºC for 20 minutes. The vials were removed from the ice bath; reactions were quenched with 1N HCl (1 mL), extracted with EtOAc (3 × 2 mL), and the organic layers were transferred to 50-mL Falcon tubes. The combined EtOAc extracts were again washed with 1 N HCl (2 × 2 mL) and water (2 × 2 mL). Carboxylic acid products (70–75) were extracted into saturated NaHCO3 (2 × 3 mL); the aqueous layers were combined, acidified to pH 3 with concentrated HCl, and extracted with EtOAc (3 × 5 mL). All products were dried over Na2SO4 and the solutions filtered in parallel into 15-mL screw-cap vials. Evaporation of the solvent using the high temperature setting of a speedvac afforded the crude sulfonamide products. All residues were triturated with 6% i-PrOH in CHCl3 (~ 2 mL) to dissolve any unreacted starting materials, and the products were suction filtered to afford the library members, 7 (13 – 79% yield). LC/MS of these products revealed that most met the 80% purity criteria; those that did not were further purified in parallel by passing through a short silica plug (5 × 1 cm) using 10-mL syringes and 6% i-PrOH in CHCl3 as eluent.
Preparation of Aminomethyl Compounds (8) (Scheme 3)
Procedure for Parallel Nitrile Reduction
To the starting nitriles 7{1–3,18–20} (0.080 – 0.26 mmol) in anhydrous THF (final concentration of V = 1.0 M) in 5-mL vials was added BH3 (1.0 M in THF; 1.3 equiv) at room temperature. The vials were capped, and the reactions stirred under nitrogen for 1 hour. Concentrated HCl was added to quench the excess hydride present in the reaction, and all solvents were removed using the high temperature setting of the speedvac. To the residue was added 2 N NaOH (1 mL), and the amines were extracted into EtOAc (3 × 2 mL). The organic extracts were combined, washed with water (2 mL) and brine (2 mL), dried over Na2SO4, and filtered in parallel into 15-mL vials. Solvent was again removed via the speedvac; residues were taken up in minimal amounts of CHCl3/i-PrOH (3:1) and purified by silica gel, eluting first with CHCl3, then gradually increasing polarity to 1:1 CHCl3/i-PrOH. Column fractions appearing to be at least 80% pure by TLC were combined into 15-mL vials and concentrated to dryness via a speedvac to yield library members 8 (10 – 63% yield).
Preparation of Aniline 9 (Scheme 4)
4-Amino-(N-(4-(((2-nitrophenyl)amino)carbonyl)aminophenyl))benzenesulfonamide (9)
The N-acetyl sulfonamide product 7{3,15} (32 mg, 0.068 mmol) was dissolved in MeOH (1 mL), and concentrated HCl (0.32 mL) was added dropwise. The reaction was stirred overnight at room temperature, and was quenched with 2 N NaOH (0.5 mL). The product was extracted into EtOAc (3 × 1 mL); the combined extracts were washed with brine (1 mL) and dried over Na2SO4. After filtering, the solvent was removed under vacuum, and TLC (15 % i-PrOH in CHCl3) revealed one major new spot. No further purification was pursued, and 9 was obtained as an oil (9.4 mg, 32%). MS (ES+): m/z 428 (M + H).
Supplementary Material
Figure 1.

Diversity reagents 2 {1–3}
Figure 3.

Diversity reagents 6{l-22}
Table 4.
Yield and purity data for chemset 8.
| Chemset 8 | Aminomethyl position | % Yield | LCMS Purity |
|---|---|---|---|
| {2, 18} | 2 | 42 | 81 |
| {3, 18} | 2 | 14 | 82 |
| {1, 19} | 3 | 23 | 88 |
| {2, 19} | 3 | 58 | 82 |
| {3, 19} | 3 | 12 | 93 |
| {1, 20} | 4 | 17 | 98 |
| {2, 20} | 4 | 10 | 100 |
| {3, 20} | 4 | 63 | 94 |
Acknowledgements
We thank Dr. Irina Protasevich for assistance with protein purification. Financial support was provided by the Department of Chemistry at UAB and by NIH (U01 AI056477 to Wayne J. Brouillette and U01 AI070386 to Christie G. Brouillette).
