

Abstract
Background
The toll like receptor (TLR) family serves an important regulatory role in the innate immune system, and recent evidence has implicated TLR signaling in the pro-inflammatory response of a variety of endogenous and exogenous stimuli within the kidney. The role of TLR signaling in fibrotic renal injury; however, remains unknown.
Materials and Methods
C3H/HeJ TLR4 hyporesponsive mice (TLR4Lps-d) or WT controls (C3H/Heou/J) underwent either sham operation or 1 week of unilateral ureteral obstruction (UUO). The kidneys were harvested and tissues were analyzed for TLR4 expression (Western Blot; RTPCR), E-cadherin and α-SMA expression (Western Blot), fibroblast accumulation (fibroblast specific protein (FSP-1+) staining), renal fibrosis (collagen I RTPCR, total collagen assay, Masson's trichrome staining), cytokine gene expression (tumor necrosis factor-α (TNF-α) and transforming growth factor-beta1 (TGF-β1) RTPCR), and pSMAD2 and integrin α1 expression (Western Blot).
Results
Mice with intact TLR4 signaling demonstrate a significant increase in TLR4 expression, α-SMA expression, fibroblast accumulation, collagen deposition, and interstitial fibrosis, and a significant decrease in E-cadherin expression in response to UUO. TLR4 deficient mice; however, exhibit a significant reduction in obstruction-induced α-SMA expression, fibroblast accumulation, and renal fibrosis, with preservation of E-cadherin expression. TLR4's influence on fibroblast accumulation and renal fibrosis occurred independent of any alterations in TNF-α,TGF-β1, or pSMAD2 expression, but did involve alterations integrin α1 expression.
Conclusion
TLR4 appears to be a significant mediator of fibrotic renal injury. While TLR4 signaling is recognized as a critical component of the innate immune response, this is the first study to demonstrate a novel role for TLR4 in renal fibroblast accumulation and tubulointerstitial fibrosis.
INTRODUCTION
Upper tract renal obstruction resulting in renal dysfunction is a significant clinical problem in both adult and pediatric populations. Pathologic manifestions of renal injury include progressive tubular dilation, renal tubular cell apoptosis, and progressive tubulointerstitial fibrosis (1). Tubulointerstitial fibrosis is a complex pathophysiological process involving inflammatory cell infiltration, the release of local cytokines and growth factors, fibroblast proliferation, and an imbalance in extracellular matrix (ECM) synthesis, deposition, and degradation (2).
The most notable growth factor involved in renal fibrosis is transforming growth factor β1 (TGF-β1). Evidence indicates that TGF-β1 is a major regulator of fibrosis via stimulation of fibroblast proliferation (3–5), extracellular matrix synthesis (ie. collagen types I, III, and IV, proteoglycans, laminin, and fibronectin) (5–8), and epithelial mesenchymal transition (EMT) (9–11). TGF-β1's pro-fibrotic effects are mediated through Smad signaling. Following TGF-β1 binding to its receptor, the receptor becomes activated and allows the Smad family of proteins to interact with the receptor to become phosphorylated and multimerize into a transcription regulating complex that exerts TGF-β1's biological effects (12, 13). The Smad family of proteins can also be activated directly by other pro-fibrotic factors, independent of TGF-β1, to stimulate renal fibrosis (14).
While the importance of EMT remains controversial, growing evidence suggests that renal tubular epithelial cells (TECs) are capable of undergoing a phenotypic transformation into matrix-producing fibroblasts (EMT) in pathologic states (15–18), with a large proportion of interstitial fibroblasts originating from TECs during renal obstruction (19, 20). Epithelial cells that have undergone EMT are characterized by the loss of epithelial cell markers, such as E-cadherin, and de novo α-SMA expression (21). The transformed epithelial cells also begin to express FSP-1, a marker specific for EMT (17, 22). TGF-β1 can initiate and complete the entire course of EMT, suggesting that induction of EMT may be a major pathway by which TGF-β1 stimulates renal fibrosis (9–11), but other mediators, such as integrins are capable of driving EMT and renal fibrosis independent of TGF-β1 (23–26).
Toll like receptors (TLR) are a key component of the mammalian innate immune system (27). Originally discovered in Drosophila melanogaster as part of the dorsoventral polarity regulatory gene cassette, Toll-like receptors (TLRs) have been found to play a crucial role in antimicrobial and antifungal responses (28–30). In humans, TLRs are expressed on a wide range of cell types, including renal epithelial cells (31). Engagement of a TLR with its specific pathogen associated molecular pattern (PAMP) initiates an intracellular signaling cascade culminating in the release of cytokines and the expression of cell surface co-stimulatory molecules capable of activating T cells and initiating an adaptive immune response (32). While PAMPs were initially studied in the context of identifying bacterial, viral, parasitic, and fungal products, certain endogenous molecules are now understood to function as PAMPs, including necrotic cells, heat shock proteins, and elements of the extracellular matrix (33–35).
The TLR family includes ten identified mammalian receptors of which TLR2 and TLR4 have been best studied and defined (36). While TLR2 and TLR4 have been implicated in renal ischemia/reperfusion injury, allograft rejection, pyelonephritis, and immune complex glomerulonephritis, their role in fibrotic renal disease and obstructive renal injury remains unknown (32, 37–40). TLR4 is considered to be a critical component of the LPS receptor complex, but TLR4 can also bind a variety of other ligands, including the extracellular matrix breakdown products hyaluronan, heparan sulfate, and fibrinogen (35, 41, 42). Given the increased synthesis of extracellular matrix products during tubulointerstitial fibrosis, we hypothesized that TLR4 activation may contribute to fibrotic renal disease. Using an in vivo model of unilateral ureteral obstruction, we sought to determine: 1) the expression of renal TLR4 after one week of unilateral ureteral obstruction, 2) the effect of TLR4 on obstruction-induced renal fibroblast accumulation and activity, 3) the impact of TLR4 expression on obstruction-induced renal fibrosis and collagen deposition, and 4) the potential mechanisms of TLR4-mediated fibrosis.
