

Abstract
A novel single-walled carbon nanotube (SWNT)-based tumor-targeted drug delivery system (DDS) has been developed, which consists of a functionalized SWNT linked to tumor-targeting modules as well as prodrug modules. There are three key features of this nanoscale DDS: (a) use of functionalized SWNTs as a biocompatible platform for the delivery of therapeutic drugs or diagnostics, (b) conjugation of prodrug modules of an anticancer agent (taxoid with a cleavable linker) that is activated to its cytotoxic form inside the tumor cells upon internalization and in situ drug release, and (c) attachment of tumor-recognition modules (biotin and a spacer) to the nanotube surface. To prove the efficacy of this DDS, three fluorescent and fluorogenic molecular probes were designed, synthesized, characterized and subjected to the analysis of the receptor-mediated endocytosis and drug release inside the cancer cells (L1210FR leukemia cell line) by means of confocal fluorescence microscopy. The specificity and cytotoxicity of the conjugate have also been assessed and compared with L1210 and human non-cancerous cell lines. Then, it has unambiguously been proven that this tumor-targeting DDS works exactly as designed and shows high potency towards specific cancer cell lines, thereby forming a solid foundation for further development.
Introduction
Recently, nanomaterials have effectively been employed to deliver biologically active cargo into living systems for the purposes of disease diagnosis and therapy.1,2 Among diverse classes of nanomaterials, carbon nanotubes (CNTs) have attracted particular attention as carriers of biologically relevant molecules due to their unique physical, chemical and physiological properties.3-5 It has been shown, for example, that CNTs can serve as a highly efficient vehicle to transport a wide range of molecules across membranes into living cells.6-14 In addition, the intrinsic stability and structural flexibility of CNTs may prolong the circulation time as well as the bioavailability of drug molecules conjugated to CNTs.15-18 Radiolabeled functionalized SWNTs (f-SWNTs) have been found to exhibit a blood circulation half-life of 1-3 h, depending on the radiolabels used.17,19 For instance, [111In]-f-SWNTs exhibited a slightly longer blood circulation half-life as compared with [86Y]-f-SWNTs.19 When SWNTs were noncovalently wrapped with a linear polyethylene glycol (PEG) chain, the blood circulation time of the PEG-SWNT was prolonged with increasing molecular weight of PEG chain, e.g., from 1.2 h for 2 kDa PEG-wrapped SWNTs to 5 h for 5 kDa PEG-wrapped SWNTs.15 However, a further increase in molecular weight of linear PEG to 7 kDa and even to 12 kDa resulted in almost no effect on the blood circulation time. Nonetheless, when 7kDa branched PEG was used the blood circulation time dramatically increased up to 24 h.15 In a related study, covalently conjugated PEG-SWNTs showed a blood circulation time of 10 h.20 When targeting moieties, such as amino acids and antibodies, were conjugated to SWNTs, the blood circulation time was found to increase from roughly 24 h to days.16,21 On the other hand, when paclitaxel molecules were conjugated to SWNTs, the blood circulation time was found to decrease from ∼3 h to ∼1 h, presumably due to the hydrophobic nature of the bound drug molecules i.e., increased nonspecific protein absorption that would accelerate the uptake by macrophages in reticutoendothelial (RES) organs.22
A number of approaches to the functionalization of CNTs with biomolecules on their external surface have been reported for potential applications to drug delivery.6-14 As an example of their versatility, single-walled carbon nanotubes (SWNTs) noncovalently bound to proteins or genes mediated by phospholipids were internalized into cells through endocytosis.23-29 SWNTs can also be covalently functionalized with small molecules linked to the carboxylic acid sites localized at the ends and defect sites on the sidewall.10 Thus, amino acids,30 oligopeptides,31-33 genes,34-37 and antibiotics38 have been transported into different types of cells via appropriately functionalized SWNTs.39
More recently, multiple cytotoxic platinum(IV) complex units were conjugated to SWNTs for delivering those anticancer drugs to tumor cells, in a process mediated by phospholipids, wherein the active platinum(II) species (cis-platin) was released upon reduction at a low pH environment within the cancer cells.40 In addition, groups led by Prato, Bianco and Kostarelos41 as well as by Dai42 have independently conjugated CNTs with doxorubicin by means of π-π stacking interaction. The release of the doxorubicin was facilitated by the lower pH in lysosome. These CNT-doxorubicin conjugates have shown high cytotoxicity to cancer cells.
However, it would be much more desirable if SWNT-anticancer drug conjugates were equipped with tumor-targeting ligand units that could specifically recognize cancer-specific receptors on the cell surface and induce receptor-mediated endocytosis. Such a tumor-targeting strategy, exploiting cancer specific biomarkers and efficient internalization, would minimize systemic toxicity, and thereby, undesirable side effects typically associated with conventional chemotherapy.
