

Abstract
Existing microfluidic devices, e.g. parallel plate flow chambers, do not accurately depict the geometry of microvascular networks in vivo. We have developed a synthetic microvascular network (SMN) on a polydimethalsiloxane (PDMS) chip that can serve as an in vitro model of the bifurcations, tortuosities, and cross-sectional changes found in microvascular networks in vivo. Microvascular networks from a cremaster muscle were mapped using a modified Geographical Information System, and then used to manufacture the SMNs on a PDMS chip. The networks were cultured with bovine aortic endothelial cells (BAEC), which reached confluency 3-4 days after seeding. Propidium Iodide staining indicated viable and healthy cells showing normal behavior in these networks. Anti-ICAM-1 conjugated 2-μm microspheres adhered to BAEC cells activated with TNF-α in significantly larger numbers compared to control IgG conjugated microspheres. This preferential adherence suggests that cultured cells retain an intact cytokine response in the SMN. This microfluidic system can provide novel insight into characterization of drug delivery particles and dynamic flow conditions in microvascular networks.
Keywords: Microfluidic Device, Microvascular Network, Flow Chamber, Adhesion Molecules
Introduction
Microvascular networks are the primary site of oxygen and nutrient delivery and waste product removal in normal tissue. These networks are not simply a collection of random vessels connected together; rather they are organized according to function, with a high degree of interconnectivity, integration, and feedback (Gaehtgens 1992). Understanding flow conditions and interaction of cells/particles traveling in the blood with endothelial cells is critical in our attempts to develop a more comprehensive theory of blood flow in microvascular networks. For example, during the past two decades it has been realized that the vascular surface of the endothelium in diseased tissue is significantly altered. Recognition of these drastically different endothelial surfaces has led to the concept of endothelial cell adhesion molecule (ECAM) mediated targeted drug delivery (Prabhakarpandian et al. 2001; Sakhalkar et al. 2003). These strategies often aim to mimic leukocyte adhesion to the endothelium after upregulation of the inflammatory response, and have shown promise in selectively targeting therapeutic agents to infarcted myocardium (Scott et al. 2007), tumors (Pattillo et al. 2005), and other diseased tissues (Wilson et al. 2008; Fiehn et al. 2008; Sakhalkar et al. 2005). However, further improvement in these targeting schemes requires a better understanding of the dynamic flow conditions and cell-particle interaction physics in microvascular networks.
Microfluidic devices, such as parallel plate flow chambers, are commonly used to investigate cell-particle interaction under dynamic conditions. These devices can be used to study particle behavior under various flow conditions (Schaff et al. 2007; Smith et al. 2007; Kiani et al. 2002). In addition, temperature and particle convection/diffusion rates for a large range of Reynolds numbers can be controlled in these devices (Zhang and Haswell 2006; El Ali, Sorger, and Jensen 2006). Portability and low energy consumption allow experiments to be performed outside the normal laboratory settings (El Ali et al. 2006). Microfluidic devices not only provide a well controlled environment for the study of particle-cell interactions, but also have the potential to replace some of the laborious and expensive in vivo studies of microvascular blood flow.
However, traditional microfluidic devices, such as parallel plate, radial, or capillary flow chambers, do not accurately reproduce the tortousities, bifurcations, and cross-sectional changes of in vivo microvascular networks. In vivo local flow conditions and cell/particle interactions with the endothelium are strongly dependent on local vascular geometry and flow conditions in surrounding vessels (Caputo et al. 2007; Gaehtgens 1992). For example, the path (Decuzzi and Ferrari 2008), velocity, and number of adherent particles (Mott and Helmke 2007) in a microvascular network is determined in part by its topology. Furthermore, traditional microfluidic devices only mimic flow in larger vessels, and not small post-capillary venules which are the major focal points of leukocyte-endothelial interaction (Springer 1994). A synthetic microfluidic device that accurately reproduces in vivo microvascular geometries and cellular morphology would provide a more realistic model of in vivo flow conditions in capillary networks.