MATERIALS AND METHODS
Animals, experimental groups and operative techniques
The animal protocol was reviewed and accepted by the Animal Care and Research Committee of the Indiana University School of Medicine. C3H/HeJ TLR4 hyporesponsive mice (TLR4Lps-d) and control C3H/HeOuJ (WT) mice were acclimated and maintained on a standard pellet diet for 1 week before initiation of the experiment. Mice were anesthetized using isoflurane inhalation. Following induction of anesthesia the left ureter was isolated and completely ligated via a midline laparotomy (5 animals per group). Sham treated animals underwent an identical surgical procedure without ureteral ligation (5 animals per group). At the completion of the experiment the animals were re-anesthetized, the left kidneys were removed and snap frozen in liquid nitrogen, and the animals were subsequently euthanized.
Tissue Homogenization
A portion of the renal cortex from each kidney was homogenized after the tissue samples had been diluted in 5 vol of homogenate buffer [10mM HEPES (pH 7.9), 10mM KCL, 0.1 mM EGTA, 1mM DTT, and 0.5 mM phenylmethanesulfonyl fluoride] using a vertishear tissue homogenizer. Renal homogenates were centrifuged at 3,000 g for 15 min a 4°C. The supernatants were subsequently stored at −80°C until testing could be performed.
Western Blot Analysis
The protein extracts from homogenized samples (50μg/lane) were electrophoresed on a Tris-glycine gel (Invitrogen, Carlsbad, CA) and transferred to a PVDF membrane. Immunoblotting was performed by incubating each membrane in 5% dry milk overnight at 4°C, followed by incubation with an anti-E-cadherin (1:200; overnight at 4°C, BD Biosciences, San Jose, CA), anti-α-SMA (1:500; overnight at 4°C, Sigma, St. Louis, MO), anti-TLR4 (1:200; overnight at 4°C, Santa Cruz Biotechnology, Santa Cruz, CA) antibody, anti-integrin α1 (1:200; overnight at 4°C Santa Cruz Biotechnology, Santa Cruz, CA) antibody, or anti-pSMAD2 (1:200; overnight at 4°C, Cell Signaling, Danvers, MA) antibody. After being washed three times in T-PBS, each membrane was incubated for 1 hour at RT with a peroxidase-conjugated secondary antibody (1:6000 for E-cadherin, 1:2000 for α-SMA, 1:1000 for TLR4; 1:5000 for integrin α1, and 1:4000 for pSMAD2). The membranes were then developed using enhanced chemiluminescence (Amersham Pharmacia Biotech, Piscataway, NJ). Equivalent protein loading for each lane was confirmed by stripping and re-blotting each membrane for GAPDH (1:20000 for 30 min at RT, secondary 1:20,000 for 30 min at RT; Biodesign International, Saco, ME). The analysis was repeated in triplicate to assure reproducibility of results. The membranes were developed using enhanced chemiluminescence (Amersham Pharmacia Biotech Inc, Piscataway, NJ), and the density of each band determined using NIH image analysis software and expressed as a percentage of GAPDH density.
Real time PCR for TLR4, Collagen 1, TNF-α, and TGF-β1
Total RNA was extracted from the renal cortical tissue by homogenization in Trizol (Gibco BRL, Gaithersburg, MD), then isolated by precipitation with chloroform and isopropanol. Total RNA (0.5 μg) was subjected to cDNA synthesis using iScript (Bio-Rad, Hercules, CA). cDNA from each sample was analyzed for TLR4 (Mm00445273_m1), Collagen 1 (Mm01302047_g1), TGF-β1 (Mm01178820_m1), and TNF-α (Mm00443258_m1) using a TaqMan gene expression assay (RT-PCR; Applied Biosystems, Foster City, CA). FAM Dye/MGB labeled probes for either mouse β-actin or GAPDH (Applied Biosystems, Foster City, CA) served as endogenous controls.
Fibroblast (FSP-1) Accumulation
Renal tissue sections were analyzed for the presence of fibroblasts utilizing an S100-A4 antibody (The S100-A4 antigen is also known as FSP-1) (17, 20, 43, 44). Transverse 4 μm renal tissue sections were deparaffinized and dehydrated with xylene and alcohol. Antigen was retrieved by incubating the cells with proteinase K for 20 min in an oven. The tissues were then blocked with 1% bovine serum albumin (BSA) and incubated with a polyclonal rabbit antibody directed against S100-A4 (FSP-1; 1:200, DakoCytomation, Carpinteria, CA) for one hour at 37°C. Sections were washed and incubated with a secondary antibody (goat anti-rabbit (1:50)) for 30 minutes. Peroxidase-stained sections were then developed with 3,3”-diaminobenzidine (DAB) and counterstained with hematoxylin (Sigma-Aldrich). Sections incubated without primary antibody exhibited no staining. Spindle shaped FSP-1 positive cells were counted in 10 high powered fields (X400) in a blinded fashion and averaged.