Numbers of tumor-targeting drug conjugates have been investigated to date. Those drug conjugates consist of a cytotoxic drug warhead and a tumor-targeting moiety either directly linked or attached through a suitable linker.43,44 Such a drug conjugate should be stable and innocuous in blood circulation but should also be designed to activate its cytotoxic drug warhead by chemical or biochemical transformation inside cancer cells.44 The efficacy of a tumor-targeting drug conjugate, therefore, depends not only on its tumor-targeting specificity, but also on the efficiency of the cleavable linker needed to release an anticancer drug inside the cancer cells. Typical tumor-targeting molecules used in those drug conjugates include monoclonal antibodies, polyunsaturated fatty acids, polysaccharides (e.g. hyaluronic acid), peptides, and vitamins (e.g. folic acid or biotin).44 In one of these laboratories, tumor-targeting drug conjugates, comprising 2nd-generation taxoids (i.e., cytotoxic drug warheads) with docosahexaenoic acid (DHA) as well as monoclonal antibodies, have recently been successfully developed as promising drug candidates. These tumor-targeting drug conjugates have exhibited remarkable efficacy against human tumor xenografts in mouse models.45-47 In addition, highly efficient self-immolative disulfide linkers have also been designed an developed in one of these laboratories, which are stable in blood plasma but readily cleavable by intracellular thiols,43,48 e.g., glutathione, thioredoxin, glutaredoxin, etc., to release a highly cytotoxic anticancer agent inside the cancer cells. Various disulfide linkers have been extensively studied and successfully used for monoclonal antibody – drug conjugates as well as folate-drug conjugates.44,46,49-55 Although the detailed mechanism of drug release through cleavage of these disulfide linkers needs further investigation,56 high in vivo efficacy of those drug conjugates using disulfide linkers has been confirmed.
The aim of this study is to devise a novel SWNT-based drug delivery system (DDS) consisting of (a) a cytotoxic drug warhead with a strategically designed cleavable linker module that can be specifically activated inside tumor cells and (b) tumor-targeting modules on the SWNT that would greatly increase the uptake of the conjugate specifically by tumor cells. We have successfully developed functionalized SWNTs as versatile vehicles to selectively and efficaciously deliver anticancer drugs to tumor cells. This SWNT-based DDS possesses the following advantages: (a) multivalent ligand-receptor interactions on the tumor cell surface,57,58 (b) increased drug loading capacity, and (c) capability of controlled drug release inside tumor cells. Our DDS is a multi-functional, multi-component system, comprised of tumor-targeting modules (e.g., biotin59) and cytotoxic drug warheads (e.g. a second-generation taxoid) covalently connected to a SWNT scaffold to form a biotin-SWNT-linker-taxoid conjugate. The warhead-linker module consists of a disulfide-containing linker that is attached to the C2′ position of a taxoid molecule. This chemical modification is known to cause a substantial loss of potency of the taxoid, rendering the systemic toxicity of the SWNT-drug-conjugate practically negligible in blood circulation.45,47 However, upon internalization into cancer cells, the conjugate is readily cleaved in situ to release the active cytotoxic agent, i.e., free taxoid, which leads to tumor cell death.
Scheme 1 illustrates the three key steps involved in the nanotube-based DDS. First, the biotin-SWNT-linker-taxoid conjugate is internalized into the tumor cells through receptor-mediated endocytosis. We chose biotin as the tumor-targeting module based on the finding of Russel-Jones et al.59,60 that biotin (vitamin H or vitamin B7) receptors on a wide rage of tumor types would serve as newer tumor-specific target in a manner similar to the widely recognized folate receptors.61 The biotin moieties covalently attached to the ends and sidewall defect sites of the SWNT efficiently recognize the biotin receptors overexpressed on the tumor cell surfaces. The presence of multiple biotin moieties, localized at the ends and sidewall defect sites of the SWNT, would enhance the internalization of the conjugate via increased probability for receptor binding or via multivalent binding.58 Second, the active drug is released through cleavage of the disulfide bond in the linker moiety, connecting the taxoid to the SWNT. The disulfide bond is readily cleaved by endogenous thiols such as glutathione (GSH), thioredoxin or other intracellular thiols to generate a sulfhydryl group, which subsequently undergoes a thiolactonization process to form benzothiophen-2-one and regenerates a free taxoid in its active form. Concentrations of GSH are typically 1-2 μM in circulating human blood plasma, but are in the range of 2-8 mM in tumor tissue.62,63 Thus, the adventitious activation of the cytotoxic drug warhead should be minute at best in the blood circulation, whereas the activation process would be facile in the tumor cells.44,46,49,50,53 Third, the released taxoid binds to tubulins/microtubules, inhibiting cell mitosis at the G2/M stage by stabilizing microtubules, which triggers signaling to cause apoptosis.64,65
Scheme 1.