Several investigators have recently developed more accurate microfluidic devices using either a better representation of cross-sectional areas in vessels (Camp, Stokol, and Shuler 2007; Frame and Sarelius 1995; Cokelet et al. 1993) or computer generated ideal geometries that roughly approximate microvascular networks (Shin et al. 2004). Previously, we have shown that antibody-coated microspheres adhere in a non-uniform pattern on protein coated synthetic microvascular networks (SMN) similar to that seen in microvascular networks in vivo(Prabhakarpandian et al. 2008). In the present study, we have developed a physiologically realistic endothelial cell coated synthetic microvascular network system. The endothelial cell lining in these networks was shown to possess normal physiological characteristics observed in vivo.
Materials and Methods
Antibodies and reagents
A mouse monoclonal anti-ICAM-1 (cat. No. NB500-318) which cross-reacts with bovine was purchased from Novus Biologicals. Human IgG1 Kappa (cat. No. PHP010) purified from myeloma serum was obtained from Serotec, MorphoSys (Raleigh, NC). Human fibronectin was obtained from BD Biosciences (San Jose, CA). Recombinant Human TNF-α was purchased from BioVision (Mountain View, CA). Protein A was purchased from Zymed (San Fransisco, CA). Fluorescent 2-μm microspheres (red: excitation 543 nm, emission 612 nm; blue: excitation 412 nm, emission 473 nm) were purchased from Duke Scientific (Palo Alto, CA). Bovine aortic endothelial cells (BAEC), EBM cell media, and EGM SingleQuots media supplements were purchased from Lonza (Allendale, NJ).
Creating synthetic microvascular networks
The microvascular network of a hamster cremaster muscle was mapped using a modified Geographic Information Systems (GIS) approach (Roth and Kiani 1999). Polydimethylsiloxane (PDMS) microvascular networks were created using soft lithography techniques described previously (Sia and Whitesides 2003; Prabhakarpandian B, 2008). In brief, the digitized networks were printed at high resolution on Mylar film, which served as a photo mask for ultraviolet light for patterning a positive photoresist mold on top of a silicon wafer. Sylgard 184 PDMS was prepared according to manufacturer's instructions (Dow Corning, Midland, MI) and cast over the photoresist mold. A beveled 16-gauge needle was used to punch the inlets and outlets in the molded PDMS. The bonding surfaces of the PDMS and a pre-cleaned 1×3 inches glass slide (Fisher Scientific, Pittsburgh, PA) were treated with oxygen plasma (200 mTorr, 18W, 30 s) produced in a plasma cleaner (Harrick Scientific, Ithaca, NY). The assembly was heated at 75°C for 10 minutes on a hotplate to achieve a good seal between PDMS and glass. Tygon Microbore tubing with an outside diameter of 0.06 inch and inner diameter of 0.02 inch connected to a 22 gauge stainless steel needle served as the connecting port to the SMN. The devices were sterilized by autoclaving following bonding. This study was performed on networks with constant depths of either 50 or 100 μm.
Seeding of bovine aortic endothelial cells in synthetic microvascular networks
Prior to cell seeding, each SMN was flushed with sterile EBM cell culture medium to prepare the device for seeding, then perfused with fibronectin (50 μg/ml) and allowed to incubate at room temperature for 30 minutes. The SMN was flushed again with supplemented with EGM SingleQuots and 20% FBS to remove excess fibronectin. The fibronectin-coated SMN was kept in an incubator at 37 °C, 5% CO2, while BAEC in flasks were prepared for injection
Bovine aortic endothelial cells (BAEC) were cultured in treated 75 cm2 polystyrene flasks. Cells were maintained using endothelial cell basal medium (EBM) supplemented with EGM SingleQuots. The cultured BAEC were lifted from the flasks with trypsin and diluted to 107 cells/ml with EBM medium supplemented with EGM SingleQuots and 20% FBS. The SMN was seeded through an inlet via syringe using approximately 0.1 ml of the 107 cells/ml suspension. A Nikon Eclipse TE200 inverted microscope was used to observe cell injection to ensure an even spread of cells throughout the network. The seeded SMN was incubated at 37 °C and fresh cell medium was perfused through the SMN once per day, starting one day after seeding. To change the medium, room temperature EBM medium was perfused through single inlet of the SMN at a flow rate of 10 μl/min via a syringe pump.