Masson's Trichrome Staining
Tissue sections were deparaffinized and rehydrated with alcohol. The slides were then washed in distilled water and stained in Weigert's iron hematoxylin working solution for 10 minutes. The slides were washed and stained in Biebrich scarlet-acid fuchsin solution for 15 minutes. The slides were then rewashed and differentiated in phosphomolybdic-phosphotungstic acid solution for 15 minutes. The tissue sections were then transferred directly to aniline blue solution for 5–10 minutes, rinsed, and differentiated in 1% acetic acid solution for 2–5 minutes. The slides were dehydrated, cleared in xylene, and mounted.
Tissue Collagen
The total soluble collagen concentration within each renal cortical tissue sample was measured with the Sircol collagen assay kit (Accurate Chemical and Scientific) according to the manufacturer's protocol. Tissue samples were dissolved in 0.5 M acetic acid and heated for 120 min at 60°C. Tissue suspensions were centrifuged, supernatants collected, and the total collagen concentration measured at 540nm.
Statistical Analysis
Data are presented as means ± SD. Differences at the 95% confidence intervals were considered significant. The experimental groups were compared using ANOVA with post hoc Bonferroni-Dunn (JMP Statistical Software version 5.0, Berkeley, CA).
RESULTS
TLR4 expression
Renal cortical tissue samples were analyzed for TLR4 protein and gene expression. One week of unilateral ureteral obstruction induced a significant increase in TLR4 protein production in WT mice as compared to sham treated animals (WT OB = 0.85 ± 0.13 vs. WT Sham = 0.02 ± 0.008% GAPDH; P < 0.005; Figure 1), while TLR4 protein production remained at sham levels in TLR4Lps-d mice exposed to 1 week of UUO. Similarly, mice with intact TLR4 signaling demonstrated a significant increase in TLR4 mRNA expression in response to UUO (WT OB = 0.96 ± 0.08 vs. WT Sham = 0.25 ± 0.06, P < 0.005). TLR4 deficient mice demonstrated a marked reduction in obstruction-induced TLR4 mRNA expression as compared to WT mice (0.52 ± 0.03; P <0.005), but mRNA levels were significantly higher than that observed in TLR4 deficient shams (0.21 ± 0.03, P <0.005).
Figure 1. TLR4 production and quantitative mRNA expression following UUO.
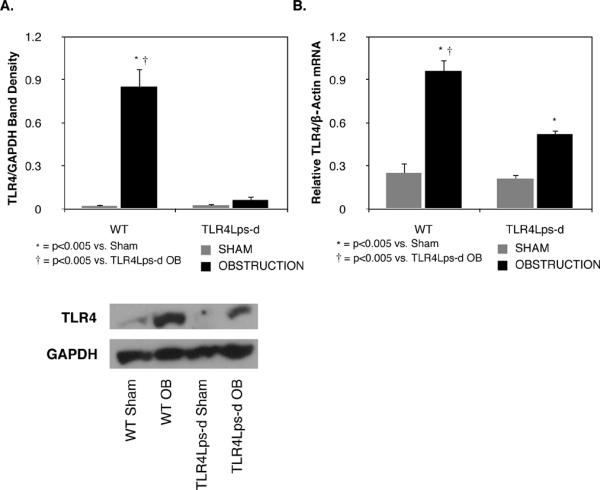
A. Gel photograph and densitometric analysis of TLR4 expression represented as a percentage of GAPDH in WT and TLR4 deficient (TLR4Lps-d) animals exposed to sham operation (WT Sham; TLR4Lps-d Sham) or 1 week of UUO (WT OB; TLR4Lps-d OB). B. Quantitative TLR4mRNA expression relative to GAPDH mRNA.
α-SMA and E-cadherin expression
In order to assess TLR4's role in obstruction-induced myofibroblast activity, renal cortical tissue was subjected to Western blot analysis for α-SMA and E-cadherin. A significant increase in α-SMA expression was detected in WT mice subjected to UUO as compared to sham-operated animals (WT OB = 0.65 ± 0.05 vs. WT Sham = 0.007 ± 0.007% GAPDH, P < 0.0001, Figure 2). TLR4 deficient mice subjected to the same degree of obstruction; however, demonstrated a significant reduction in α-SMA expression (TLR4Lps-d OB = 0.35 ± 0.04% GAPDH, P <0.005). A marked reduction in E-cadherin expression was simultaneously detected in WT mice subjected to UUO (WT OB = 0.03 ± 0.02 vs. WT Sham = 1.3 ± 0.05%GAPDH, P < 0.0001); however, no significant reduction in E-cadherin expression was detected in obstructed TLR4Lps-d mice (TLR4Lps-d OB = 0.93 ± 0.26 vs. TLR4Lps-d Sham = 1.05 ± 0.13% GAPDH). These findings demonstrate that TLR4 signaling contributes to myofibroblast activity, and suggest that the process of epithelial-mesenchymal transition may be involved.
Figure 2. Renal cortical α-SMA and E-cadherin expression following UUO.

A. Gel photograph depicting E-cadherin, α-SMA, and GAPDH expression in WT and TLR4 deficient animals exposed to sham operation (WT sham; TLR4Lps-d sham) or 1 week of UUO (WT OB; TLR4Lps-d OB). B. Densitometric analysis of E-cadherin and α-SMA expression represented as a percentage of GAPDH.