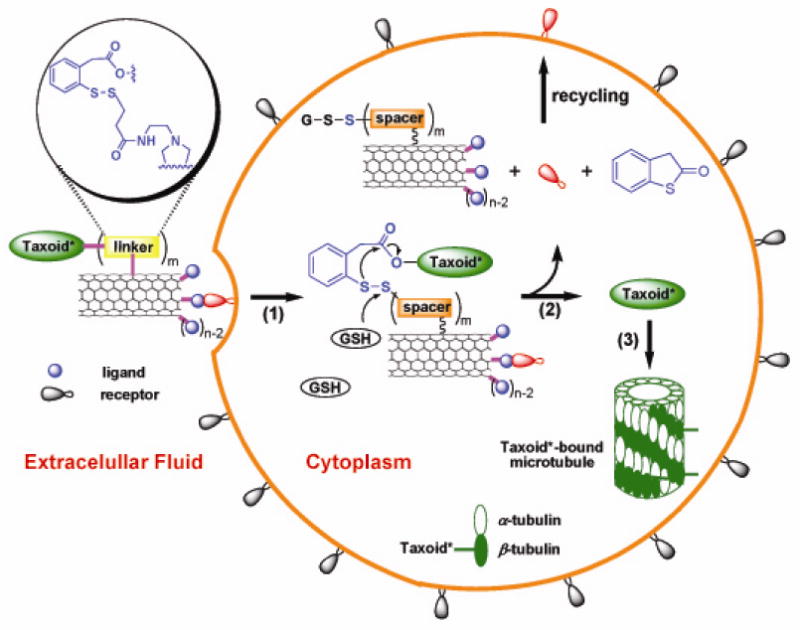
Schematic illustration of three key steps involved in the tumor-targeting drug delivery of biotin-SWNT-taxoid conjugate 3: (1) internalization of the whole conjugate via receptor-mediated endocytosis; (2) drug release through cleavage of the disulfide linker moiety by intracellular thiol, e.g., GSH; (3) binding of the free taxoid molecules to tubulins/microtubules, forming stabilized microtubules that block cell mitosis and trigger apoptosis. [Note: Since each taxoid molecule is fluorescently labeled with fluorescein, the internalized conjugates 3 in the cytoplasm and the taxoid-bound microtubules are fluorescent.]
Results and Discussion
Synthesis and Characterization
To evaluate the specificity and efficacy of the SWNT-based DDS, we designed and synthesized three fluorescently labeled SWNT-conjugates (1, 2, and 3), as shown in Schemes 2-4. Conjugates 1 and 2 were labeled with fluorescein isothiocyanate (FITC). The resulting fluorescent conjugates, SWNT-FITC (1) and biotin-SWNT-FITC (2), were used to track the internalization of SWNT and biotin-SWNT, respectively, into the tumor cells. Biotin-SWNT-linker-taxoid-fluorescein (3) was designed as a fluorescent molecular probe of the SWNT-based DDS for receptor-mediated endocytosis and intracellular drug release, as illustrated in Scheme 1.
Scheme 2.
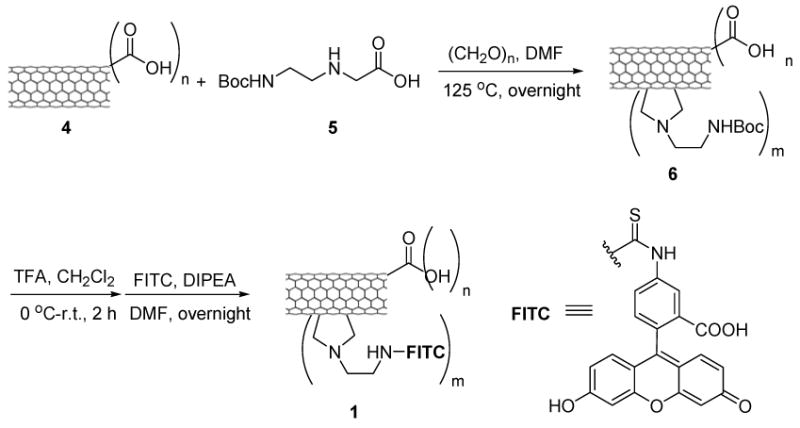
Synthesis of SWNT-FITC conjugate 1.
Scheme 4.