Footnotes
Supporting Information Available: Proton spectra of selected library members. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Marty AM. History of the development and use of biological weapons. Clinics in Laboratory Medicine. 2001;21:421–434. [PubMed] [Google Scholar]; (b) Marty AM, Conran RM, Kortepeter MG. Recent challenges in infectious diseases. Biological pathogens as weapons and emerging endemic threats. Clinics in Laboratory Medicine. 2001;21:411–420. and references cited therein. [PubMed] [Google Scholar]; (c) Friedlander AM. Tackling anthrax. Nature. 2001;414:160–161. doi: 10.1038/35102660. [DOI] [PubMed] [Google Scholar]; (d) Cohen J. Bioterrorism: blocking smallpox: a second defense. Science. 2001;294:500. doi: 10.1126/science.294.5542.500. [DOI] [PubMed] [Google Scholar]
- 2.(a) Smiley ST. Cell-mediated defense against Yersinia pestis infection. Adv. Exp. Med. Biol. 2007;603:376. doi: 10.1007/978-0-387-72124-8_35. [DOI] [PubMed] [Google Scholar]; (b) Santic M, Molmeret M, Klose KE, Abu Kwaik Y. Francisella tularensis travels a novel, twisted road within macrophages. Trends Microbiol. 2006;14:37. doi: 10.1016/j.tim.2005.11.008. [DOI] [PubMed] [Google Scholar]; (c) Knight J. Bioweapons: delivering death in the mail. Nature. 2001;414:837–838. doi: 10.1038/414837a. [DOI] [PubMed] [Google Scholar]
- 3.Walsh CT, Wright GD. Introduction: antibiotic resistance. Chem. Rev. 2005;105:391–394. doi: 10.1021/cr030100y. [DOI] [PubMed] [Google Scholar]
- 4.(a) Velu SE, Cristofoli W, Garcia GJ, Brouillette CG, Pierson M, Luan C-H, DeLucas LJ, Brouillette WJ. Tethered dimers as NAD synthetase inhibitors with antibacterial activity. J. Med. Chem. 2003;46:3371–3381. doi: 10.1021/jm030003x. [DOI] [PubMed] [Google Scholar]; (b) Velu SE, Luan C-H, DeLucas LJ, Brouillette CG, Brouillette WJ. Tethered dimer inhibitors of NAD synthetase: parallel synthesis of an aryl-substituted SAR library. J. Comb. Chem. 2005;7:898–904. doi: 10.1021/cc050063j. [DOI] [PubMed] [Google Scholar]; (c) Velu SE, Mou L, Luan C-H, DeLucas LJ, Brouillette CG, Brouillette WJ. Antibacterial nicotinamide adenine dinucleotide synthetase inhibitors: amide- and ether-linked tethered dimers with alpha-amino acid end groups. J. Med. Chem. 2007;50:2612–2621. doi: 10.1021/jm061349l. [DOI] [PubMed] [Google Scholar]
- 5.(a) Brouillette WJ, Muccio D, Jedrzejas MJ, Brouillette CG, Devedjiev Y, Cristofoli W, DeLucas LJ, Garcia GJ, Schmitt L, Velu SE. U.S. Patent 6,500,852 B1. 2002; (b) Brouillette WJ, Brouillette CG, DeLucas LJ. U.S. Patent 6,73,827. 2004; (c) Brouillette WJ, Muccio D, Jedrzejas MJ, Brouillette CG, Devedjiev Y, Cristofoli W, DeLucas LJ, Garcia GJ, Schmitt L, Velu SE. U.S. Patent 6,727,237. 2004; (d) Brouillette WJ, DeLucas LJ, Brouillette CG, Velu SE, Kim Y-C, Mou L, Porter S. U.S. Patent 6,861,448. 2005