Fibroblast (FSP-1) accumulation
The role of TLR4 signaling in obstruction-induced fibroblast accumulation was further evaluated by staining for FSP-1+ cells in renal cortical tissue sections. While the number of FSP-1+ cells increased significantly in WT mice subjected to renal obstruction (Figure 3), a marked reduction in FSP-1+ cell accumulation was detected in TLR4 deficient mice subjected to the same degree of obstruction. These findings provide further evidence that TLR4 signaling contributes to fibroblast accumulation during renal obstruction.
Figure 3. Renal cortical Masson's trichrome and FSP+ fibroblast staining following UUO.
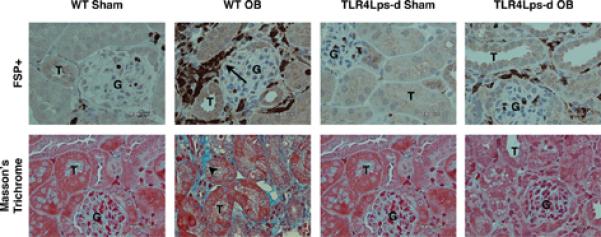
Photographs (magnification 400X) depicting renal cortical collagen deposition (blue stain; arrowheads) or FSP-1+ cells (brown stain; arrows) in WT and TLR4Lps-d mice exposed to sham operation (WT Sham; TLR4Lps-d Sham) or 1 week of UUO (WT OB; TLR4Lps-d OB). T = tubule; G = glomerulus.
Collagen expression and deposition
Renal cortical tissue samples were analyzed for real time collagen I mRNA expression. Low levels of collagen I mRNA were present in sham treated samples; however, collagen I expression significantly increased in WT mice and TLR4Lps-d mice exposed to 1 week of UUO (WT OB = 0.8 ± 0.04 vs. WT Sham = 0.09 ± 0.01, P<0.0001; TLR4Lps-d OB = 0.47 ± 0.03 vs. TLR4Lps-d Sham = 0.04 ± 0.003, P<0.0001, Figure 4A). Obstructed TLR4 deficient mice; however, exhibited a significant reduction in collagen I mRNA levels as compared to mice with intact TLR4 signaling.
Figure 4. Renal cortical collagen 1 mRNA expression and total collagen content following UUO.

A. Quantitative collagen 1 mRNA expression relative to GAPDH mRNA in WT and TLR4 deficient (TLR4Lps-d) animals exposed to sham operation (WT Sham; TLR4Lps-d Sham) or 1 week of UUO (WT OB; TLR4Lps-d OB). B. Total soluble collagen concentration in renal cortical specimens from WT and TLR4 deficient (TLR4Lps-d) animals exposed to sham operation or 1 week of UUO.
Masson's trichrome staining corroborated the collagen PCR findings. While sham-operated animals exhibited little collagen deposition in renal cortical tissue sections, a significant increase in collagen deposition was evident in the WT obstructed kidneys (Figure 3). The TLR4Lps-d samples exhibited some increase in collagen staining in response to obstruction; however, it was significantly less than that observed in the WT obstructed samples.
Total collagen content
The total collagen concentration of renal cortical tissue sections was measured as a quantitative assessment of tubulointerstitial fibrosis. Tissue collagen levels were significantly higher in WT animals exposed to 1 week of UUO as compared to WT Shams (WT OB = 45 ± 2.5 vs. WT Sham = 20.5 ± 1 μg/ml, P < 0.005, Figure 4B). In contrast, TLR4 deficient animals exhibited no increase in tissue collagen levels in response to obstruction (TLR4Lps-d OB = 24 ± 2.3 vs. TLR4Lps-d Sham = 22 ± 3.6 μg/ml).
TNF-α and TGF-β1 expression
In order to evaluate a potential mechanism of TLR4-mediated renal fibrosis, the real time gene expression of the pro-fibrotic cytokines, TNF-α and TGF-β1, was evaluated. TNF-α and TGF-β1 mRNA expression was significantly upregulated in response to obstruction in mice with both intact and deficient TLR4 signaling. (TNF: WT OB = 0.48 ± 0.05 vs. WT Sham = 0.020 ± 0.002, P < 0.0001; TLR4Lps-d OB = 0.55 ± 0.08 vs. TLR4Lps-d Sham = 0.02 ± 0.001, P < 0.0001; TGF: WT OB = 0.67 ± 0.04 vs. WT Sham = 0.2 ± 0.04, P < 0.001, TLR4Lps-d OB = 0.71 ± 0.03 vs. TLR4Lps-d Sham = 0.23 ± 0.06, P <0.001, Figure 5). Surprisingly, no significant difference in TNF-α or TGF-β1 gene expression was detected in WT or TLR4 deficient mice exposed to renal obstruction.
Figure 5. Quantitative renal cortical TNF-α and TGF-β1 mRNA expression following UUO.
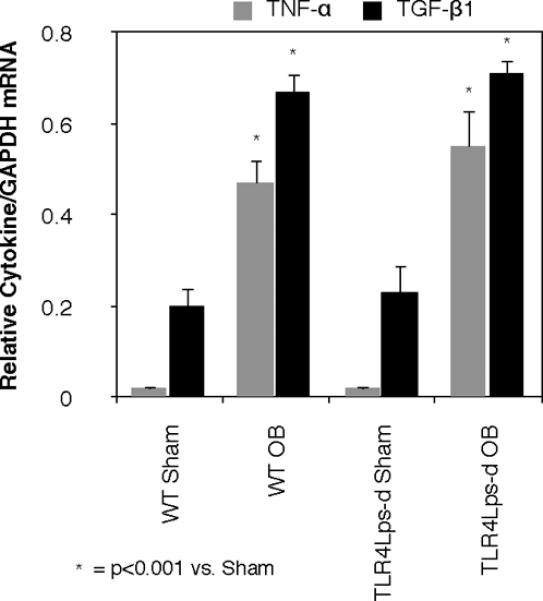
Quantitative TNF-α and TGF-β mRNA expression relative to GAPDH mRNA in WT and TLR4 deficient (TLR4Lps-d) animals exposed to sham operation (WT Sham; TLR4Lps-d Sham) or 1 week of UUO (WT OB; TLR4Lps-d OB).