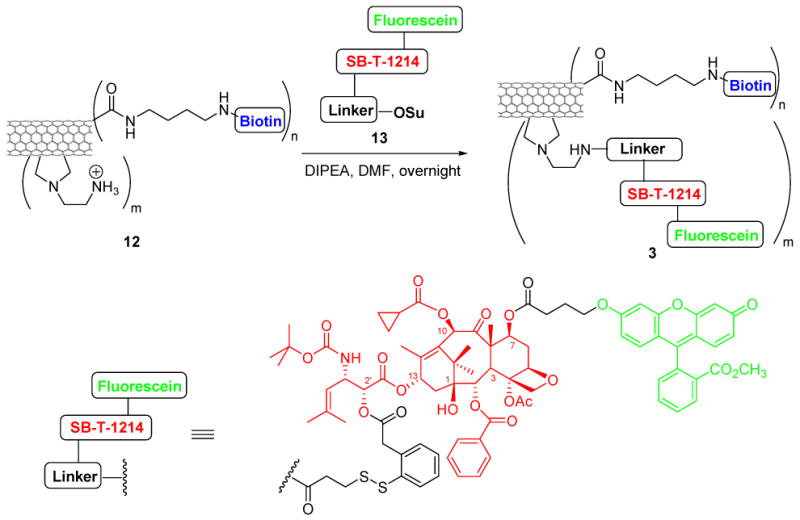
Synthesis of biotin-SWNT-linker-(taxoid-fluorescein) conjugate 3.
Syntheses of conjugates 1, 2 and 3 are detailed in the Experimental Section and Schemes 2-4. A batch of pristine HiPco SWNT (0) was first functionalized and purified by oxidation in concentrated H2SO4 : HNO3 (3:1 by volume) with sonication for 2 h, followed by refluxing at 70°C for 30 min.66 Figures 1A and 1B show the TEM images of HiPco SWNTs before and after oxidation. The ends and defect sites on the sidewall of the oxidized SWNTs were functionalized with carboxylic acid and carboxylate groups,67-69 whose presence was confirmed by ATR IR spectroscopy, showing expected relevant peaks at 1703 and 1630 cm-1 (Figure 1C). Figure 1D shows the AFM height image of oxidized SWNTs 4. The histogram of length and height measurements on 50 oxidized SWNTs 4 recorded by AFM show an average size of ∼3 nm in diameter (due to the formation of bundles under the conditions used) and ∼250 nm in length (see Figure S2, Supporting Information). These carboxylic acid and carboxylate groups were subsequently converted to the corresponding amide groups through condensation with amines. Tube sidewalls were subsequently functionalized with amine moieties through 1,3-dipolar cycloaddition of azomethine ylide that had been generated in situ.70 The extent of amine loading was estimated to be 0.50 ± 0.03 mmol/g and 0.20 ± 0.02 mmol/g at the ends/defect sites and the sidewalls of SWNTs, respectively, by means of the quantitative Kaiser test.71 Finally, biotin molecules and the fluorescein-labeled taxoid-linker moieties were conjugated to the amine moieties localized at the ends/defect sites and sidewalls of SWNTs, respectively, through standard peptide coupling reactions to yield the desired conjugate, biotin-SWNT-linker-(taxoid-fluorescein) (3). In principle, these couplings using modifiers in large excess should proceed quantitatively to provide a maximum of 178 biotin modules (at the ends and the defect sites on the side wall) and 71 taxoid modules (on the side wall) per SWNT (based on the mass of a carbon nanotube with 250 nm in length and 1 nm in diameter estimated as 5.5×10-19 g).72 Therefore, conjugate 3 at a concentration of 100 μg/mL is estimated to contain taxoid molecules in the order of 13.9 μM.
Figure 1.

TEM images of HiPco SWNTs: (A) pristine SWNTs; (B) acid oxidized SWNTs. (C) AFM height image of acid-oxidized SWNTs 4, and (D) ATR-IR spectra of SWNTs vs. acid oxidized SWNTs. Note that the peak at 2349 cm-1 can be attributed to the asymmetric stretching mode of CO2 molecules in the atmosphere.
Conjugate 3 was analyzed by UV-visible spectroscopy (Figure 2). The peak at ∼280 nm can be attributed to the sum of contributions arising from taxoid (SB-T-1214) molecules and dye molecules (fluorescein). Conjugate 1 was prepared from functionalized SWNT 6 by attaching FITC groups to its sidewall through deprotection of the Boc group and the addition of FITC. Conjugate 2 was prepared from conjugate 1 by introducing the biotin moiety in the same manner as that used for the synthesis of conjugate 3. Conjugates 1 and 2 were also characterized by UV-visible spectroscopy (see Figure S3, Supporting Information). Absorption peaks at ∼450 and ∼490 nm confirmed the presence of FITC in these conjugates. It is noteworthy that the solubility of the functionalized SWNTs (1, 2, and 3) in dichloromethane was greatly enhanced as compared with pristine tubes (0), as shown in Figure 3A. In addition, conjugate 3 are also well dispersed in the cell culture medium at the final concentration of 50 μg/mL for months (Figure 3B).
Figure 2.