Propidium iodide staining
Propidium iodide (PI) staining of live cells was used to show the viability of confluent BAECs in the SMNs. Viable cells with intact membrane exclude PI, whereas permeabolized cells stain positive with PI. Briefly, 4 days after seeding, live endothelial cells were stained with 1.5μg/ml of PI for 20 min. This was performed without washing to avoid loss of floating nonviable cells. For positive controls, endothelial cells were permeabolized with 80 % ethanol for 5 minutes before being stained with PI. PI positive cells were observed under a Nikon Eclipse TE200 inverted microscope equipped with UV fluorescence. Images of PI stained cells were taken using a Q Imaging Retiga 1300 (Surrey, BC, Canada) camera and Image-Pro Plus (Bethesda, MD) imaging software (Kelly et al. 2003).
Antibody conjugated microspheres
Antibody conjugated microspheres were prepared as described previously (Yuan et al. 2005). Briefly, 2 μm fluorescent polystyrene microspheres were washed with a sodium bicarbonate buffer and coated with protein A via passive adsorption and incubated overnight at room temperature. The microspheres were then washed and incubated in a blocking buffer (Hank's balanced saline solution supplemented with 1% bovine serum albumin) at room temperature. The microspheres were conjugated with a mAb solution of either anti-ICAM-1 (red microspheres) or IgG1 (blue microspheres) and stored at 4 °C until use. IgG conjugated microspheres served as a control for non-specific binding. Immediately before use, the microspheres were resuspended in EGM media supplemented with 20% FBS, and diluted to a concentration of 107 microspheres/ml.
Microsphere adhesion to upregulated endothelial cells
The SMN (80 to 90 percent confluent with BAEC) was perfused with EBM medium containing 10 U/ml TNF-α to activate the endothelial cell layer; non-activated SMNs were used as negative control. The SMN was incubated for either 4 or 24 hours. Equal numbers of anti-ICAM-1 and IgG conjugated microspheres were diluted together in EBM medium to 107 microspheres/ml and injected at a flow rate of 0.5 μl/min using a syringe pump and allowed to settle for 30 minutes in an incubator at 37 °C. EBM medium was perfused through the SMN to remove non-adhered microspheres.
Statistics
Data are presented as mean ± standard error of the mean (SEM). The level of anti-ICAM-1 conjugated microsphere adhesion compared to control IgG conjugated microsphere adhesion was tested using analysis of variance (ANOVA) for each time point. The difference between time points and the control was also tested using ANOVA. The differences were considered to be significant if P<0.05.
Results
Our GIS-based system (Roth and Kiani 1999) was used to map microvascular networks of the hamster cremaster muscle from a montage of fluorescent images (Figure 1A) which were then digitized (Figure 1B) and used as a geometric blueprint for fabrication of the PDMS synthetic microvascular networks (Figure 1C). Endothelial cells readily attached to the walls of the SMN within two hours after seeding. Figure 2 shows the progression of BAEC growth in the SMN towards confluency, peaking at four days after seeding. Cells grew as monolayers and exhibited a cobblestone morphology, elongated in the direction of fluid flow, typical of endothelial cells found in vivo (Levesque and Nerem 1985).
Figure 1.