pSmad2 and Integrin α1 expression
In order to further evaluate the TGF-β1 signaling pathway as a potential mechanism of TLR4-mediated renal fibrosis, the downstream Smad family member, Smad2, was also evaluated. Active phosphorylated Smad2 expression was significantly increased in response to obstruction in mice with both intact and deficient TLR4 signaling (WT OB = 0.80 ± 0.1 vs. WT Sham = 0.11 ± 0.05%GAPDH, P < 0.005; TLR4Lps-d OB = 0.59 ± 0.08 vs. TLR4Lps-d Sham = 0.15 ± 0.1%GAPDH, P < 0.05, Figure 6A), and no significant difference in pSmad2 expression was detected in WT or TLR4 deficient mice exposed to renal obstruction.
Figure 6. Renal cortical pSmad2 and Integrin α1 expression following UUO.
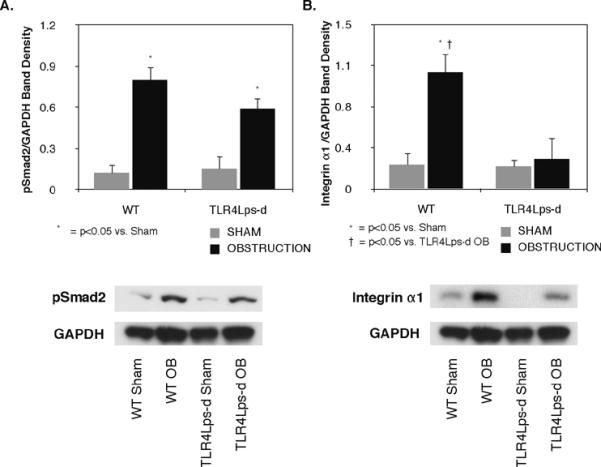
A. Gel photograph and densitometric analysis of pSmad2 expression represented as a percentage of GAPDH in WT and TLR4 deficient animals exposed to sham operation (WT sham; TLR4Lps-d sham) or 1 week of UUO (WT OB; TLR4Lps-d OB). B. Gel photograph and densitometric analysis of integrin α1 expression represented as a percentage of GAPDH in WT and TLR4 deficient animals exposed to sham operation or 1 week of UUO.
Integrin α1 was evaluated as an alternative mechanism of TLR4-mediated renal fibrosis, and possibly, EMT. Integrin α1 expression was significantly higher in WT animals exposed to 1 week of UUO as compared to WT Shams (WT OB = 1.06 ± 0.2 vs. WT Sham = 0.23 ± 0.1%GAPDH, P < 0.05, Figure 6A). In contrast, TLR4 deficient animals exhibited no increase in integrin α1 expression in response to obstruction (TLR4Lps-d OB = 0.27 ± 0.2 vs. TLR4Lps-d Sham = 0.2 ± 0.07%GAPDH).
DISCUSSION
Prolonged upper tract renal obstruction results in permanent renal dysfunction as a consequence of progressive tubulointerstitial fibrosis and apoptotic cell death. TGF-β1 is believed to be the primary mediator of obstruction-induced renal fibrosis, stimulating fibroblast activity, EMT, and increased extracellular matrix deposition (5, 7). It is apparent; however, that other signaling pathways are involved in obstruction-induced fibrosis. Toll-like receptors have an essential role in the innate immune system and are expressed in a wide variety of cells, including renal epithelial cells (31). Since TLR4 has been shown to bind the extracellular matrix breakdown products hyaluronan, heparan sulfate, and fibrinogen (35, 41, 42), we hypothesized that TLR4 activation may contribute to fibrotic renal disease. This is the first study to demonstrate that TLR4 contributes to obstruction-induced fibroblast accumulation and renal fibrosis independent of downstream TGF-β1 and TNF-α production.
TLR4 expression and mRNA levels were evaluated in the renal cortices of WT mice exposed to sham operation or 1 week of UUO. Obstruction induced a dramatic increase in TLR4 protein expression and mRNA levels as compared to sham treatment animals. In contrast, the TLR4 deficient mice exhibited no increase in TLR4 protein production and a significant reduction in TLR4 mRNA levels in response to obstruction. These results demonstrate that obstruction induces TLR4 expression and suggest that a positive feedback loop may exist in which obstruction-induced TLR4 activation stimulates further increases in TLR4 expression.
In order to evaluate TLR4's role in obstruction-induced tubulointerstitial fibrosis, renal samples were analyzed for evidence of fibroblast accumulation and activity, collagen I gene expression, and renal cortical collagen deposition. One week of renal obstruction resulted in a significant increase in renal cortical α-SMA expression and FSP-1+ fibroblast accumulation in WT animals, while animals with deficient TLR4 signaling exhibited a dramatic inhibition of this process, with significantly reduced α-SMA expression and a marked decrease in the number of FSP-1+ fibroblasts in response to obstruction. This indicates that TLR4 signaling contributes to obstruction-induced fibroblast accumulation and activity. Interestingly, a reduction in E-cadherin expression was also observed in WT animals subjected to renal obstruction; however, the expression of E-cadherin was preserved in animals with deficient TLR4 signaling. In conjunction with our findings that α-SMA expression and FSP-1+ cell accumulation were significantly reduced in TLR4 deficient animals, these results suggest a possible role for TLR4 signaling in obstruction-induced epithelial mesenchymal transition.