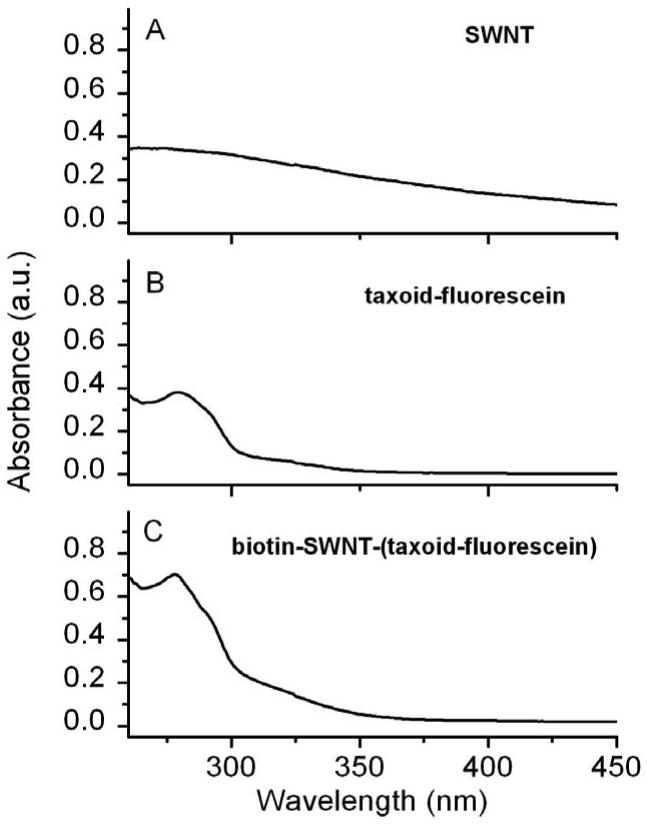
UV-visible spectra of SWNTs and their biofunctionalized conjugates plotted to scale: (A) acid oxidized SWNTs 4 at 50 μg/mL concentration; (B) taxoid-fluorescein conjugates at 15 μM concentration; and (C) biotin-SWNT-(taxoid-fluorescein) conjugate 3 at 50 μg/mL concentration.
Figure 3.

(A) Photographs of vials containing pristine SWNT 0, SWNT-FITC 1, biotin-SWNT-FITC 2, and biotin-SWNT-(taxoid-fluorescein) 3 in CH2Cl2 (∼1 mg/mL). (B) Photographs of SWNT conjugates at concentrations of 50 μg/mL in cell culture medium: SWNT-FITC conjugate 1 (left) and biotin-SWNT-(taxoid-fluorescein) conjugate 3 (right). Arrow indicates that some of the nanotubes 1 precipitated from the medium.
Internalization of SWNT conjugate 1 and biotin-SWNT conjugate 2
We examined cellular uptake of conjugates 1 and 2 using a leukemia cell line, L1210FR, overexpressing biotin receptors on its surface.59 Figures 4A and 4B show confocal fluorescence microscopy (CFM) images of L1210FR cells after treatment with 10 μg/mL (final concentration) of SWNT-FITC 1 and biotin-SWNT-FITC 2 conjugates, respectively, for 3 h at 37 °C. Treated leukemia cells were then washed with phosphate buffered saline (PBS) to remove excess fluorescent probes in extracellular medium. [Note: We did not observe fluorescent probes attached to the cell surface in the CFM images, which may indicate the occurrence of highly efficient receptor-mediated endocytosis.] L1210FR cells treated with conjugate 2 yielded far more intense fluorescence than those incubated with conjugate 1. This observation should be attributed to the markedly increased permeability of conjugate 2 into the cancer cells through highly efficient receptor-mediated endocytosis. Flow cytometry analyses (Figure 4C) on 10,000 treated live cells, in average, also supported this observation, i.e., the fluorescence intensity of biotin-SWNT-FITC conjugate 2 increased by one order of magnitude as compared with that of SWNT-FITC conjugate 1.
Figure 4.

CFM images of L1210FR cells after incubation with SWNT-FITC 1 (A) and biotin-SWNT-FITC 2 (B) at the final concentration of 10 μg/mL at 37 °C for 2 h. (C) Comparison of fluorescence intensities of L1210FR cells by flow cytometry upon treatment with pristine SWNTs 0 (purple), conjugate 1 (blue), and conjugate 2 (red) at the final concentration of 10 μg/mL in each case. Background, i.e., data for untreated cells, is plotted in black.