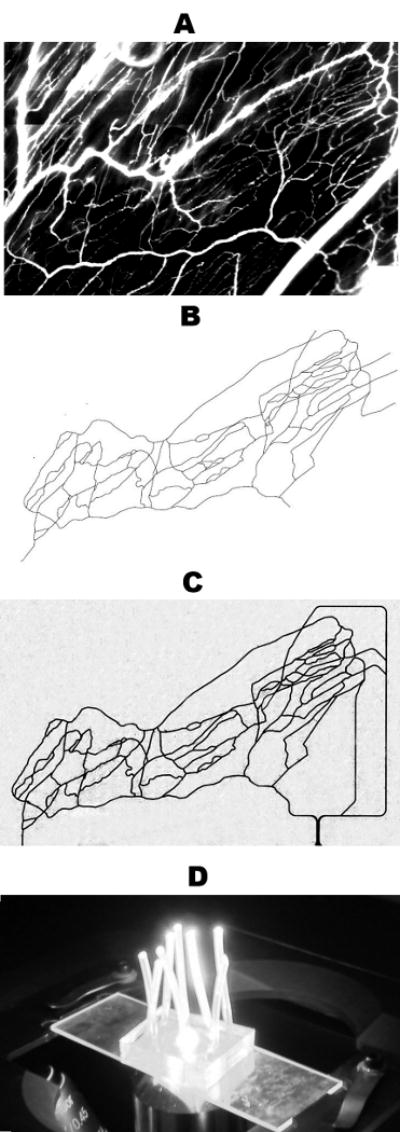
Panel A shows a montage of fluorescent images obtained in vivo from the cremaster muscle after I.V. injection of FITC-dextran. Our GIS-based system was used to digitize this microvascular network, panel B. This digitized map was then used to generate the synthetic microvascular network on PDMS, shown here perfused with trypan blue, panel C. The final “microvascular network on a chip” is shown in panel D.
Figure 2.
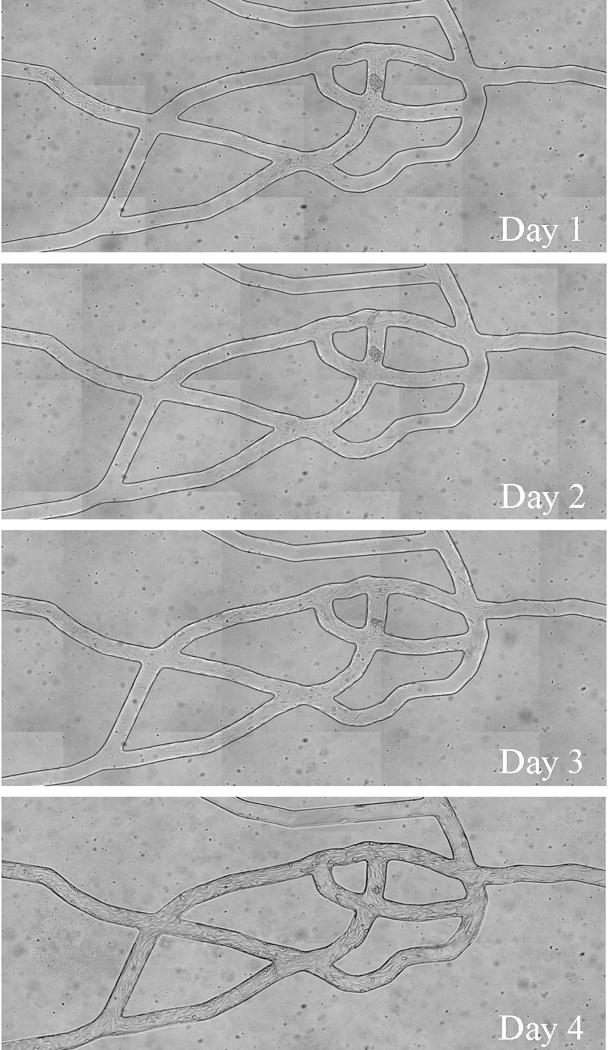
Bovine aortic endothelial cells seeded in the PDMS chambers became confluent throughout the network after 3 to 4 days. Cells exhibited the normal cobblestone configuration in the direction of flow.