Renal samples were subsequently analyzed for collagen I gene expression and renal cortical collagen deposition. Our results demonstrate a significant reduction in obstruction-induced collagen I mRNA expression in mice with deficient TLR4 signaling. Furthermore, while total collagen content was over 2 times higher in WT obstructed kidneys as compared to shams, total collagen content remained at sham treatment levels in TLR4 deficient kidneys exposed to the same degree of obstruction. Masson's trichrome staining corroborated these observations, demonstrating a marked reduction in obstruction-induced collagen deposition in mice with deficient TLR4 signaling. Based on these observations, TLR4 signaling appears to have an important role in the pathogenesis of obstruction-induced tubulointerstitial fibrosis.
In order to evaluate the mechanisms of TLR4's pro-fibrotic effect, we investigated the expression of two pro-fibrotic cytokines, TNF-α and TGF-β1, during UUO in WT and TLR4 deficient mice. TNF-α is known to stimulate obstruction-induced renal fibrosis, ECM accumulation, and the upregulation of a number of cytokines and transcription factors involved in tubulointerstitial fibrosis, including TGF-β1 and NF-κB (45, 46). TGF-β1 is a major regulator of renal fibrosis via stimulation of fibroblast proliferation and extracellular matrix synthesis, and TGF-β1 is independently capable of inducing and completing the entire EMT process (3, 5, 7, 21, 47). While we did observe an increase in renal TGF-β1 and TNF-α mRNA levels in response to obstruction, no reduction in either TGF-β1 or TNF-α gene expression was detected in the TLR4 deficient animals, despite the demonstration of a significant reduction in collagen deposition and renal fibrosis in these animals. This finding suggests that TLR4 signaling may contribute to fibroblast accumulation and renal fibrosis independent of these two mediators. Alternatively, TLR4 may exert its pro-fibrotic effect downstream of TNF-α and TGF-β1 gene regulation.
In order to investigate the possibility of downstream signaling involvement in the TGF-β1 pathway, pSmad2 expression was evaluated. Again, while we did observe an increase in renal pSmad2 expression in response to obstruction, no reduction in pSmad2 expression was detected in the TLR4 deficient animals. While these results are not conclusive and will require further validation, the data suggests that the TGF-β1/Smad signaling pathway is not the primary mechanism of TLR4's pro-fibrotic effect. Integrin α1, a member of the collagen receptor family implicated in fibrosis and EMT, was subsequently evaluated as an alternative mechanism of TLR4-mediated renal injury. While Integrin α1 expression was significantly increased in response to obstruction in WT animals, integrin α1 expression was markedly reduced in TLR4 deficient animals exposed to the same degree of obstruction. Although these results are preliminary, they do suggest that the integrin family may be involved in TLR4-mediated renal fibrosis.
CONCLUSION
While TLR4 signaling is recognized as a critical component of the innate immune response, this is the first study to demonstrate a novel role for TLR4 signaling in obstruction-induced renal fibrogenesis. TLR4 expression is significantly increased during renal obstruction and induces fibroblast accumulation and fibrotic renal injury independent of alterations in TNF-α and TGF-β1 gene expression. As the mechanisms of this novel signaling pathway become more clearly defined, new therapeutic strategies to ameliorate renal fibrosis may be realized.
Key of Definitions for Abbreviations
- TLR
Toll Like Receptor
- TLR4Lps-d
TLR4 receptor hyporesponsive mice
- WT
Wild Type
- OB
Obstructed
- UUO
Unilateral Ureteral Obstruction
- PAMP
Pathogen Associated Molecular Pattern
- EMT
Epithelial Mesenchymal Transition
- TNF
Tumor Necrosis Factor
- TGF
Transforming Growth Factor
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- SMA
Smooth Muscle Actin
- FSP-1
Fibroblast Specific Protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Misseri R, Rink RC, Meldrum DR, Meldrum KK. Inflammatory mediators and growth factors in obstructive renal injury. J Surg Res. 2004;119:149–159. doi: 10.1016/j.jss.2004.02.016. [DOI] [PubMed] [Google Scholar]
- 2.Kaneto H, Morrissey J, Klahr S. Increased expression of TGF-beta 1 mRNA in the obstructed kidney of rats with unilateral ureteral ligation. Kidney Int. 1993;44:313–321. doi: 10.1038/ki.1993.246. [DOI] [PubMed] [Google Scholar]
- 3.Kuncio GS, Neilson EG, Haverty T. Mechanisms of tubulointerstitial fibrosis. Kidney Int. 1991;39:550–556. doi: 10.1038/ki.1991.63. [DOI] [PubMed] [Google Scholar]