The mechanism for the internalization of SWNTs into cells has not been fully established. It has been proposed that SWNTs wrapped with proteins or genes can be internalized into cells via endocytosis,25 whereas SWNTs functionalized with small molecules tend to act as nanoneedles that can pierce through cell membranes, thereby allowing for their diffusion into cells.39 Endocytosis is known to be energy dependent and could be hindered at low temperature and/or in the presence of a metabolic inhibitor, such as NaN3, which is known to disrupt specifically the production of ATP in cells.73-75 To probe the mechanism for cellular uptake of the SWNT conjugates, we incubated L1210FR cells with conjugate 1 (a) at 4 °C and (b) in the presence of NaN3 (0.05% w/v) in order to compare the degree of uptake with that at 37 °C. We found that conjugate 1 was able to cross the cell membrane even at a low temperature (4 °C) after 3 h of incubation although the fluorescence intensity decreased by two-fold (Figure 5B), due to the intrinsic temperature dependence of the diffusion process. In contrast, a similar intensity of fluorescence was observed after incubation in the absence of (Figure 5A) or presence of (Figure 5C) NaN3 at 37 ° C. The control experiment, in which cells were subjected to the same concentration (10 μg/mL) of oxidized SWNT 4 (i.e., not conjugated to FITC), showed no fluorescence at all (Figure 5D). Flow cytometry analyses on 10,000 cells are consistent with the CFM observations. Accordingly, the internalization of SWNT within the cells appears to be temperature-dependent, but energy-independent. Thus, the results do not support the endocytosis mechanism for the internalization of oxidized SWNTs 4 (and its fluorescent probe SWNT-FITC 1), favoring the “pierce through” mechanism.
Figure 5.
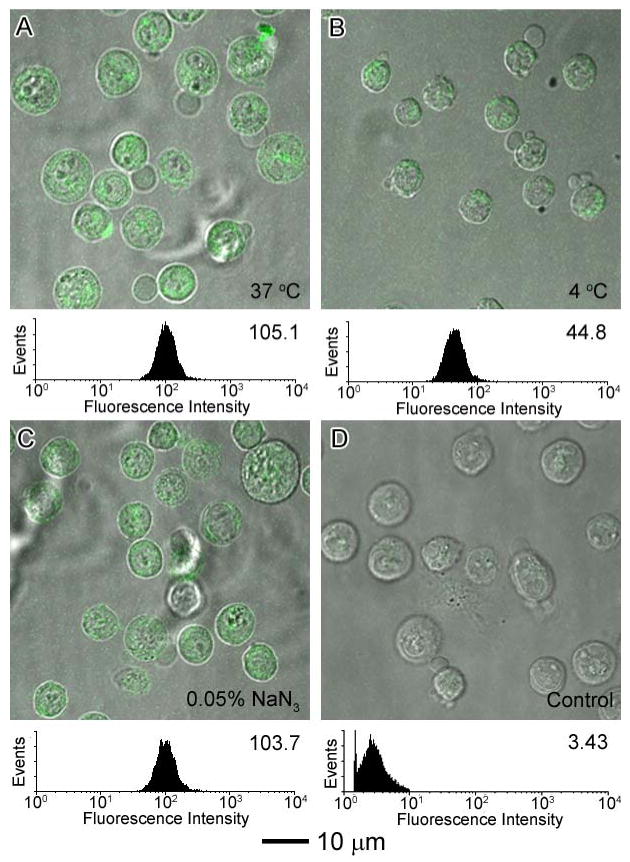
CFM images and flow cytometry analysis of L1210FR cells after incubation with SWNT-FITC 1 at the final concentration of 10 μg/mL under different conditions after 3 h of incubation: (A) at 37°C; (B) at 4 °C; and (C) at 37 °C in the presence of 0.05% (w/v) NaN3. (D) Control experiment: CFM images and flow cytometry data of L1210FR cells after treatment with oxidized SWNT 4 at the same concentrations at 37 °C for 3 h. All of the CFM images and flow cytometry data were acquired under the same conditions.
Biotin uptake is known to be a temperature- and energy-dependent receptor-mediated endocytosis process.76,77 To examine the mechanism for cellular uptake of biotin-SWNT conjugates, we incubated conjugate 2 under different conditions using L1210FR cells, overexpressing biotin receptors on their surfaces. As Figures 6A and 6B shows, the fluorescence intensity of cells incubated at 4 °C decreases by one order of magnitude as compared with that of cells treated at 37 °C. The results indicate that the internalization of conjugate 2 was hindered at 4 °C. The fluorescent intensity decreases dramatically in the presence of NaN3 as shown in Figure 6C. Thus, It has been found that endocytosis of the biotin-SWNT conjugates is energy-dependent and hindered by NaN3, indicating receptor-mediated endocytosis. To further verify the nature of the internalization of biotin-conjugate 2, we incubated L1210FR cells with excess biotin to saturate accessible biotin receptors on the surfaces of the leukemia cells, and then treated them with biotin-conjugate 2 at 37 °C for 3 h. The CFM image in Figure 6D clearly indicates a drastic reduction in the fluorescence intensity, as compared to that observed in the absence of excess biotin (Figure 6A). These results confirm that the receptor-mediated endocytosis is by far the predominant mechanism accounting for internalization, with nanotube diffusion as a minor contributing pathway.