- 6.Moro WB. Ph.D. Dissertation. Birmingham: University of Alabama; 2007. Chapter 1, Antibacterial tethered dimer inhibitors of NAD synthetase: SAR for the ammonium end group, in The Design and Synthesis of Antibacterial Inhibitors of NAD Synthetase; pp. 23–92. [Google Scholar]
- 7.Zalkin H. The amidotrasferases. Adv. Enzymol. Relat. Areas Mol. Biol. 1993;66:203–309. doi: 10.1002/9780470123126.ch5. [DOI] [PubMed] [Google Scholar]
- 8.Sutherland S. New antibiotics for anthrax? Drug Discovery Today. 2003;8:335–336. doi: 10.1016/s1359-6446(03)02665-5. [DOI] [PubMed] [Google Scholar]
- 9.Nessi C, Albertini AM, Speranza ML, Galizzi A. The outB gene of Bacillus subtilis codes for NAD synthetase. J. Biol. Chem. 1995;270:6181–6185. doi: 10.1074/jbc.270.11.6181. [DOI] [PubMed] [Google Scholar]
- 10.Moro WB. Ph.D. Dissertation. Birmingham: University of Alabama; 2007. Chapter 2, Virtual screening to identify new lead inhibitors of NAD synthetase, in The Design and Synthesis of Antibacterial Inhibitors of NAD Synthetase; pp. 93–122. [Google Scholar]
- 11.Moro WB, Yang W, Kane T, Brouillette CG, Brouillette WJ. Virtual Screening to Identify Lead Inhibitors for Bacterial NAD Synthetase (NADs) Bioorg. Med. Chem. Lett. 2009 doi: 10.1016/j.bmcl.2009.02.034. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lewis FD, Delos Santos GB, Liu W. Convergent synthesis of nonsymmetric π-stacked protophanes assembled with urea linkers. J. Org. Chem. 2005;70:2974–2979. doi: 10.1021/jo047889c. [DOI] [PubMed] [Google Scholar]
- 13.Natarajan A, Guo Y, Harbinski F, Fan Y-H, Chen H, Luus L, Diercks J, Aktas H, Chorev M, Halperin JA. Novel arylsulfoanilide-oxindole hybrid as an anticancer agent that inhibits translation initiation. J. Med. Chem. 2004;47:4979–4982. doi: 10.1021/jm0496234. [DOI] [PubMed] [Google Scholar]
- 14.Brown HC, Subba Rao BC. Hydroboration. III. The reduction of organic compounds by diborane, an acid-type reducing agent. J. Amer. Chem. Soc. 1960;82:681–686. [Google Scholar]
- 15.Van Meter CT, Lowy A. p-(p-Aminophenyl)-benzenesulfonamide and derivatives. II. J. Amer. Chem. Soc. 1941;63:1330–1331. [Google Scholar]
- 16.(a) Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria That Grow Aerobically; Approved Standard. The Clinical and Laboratory Standards Institute (CLSI, formerly NCCLS) (7th ed.) 2006 January;26(2) M7–A7. [Google Scholar]; (b) Performance Standards for Antimicrobial Susceptibility Testing; Eighteenth Informational Supplement. The Clinical and Laboratory Standards Institute (CLSI, formerly NCCLS) 2008 January;28(1) M100–S18, replaces 27 (1), M100-S117. [Google Scholar]
- 17.Lu S, Smith CD, Yang Z, Pruett PS, Nagy L, McCombs D, Delucas LJ, Brouillette WJ, Brouillette CG. Structure of nicotinic acid mononucleotide adenylyltransferase from Bacillus anthracis. Acta Crystallographica, Sect F – Struct. Biol. Cryst. Commun. 2008;64:893–898. doi: 10.1107/S1744309108029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.