- 4.Postlethwaite AE, Keski-Oja J, Moses HL, Kang AH. Stimulation of the chemotactic migration of human fibroblasts by transforming growth factor beta. J Exp Med. 1987;165:251–256. doi: 10.1084/jem.165.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roberts AB, McCune BK, Sporn MB. TGF-beta: regulation of extracellular matrix. Kidney Int. 1992;41:557–559. doi: 10.1038/ki.1992.81. [DOI] [PubMed] [Google Scholar]
- 6.Alvarez RJ, Sun MJ, Haverty TP, Iozzo RV, Myers JC, Neilson EG. Biosynthetic and proliferative characteristics of tubulointerstitial fibroblasts probed with paracrine cytokines. Kidney Int. 1992;41:14–23. doi: 10.1038/ki.1992.3. [DOI] [PubMed] [Google Scholar]
- 7.Border WA, Noble NA. Transforming growth factor beta in tissue fibrosis. N Engl J Med. 1994;331:1286–1292. doi: 10.1056/NEJM199411103311907. [DOI] [PubMed] [Google Scholar]
- 8.Miyajima A, Chen J, Lawrence C, Ledbetter S, Soslow RA, Stern J, Jha S, Pigato J, Lemer ML, Poppas DP, Vaughan ED, Felsen D. Antibody to transforming growth factor-beta ameliorates tubular apoptosis in unilateral ureteral obstruction. Kidney Int. 2000;58:2301–2313. doi: 10.1046/j.1523-1755.2000.00414.x. [DOI] [PubMed] [Google Scholar]
- 9.Xie L, Law BK, Chytil AM, Brown KA, Aakre ME, Moses HL. Activation of the Erk pathway is required for TGF-beta1-induced EMT in vitro. Neoplasia. 2004;6:603–610. doi: 10.1593/neo.04241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang J, Liu Y. Dissection of key events in tubular epithelial to myofibroblast transition and its implications in renal interstitial fibrosis. Am J Pathol. 2001;159:1465–1475. doi: 10.1016/S0002-9440(10)62533-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–968. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 12.Massague J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- 13.Schnaper HW, Hayashida T, Poncelet AC. It's a Smad world: regulation of TGF-beta signaling in the kidney. J Am Soc Nephrol. 2002;13:1126–1128. doi: 10.1681/ASN.V1341126. [DOI] [PubMed] [Google Scholar]
- 14.Wang W, Koka V, Lan HY. Transforming growth factor-beta and Smad signalling in kidney diseases. Nephrology (Carlton) 2005;10:48–56. doi: 10.1111/j.1440-1797.2005.00334.x. [DOI] [PubMed] [Google Scholar]
- 15.Healy E, Brady HR. Role of tubule epithelial cells in the pathogenesis of tubulointerstitial fibrosis induced by glomerular disease. Curr Opin Nephrol Hypertens. 1998;7:525–530. doi: 10.1097/00041552-199809000-00007. [DOI] [PubMed] [Google Scholar]
- 16.Strutz F, Muller GA, Neilson EG. Transdifferentiation: a new angle on renal fibrosis. Exp Nephrol. 1996;4:267–270. [PubMed] [Google Scholar]
- 17.Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, Neilson EG. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130:393–405. doi: 10.1083/jcb.130.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeisberg M, Bonner G, Maeshima Y, Colorado P, Muller GA, Strutz F, Kalluri R. Renal fibrosis: collagen composition and assembly regulates epithelialmesenchymal transdifferentiation. Am J Pathol. 2001;159:1313–1321. doi: 10.1016/S0002-9440(10)62518-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Iwano M, Neilson EG. Mechanisms of tubulointerstitial fibrosis. Curr Opin Nephrol Hypertens. 2004;13:279–284. doi: 10.1097/00041552-200405000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110:341–350. doi: 10.1172/JCI15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rastaldi MP. Epithelial-mesenchymal transition and its implications for the development of renal tubulointerstitial fibrosis. J Nephrol. 2006;19:407–412. [PubMed] [Google Scholar]
- 22.Rossini M, Cheunsuchon B, Donnert E, Ma LJ, Thomas JW, Neilson EG, Fogo AB. Immunolocalization of fibroblast growth factor-1 (FGF-1), its receptor (FGFR-1), and fibroblast-specific protein-1 (FSP-1) in inflammatory renal disease. Kidney Int. 2005;68:2621–2628. doi: 10.1111/j.1523-1755.2005.00734.x. [DOI] [PubMed] [Google Scholar]
- 23.Cosgrove D, Rodgers K, Meehan D, Miller C, Bovard K, Gilroy A, Gardner H, Kotelianski V, Gotwals P, Amatucci A, Kalluri R. Integrin alpha1beta1 and transforming growth factor-beta1 play distinct roles in alport glomerular pathogenesis and serve as dual targets for metabolic therapy. Am J Pathol. 2000;157:1649–1659. doi: 10.1016/s0002-9440(10)64802-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kagami S, Kondo S, Loster K, Reutter W, Kuhara T, Yasutomo K, Kuroda Y. Alpha1beta1 integrin-mediated collagen matrix remodeling by rat mesangial cells is differentially regulated by transforming growth factor-beta and platelet-derived growth factor-BB. J Am Soc Nephrol. 1999;10:779–789. doi: 10.1681/ASN.V104779. [DOI] [PubMed] [Google Scholar]
- 25.Norman JT, Fine LG. Progressive renal disease: fibroblasts, extracellular matrix, and integrins. Exp Nephrol. 1999;7:167–177. doi: 10.1159/000020597. [DOI] [PubMed] [Google Scholar]