Figure 6.

CFM images and flow cytometry analysis of L1210FR cells after incubation with biotin-SWNT-FITC 2 at a final concentration of 10 μg/mL under different conditions after an incubation period of 3 h: (A) at 37°C; (B) at 4 °C; (C) at 37 °C in the presence of 0.05% (w/v) NaN3; (D) at 37 °C after pretreatment with excess biotin. All of the CFM images and flow cytometry data were acquired under the same conditions.
Release of Taxoid from Conjugate 3 in vitro
Building upon the promising results with biotin-SWNT conjugate 2 as a potentially versatile vehicle for tumor-targeted drug delivery, we investigated the efficacy of biotin-SWNT-linker-taxoid-fluorescein (3) for cellular uptake and drug release inside the leukemia cells. As the anticancer drug warhead, we used a highly potent 2nd-generation taxoid, SB-T-1214. [Note: 2nd-generation taxoids exhibit 2-3 orders of magnitude higher potency against multidrug-resistant (MDR) cancer cell lines as compared with paclitaxel (Taxol®), which is the most widely used anticancer drug in current chemotherapy treatments.78,79] To evaluate the efficacy of biotin-SWNT-taxoid conjugate 3 for its internalization and drug release, we incubated conjugate 3 with L1210FR cells at 50 μg/mL concentration for 3 h at 37°C and washed the treated cells with PBS.
CFM analysis
As Figure 7A shows, internalization of conjugate 3 was confirmed by the bright fluorescence of the L1210FR cells observed by CFM. Next, the leukemia cells were treated with glutathione ethyl ester for an additional 2 h at 37 °C in order to ensure cleavage of the disulfide linkage covalently connecting the taxoid to the biotin-SWNT moiety. It is anticipated that the fluorescein-labeled taxoid released from the conjugate inside the leukemia cells should bind to tubulin/microtubule that is the target protein of the drug. In fact, Figure 7B clearly shows that the fluorescent taxoid does bind to the target protein to light up the large bundles of microtubules, which provides ultimate proof of the designed drug release.
Figure 7.
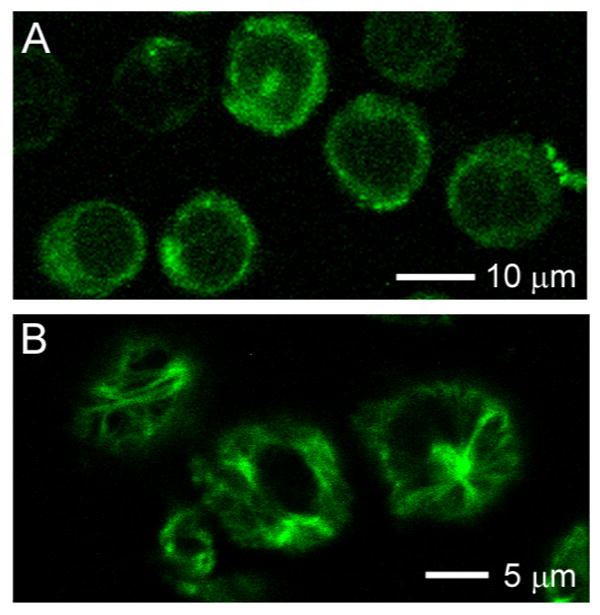
CFM images of L1210FR cells treated with biotin-SWNT-(taxoid-fluorescein) 3 incubated before (A) and after (B) addition of GSH-ethyl ester. Image (B) clearly highlights the presence of fluorescent microtubule networks in the living cells generated by the binding of the fluorescent taxoid (SB-T-1214-fluorescein) upon cleavage of the disulfide bond in the linker by either GSH or GSH-ethyl ester.
It should be noted that the intracellular glutathione in the leukemia cells should be able to cleave the disulfide linkage over longer incubation times, but the endogenous glutathione level in cancer cells varies due to the significant difference in physiological conditions between cultivated cancer cells and those in the actual leukemia or solid tumors in vivo. Accordingly, the extracellular addition of excess glutathione ethyl ester is beneficial for a rapid visualization of the drug release inside the leukemia cells. This acceleration is evident by comparing Figure 7A and Figure 7B, i.e., before and after addition of GSH-ethyl ester, respectively. To further confirm the presence of the microtubule network, we incubated the cells with conjugate 3 overnight, followed by removal of excess conjugate 3, fixation of the cells, and staining microtubules with a “fluorescent red”-labeled antibody. We also confirmed that the green fluorescence associated with conjugate 3 and the “fluorescence-red” of the antibody for microtubules are clearly overlapped (see Figure S4, Supporting Information). [Note: In the double-staining experiment, no GSH-ethyl ester was added. Thus, the endogenous GSH cleaved the disulfide bond, releasing the taxoid-fluorescein molecules. Exposure to the endogenous GSH overnight might not be enough to release all taxoid-fluorescein molecules in conjugate 3, but obviously sufficient amounts of the taxoid molecules were released for this purpose.]