No detectable cell death in BAECs cultured in the SMN was observed, as demonstrated by negative labeling of live endothelial cells with propidium iodine (PI) which labels membrane compromised cells. As shown in Figure 3A, confluent BAECs cultured in the SMN showed efficient exclusion of PI, indicating their viability at confuency. On the other hand, BAECs permeabolized with ethanol, used as positive controls, demonstrated PI fluorescence, see Figure 3B. The level of adhesion of anti-ICAM-1 conjugated microspheres to the endothelial lining of the SMNs was quantified after activation of BAECs with TNF-α and compared to the level of adhesion to activated cells in cell culture flasks. As shown in Figure 4, a significantly larger number of anti-ICAM-1 conjugated microspheres (shown in red) adhered to activated endothelial cells as compared to IgG conjugated microspheres (green), indicating preferential adherence of anti-ICAM-1 microspheres. In agreement with our previous findings (Sakhalkar et al. 2003), we observed a basal level of adhesion of anti-ICAM-1 conjugated microspheres to non-activated endothelial cells (Figure 5). However, the adhesion patterns of these antibody-conjugated microspheres were not uniform throughout these SMNs similar to our previous observations on protein coated SMN (Prabhakarpandian et al. 2008). Furthermore, as shown in Figure 5, a significant increase in the number of anti-ICAM-1 conjugated microspheres adhering to activated endothelial cells at 4 and 24 hours after activation, compared to non-activated control cells, was observed (79% and 161% increase in microsphere adhesion to activated cells compared to non-activated cells, respectively). Endothelial cell adhesion molecules (ECAMs) of endothelial cells lining the SMN, upregulated by either 4 or 24 hours of incubation with TNF-α, demonstrated an adhesion molecule profile consistent with other studies both in vitro and in vivo (Mulligan et al. 1993; Gaehtgens 1992). In addition, the increase in the number of anti-ICAM-1 microsphere adhesion 24 hours after activation of endothelial cells is in general agreement with previous finding from our group and others (Kiani et al. 2002; Werner et al. 1998).
Figure 3.
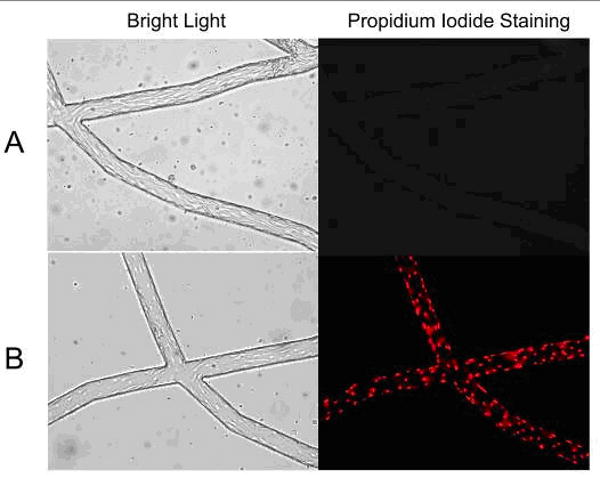
Endothelial cells cultured in the synthetic microvascular network were stained with propidium iodide (PI) to examine cell viability. A: The left panel demonstrates representative phase image of endothelial cells cultured in the SMN. Live endothelial cells cultured in the SMN were stained with PI at confluency. The right panel illustrates exclusion of PI in these cells, demonstrating their viability. B: The left panel shows representative phase images of control endothelial cells following fixation with ethanol. The right panel represents uptake of PI in positive controls, following fixation/permeabolization of these cells with ethanol.
Figure 4.

Anti-ICAM-1 (red) and IgG1 (green) conjugated microspheres attached to activated bovine aortic endothelial cells in one region of the network. Anti-ICAM-1 conjugated microspheres show preferential adhesion to endothelial cells as compared to IgG conjugated microspheres.
Figure 5.