- 26.Pozzi A, Wary KK, Giancotti FG, Gardner HA. Integrin alpha1beta1 mediates a unique collagen-dependent proliferation pathway in vivo. J Cell Biol. 1998;142:587–594. doi: 10.1083/jcb.142.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 28.Gay NJ, Keith FJ. Drosophila Toll and IL-1 receptor. Nature. 1991;351:355–356. doi: 10.1038/351355b0. [DOI] [PubMed] [Google Scholar]
- 29.Lemaitre B, Nicolas E, Michaut L, Reichhart JM, Hoffmann JA. The dorsoventral regulatory gene cassette spatzle/Toll/cactus controls the potent antifungal response in Drosophila adults. Cell. 1996;86:973–983. doi: 10.1016/s0092-8674(00)80172-5. [DOI] [PubMed] [Google Scholar]
- 30.Rosetto M, Engstrom Y, Baldari CT, Telford JL, Hultmark D. Signals from the IL-1 receptor homolog, Toll, can activate an immune response in a Drosophila hemocyte cell line. Biochem Biophys Res Commun. 1995;209:111–116. doi: 10.1006/bbrc.1995.1477. [DOI] [PubMed] [Google Scholar]
- 31.Samuelsson P, Hang L, Wullt B, Irjala H, Svanborg C. Toll-like receptor 4 expression and cytokine responses in the human urinary tract mucosa. Infect Immun. 2004;72:3179–3186. doi: 10.1128/IAI.72.6.3179-3186.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gay NJ, Gangloff M. Structure and function of Toll receptors and their ligands. Annu Rev Biochem. 2007;76:141–165. doi: 10.1146/annurev.biochem.76.060305.151318. [DOI] [PubMed] [Google Scholar]
- 33.Li M, Carpio DF, Zheng Y, Bruzzo P, Singh V, Ouaaz F, Medzhitov RM, Beg AA. An essential role of the NF-kappa B/Toll-like receptor pathway in induction of inflammatory and tissue-repair gene expression by necrotic cells. J Immunol. 2001;166:7128–7135. doi: 10.4049/jimmunol.166.12.7128. [DOI] [PubMed] [Google Scholar]
- 34.Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- 35.Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Akira S. Toll-like receptor signaling. J Biol Chem. 2003;278:38105–38108. doi: 10.1074/jbc.R300028200. [DOI] [PubMed] [Google Scholar]
- 37.Cunningham PN, Wang Y, Guo R, He G, Quigg RJ. Role of Toll-like receptor 4 in endotoxin-induced acute renal failure. J Immunol. 2004;172:2629–2635. doi: 10.4049/jimmunol.172.4.2629. [DOI] [PubMed] [Google Scholar]
- 38.Deng JF, Geng L, Qian YG, Li H, Wang Y, Xie HY, Feng XW, Zheng SS. The role of toll-like receptors 2 and 4 in acute allograft rejection after liver transplantation. Transplant Proc. 2007;39:3222–3224. doi: 10.1016/j.transproceed.2007.02.102. [DOI] [PubMed] [Google Scholar]
- 39.Wolfs TG, Buurman WA, van Schadewijk A, de Vries B, Daemen MA, Hiemstra PS, van 't Veer C. In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-gamma and TNF-alpha mediated up-regulation during inflammation. J Immunol. 2002;168:1286–1293. doi: 10.4049/jimmunol.168.3.1286. [DOI] [PubMed] [Google Scholar]
- 40.Zhang B, Ramesh G, Uematsu S, Akira S, Reeves WB. TLR4 signaling mediates inflammation and tissue injury in nephrotoxicity. J Am Soc Nephrol. 2008;19:923–932. doi: 10.1681/ASN.2007090982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168:5233–5239. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- 42.Okamura Y, Watari M, Jerud ES, Young DW, Ishizaka ST, Rose J, Chow JC, Strauss JF., 3rd The extra domain A of fibronectin activates Toll-like receptor 4. J Biol Chem. 2001;276:10229–10233. doi: 10.1074/jbc.M100099200. [DOI] [PubMed] [Google Scholar]
- 43.Barraclough R. Calcium-binding protein S100A4 in health and disease. Biochim Biophys Acta. 1998;1448:190–199. doi: 10.1016/s0167-4889(98)00143-8. [DOI] [PubMed] [Google Scholar]
- 44.El Chaar M, Chen J, Seshan SV, Jha S, Richardson I, Ledbetter SR, Vaughan ED, Jr., Poppas DP, Felsen D. Effect of combination therapy with enalapril and the TGF-beta antagonist 1D11 in unilateral ureteral obstruction. Am J Physiol Renal Physiol. 2007;292:F1291–1301. doi: 10.1152/ajprenal.00327.2005. [DOI] [PubMed] [Google Scholar]
- 45.Guo G, Morrissey J, McCracken R, Tolley T, Liapis H, Klahr S. Contributions of angiotensin II and tumor necrosis factor-alpha to the development of renal fibrosis. Am J Physiol Renal Physiol. 2001;280:F777–785. doi: 10.1152/ajprenal.2001.280.5.F777. [DOI] [PubMed] [Google Scholar]
- 46.Meldrum KK, Misseri R, Metcalfe P, Dinarello CA, Hile KL, Meldrum DR. TNF-alpha neutralization ameliorates obstruction-induced renal fibrosis and dysfunction. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1456–146. doi: 10.1152/ajpregu.00620.2005. [DOI] [PubMed] [Google Scholar]
- 47.Fan JM, Ng YY, Hill PA, Nikolic-Paterson DJ, Mu W, Atkins RC, Lan HY. Transforming growth factor-beta regulates tubular epithelialmyofibroblast transdifferentiation in vitro. Kidney Int. 1999;56:1455–1467. doi: 10.1046/j.1523-1755.1999.00656.x. [DOI] [PubMed] [Google Scholar]