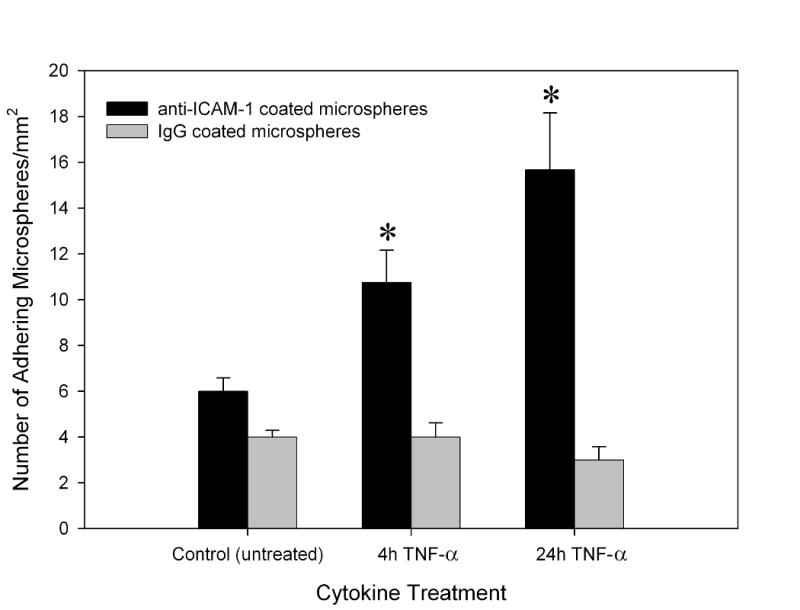
Anti-ICAM-1 conjugated microspheres were found to adhere preferentially to bovine aortic endothelial cells within the synthetic microvascular network (SMN) after 4 and 24 hour activation with TNF-α. Mean ± SEM; n=4 SMN per TNF-α treated groups and n=3 SMN for untreated control group, * P < 0.05 indicates significant differences in anti-ICAM-1 adhesion in SMN compared to the non-activated control group.
Discussion
We have developed a methodology for reproducing complete microvascular networks in vitro on a PDMS chip in a physiologically realistic environment comprising of cultured endothelial cells. Confluent endothelial cells were shown to be viable, healthy, and exhibited ICAM-1 upregulation in response to TNF-α treatment. Although there have been previous studies for culture of endothelial cells in linear microchannels (Frame and Sarelius 1995) and on generated networks (Shin et al. 2004; Fidkowski et al. 2005), this is the first study to successfully culture confluent layer of endothelial cells in an anatomically realistic microvascular network derived from in vivo images. In addition, the endothelial cells were found to orient themselves in the direction of flow as observed in vivo under physiological flow conditions.
The physiologically realistic in vitro microvascular network developed in this study can be used to study the effects of various stimuli, including cytokines (Sakhalkar et al. 2003) or ionizing radiation (Kiani et al. 2002), on microvascular networks. The quantification of level of adhesion of antibody-conjugated microspheres on activated BAEC in the SMN allows the use of this network to study both cell signaling and targeted drug delivery. For example, the upregulation of adhesion molecules following activation of endothelial cells lining the SMN can be used to study cellular interactions of leukocytes and platelets. Alternatively, a co-culture of endothelial cells with tumor cells in the SMN can be used as a model to specifically target tumor cells in a realistic scenario. In addition, non-uniform patterns of adhesion observed in the SMN allow the investigation of the influence of topological features on cell-particle interaction—a feature that cannot be observed using more traditional fluidic devices such as parallel plate flow chambers. Another key feature of the SMN is the ability of a single experiment to extract a shear based adhesion map (Prabhakarpandian, et al., 2008). Another advantage of this system, compared to traditional microfluidic devices and cells cultured in flasks, is the significant reduction of reagent costs due to the small volume of the SMN (Prabhakarpandian et al. 2008). Furthermore, PDMS has the advantage of optical transparency and rapid diffusion of oxygen and carbon dioxide (Whitesides 2006; El Ali et al. 2006) for long term cell culture.
In this study, the geometry of an in vivo microvascular network was regenerated on a PDMS chip. Although the device used in the current study had a rectangular cross-sectional area, we have made semi-circular channels for these networks in a previous study (Prabhakarpandian et al. 2008). Rectangular channels were used in this study primarily due to the technical difficulty of maintaining a semi-circular profile for depths greater than 30 μm. Also, most of the particle adhesion occurs in post-capillary venules, which normally have a diameter larger than 40 μm. Culturing cells in smaller diameter channels also tends to be more challenging due in part to the limited volume of cell media that is available to the cells at a given time. Although the SMNs used in this study had a uniform cross-sectional area throughout the networks, it is possible to produce a device with non-uniform cross-sectional areas and/or diameters. We have previously fabricated channels (data not shown) as small as 10 μm and as large as 200μm to encompass the microcirculation regime.
We are currently extending our studies in these SMNs to include the culture of different cell lines, such as fibroblasts, and co-cultures of endothelial and cancer cells which approximates the often incomplete lining of tumor vasculature with endothelial cells in vivo. BAECs were used in this study due to their low cost and ease of culture. However, in future studies, endothelial cell lines derived from the microvasculature may provide a more physiologically relevant model. We believe that the development of an automated methodology for culturing cells in SMNs will lead to the wider use and application of such devices. Recent studies have shown that the behavior of endothelial cells under shear is very much influenced by the composition of the extracellular matrix. The use of extracellular matrix, such as collagen or matrigel, will allow growth of cells around the inner diameter of the channels to better approximate the 3D in vivo situation. Furthermore, the inclusion of smooth muscles cells, which play a critical role in vivo through cross-talk with endothelial cells, will be addressed in future studies.
Acknowledgments
This work was supported by NIH (2R44HL076034-02) and the Pennsylvania Department of Health and the Nanotechnology Institute.
References
- Camp JP, Stokol T, Shuler ML. Biomed Microdevices. 2007;10(2) doi: 10.1007/s10544-007-9123-x. PMCID: 17891456. [DOI] [PubMed] [Google Scholar]
- Caputo KE, Lee D, King MR, Hammer DA. Biophys J. 2007;92(3) doi: 10.1529/biophysj.106.082321. PMCID: 17085490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokelet GR, Soave R, Pugh G, Rathbun L. Microvasc Res. 1993;46(3) doi: 10.1006/mvre.1993.1062. PMCID: 8121322. [DOI] [PubMed] [Google Scholar]
- Decuzzi P, Ferrari M. Biomaterials. 2008;29(3) doi: 10.1016/j.biomaterials.2007.09.025. PMCID: 17936897. [DOI] [PubMed] [Google Scholar]
- El Ali J, Sorger PK, Jensen KF. Nature. 2006;442(7101) doi: 10.1038/nature05063. PMCID: 16871208. [DOI] [PubMed] [Google Scholar]
- Fidkowski C, Kaazempur-Mofrad MR, Borenstein J, Vacanti JP, Langer R, Wang Y. Tissue Eng. 2005;11(1-2) doi: 10.1089/ten.2005.11.302. PMCID: 15738683. [DOI] [PubMed] [Google Scholar]
- Fiehn C, Kratz F, Sass G, Muller-Ladner U, Neumann E. Ann Rheum Dis. 2008;67(8) doi: 10.1136/ard.2007.086843. PMCID: 18408252. [DOI] [PubMed] [Google Scholar]
- Frame MD, Sarelius IH. Microcirculation. 1995;2(4) doi: 10.3109/10739689509148282. PMCID: 8714819. [DOI] [PubMed] [Google Scholar]
- Gaehtgens P. Int J Microcirc Clin Exp. 1992;11(2) PMCID: 1612826. [PubMed] [Google Scholar]
- Kelly KJ, Sandoval RM, Dunn KW, Molitoris BA, Dagher PC. Am J Physiol Cell Physiol. 2003;284(2) doi: 10.1152/ajpcell.00353.2002. PMCID: 12676658. [DOI] [PubMed] [Google Scholar]
- Kiani MF, Yuan H, Chen X, Smith L, Gaber MW, Goetz DJ. Pharm Res. 2002;19(9) doi: 10.1023/a:1020350708672. PMCID: 12403068. [DOI] [PubMed] [Google Scholar]
- Levesque MJ, Nerem RM. J Biomech Eng. 1985;107(4) doi: 10.1115/1.3138567. PMCID: 4079361. [DOI] [PubMed] [Google Scholar]
- Mott RE, Helmke BP. Am J Physiol Cell Physiol. 2007;293(5) doi: 10.1152/ajpcell.00457.2006. PMCID: 17855768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan MS, Vaporciyan AA, Miyasaka M, Tamatani T, Ward PA. Am J Pathol. 1993;142(6) PMCID: 7685152. [PMC free article] [PubMed] [Google Scholar]
- Pattillo CB, Sari-Sarraf F, Nallamothu R, Moore BM, Wood GC, Kiani MF. Pharm Res. 2005;22(7) doi: 10.1007/s11095-005-5646-0. PMCID: 16028012. [DOI] [PubMed] [Google Scholar]
- Prabhakarpandian B, Goetz DJ, Swerlick RA, Chen X, Kiani MF. Microcirculation. 2001;8(5) doi: 10.1038/sj/mn/7800105. PMCID: 11687947. [DOI] [PubMed] [Google Scholar]
- Prabhakarpandian B, Pant K, Scott RC, Patillo CB, Irimia D, Kiani MF, Sundaram S. Biomed Microdevices. 2008;10(4) doi: 10.1007/s10544-008-9170-y. PMCID: 18327641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roth NM, Kiani MF. Ann Biomed Eng. 1999;27(1) doi: 10.1114/1.204. PMCID: 9916759. [DOI] [PubMed] [Google Scholar]
- Sakhalkar HS, Dalal MK, Salem AK, Ansari R, Fu J, Kiani MF, Kurjiaka DT, Hanes J, Shakesheff KM, Goetz DJ. Proc Natl Acad Sci U S A. 2003;100(26) doi: 10.1073/pnas.2631433100. PMCID: 14668435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakhalkar HS, Hanes J, Fu J, Benavides U, Malgor R, Borruso CL, Kohn LD, Kurjiaka DT, Goetz DJ. FASEB J. 2005;19(7) doi: 10.1096/fj.04-2668fje. PMCID: 15764649. [DOI] [PubMed] [Google Scholar]
- Schaff UY, Xing MM, Lin KK, Pan N, Jeon NL, Simon SI. Lab Chip. 2007;7(4) doi: 10.1039/b617915k. PMCID: 17389960. [DOI] [PubMed] [Google Scholar]
- Scott RC, Wang B, Nallamothu R, Pattillo CB, Perez-Liz G, Issekutz A, Valle LD, Wood GC, Kiani MF. Biotechnol Bioeng. 2007;96(4) doi: 10.1002/bit.21233. PMCID: 17051598. [DOI] [PubMed] [Google Scholar]
- Shin M, Matsuda K, Ishii O, Terai H, Kaazempur-Mofrad M, Borenstein J, Detmar M, Vacanti JP. Biomed Microdevices. 2004;6(4) doi: 10.1023/B:BMMD.0000048559.29932.27. PMCID: 15548874. [DOI] [PubMed] [Google Scholar]
- Sia SK, Whitesides GM. Electrophoresis. 2003;24(21) doi: 10.1002/elps.200305584. PMCID: 14613181. [DOI] [PubMed] [Google Scholar]
- Smith LA, Aranda-Espinoza H, Haun JB, Hammer DA. Biophys J. 2007;92(2) doi: 10.1529/biophysj.105.079418. PMCID: 17071667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Springer TA. Cell. 1994;76(2) doi: 10.1016/0092-8674(94)90337-9. PMCID: 7507411. [DOI] [PubMed] [Google Scholar]
- Werner A, Kloss CU, Walter J, Kreutzberg GW, Raivich G. J Neurocytol. 1998;27(4) doi: 10.1023/a:1006928830251. PMCID: 10640181. [DOI] [PubMed] [Google Scholar]
- Whitesides GM. Nature. 2006;442(7101) doi: 10.1038/nature05058. PMCID: 16871203. [DOI] [PubMed] [Google Scholar]
- Wilson B, Samanta MK, Santhi K, Kumar KP, Paramakrishnan N, Suresh B. T Eur J Pharm Biopharm. 2008;70(1) doi: 10.1016/j.ejpb.2008.03.009. PMCID: 18472255. [DOI] [PubMed] [Google Scholar]
- Yuan H, Goetz DJ, Gaber MW, Issekutz AC, Merchant TE, Kiani MF. Radiat Res. 2005;163(5) doi: 10.1667/rr3361. PMCID: 15850416. [DOI] [PubMed] [Google Scholar]
- Zhang X, Haswell SJ. Ernst Schering Found Symp Proc. 2006;3 doi: 10.1007/2789_2007_026. PMCID: 17695708. [DOI] [PubMed] [Google Scholar]