Evaluation of tumor-targeting specificity through receptor-mediated endocytosis
(a) CFM analysis
To evaluate the specificity of biotin-SWNT-taxoid-fluorescein (3) to cell lines, overexpressing biotin receptors on their surface, we chose two other cell lines, i.e., murine leukemia L1210 cell line and WI38 human lung fibroblast cell line, which do not overexpress biotin receptors. Conjugate 3 is expected to evince a much higher level of cellular uptake into L1210FR as compared with L1210 and WI38 cells. Figure 8 shows that the L1210FR cells (Figure 8A) indeed yield a much stronger fluorescence intensity than L1210 (Figure 8B) or WI38 (Figure 8C) cells upon incubation with conjugate 3 under identical conditions, as expected.
Figure 8.

CFM images and flow cytometry analysis of different cell types upon incubation with biotin-SWNT-taxoid conjugate 3 at a final concentration of 50 μg/mL at 37 °C for an incubation period of 3 h: (A) L1210FR leukemia cell line overexpressing biotin receptors; (B) L1210 leukemia cell line; and (C) WI38 human lung fibroblast cell line. All of the CFM images and flow cytometry data were taken under identical conditions.
(b) Cytotoxicity assay
We also performed the cytotoxicity assessment for conjugate 3 against these three cell lines by means of the MTT assay. As Table 1 shows, after 72 h incubation, the IC50 value of the conjugate 3 against L1210FR cell line was measured to be 0.36 μg/mL, whereas the IC50 values for L1210 and WI38 cell lines were more than 50 μg/mL (see Figure S5, Supporting Information, for data plot and graphs).
Table 1.
IC50 values of biotin-SWNT-taxoid conjugate 3 corresponding to different cell lines
| Cell Line | L1210FR | L1210 | WI38 |
|---|---|---|---|
| IC50 (μg/mL) | 0.36 ± 0.04 | >50 | >50 |
In control experiments, the IC50 values of oxidized SWNT 4, conjugate 1 (SWNT-FITC), and conjugate 2 (biotin-SWNT-FITC) against all three cell lines were found to be consistently higher than 100 μg/mL. It is suggested, therefore, that the observed cytotoxicity is attributed to taxoid-fluorescein molecules released from conjugate 3 (biotin-SWNT-taxoid-fluorescein) internalized. It is worthy of note that taxoid-fluorescein molecules appear to be mostly released by endogenous GSH in L1210FR cells after 72 h of incubation time.
According to calculations, the IC50 value (0.36 ± 0.04 μg/mL) of conjugate 3 corresponds to ∼51 nM of taxoid-fluorescein molecules in L1210FR cell line by assuming that all taxoid-flurorescein molecules attached to SWNTs are released. If the drug release is not complete, the IC50 values should be even smaller (i.e., more potent). This means that the apparent cytotoxicity per taxoid is substantially increased by using the biotin-SWNT-based drug delivery system, i.e., 87.6 ± 4.2 nM for drug itself (i.e., SB-T-1214-fluorescein) determined in a parallel control experiment vs. ∼51 nM for conjugate 3. The results clearly indicate that the mass drug delivery into the cytosol of the cancer cells using this drug delivery system is superior than the simple exposure of the drug itself to the same cancer cells. The latter is very likely to include a concentration-dependent cell penetration efficiency factor (i.e., not all extracellular taxoids can be internalized). When taxoids get into the cancer cells through the mass drug delivery system, the released taxoids can quickly and tightly bind to the target protein (tubulins/microtubules) so that the effective intracellular drug concentration is substantially higher than that achieved by extracellular exposure of the drug.
Conclusions
Biotin-functionalized SWNT conjugates have been successfully designed and synthesized as a novel and efficient DDS for potential use in tumor-targeting chemotherapy. We have unambiguously observed the occurrence of the designed cancer-specific receptor-mediated endocytosis of the whole conjugate, followed by efficient drug release and binding of the drug to the target microtubules by CFM analysis. The conjugate shows the specificity to the cancer cells, overexpressing biotin receptors on their surface, and the cytotoxicity of the conjugate is solely ascribed to the released taxoid molecules inside the cancer cells. We also have found that the mass drug delivery into the cytosol of the cancer cells using this drug delivery system is superior than the simple extracellular exposure of the drug itself to the same cancer cells. These results strongly suggest that the functionalized SWNT-based DDS can serve as a highly promising drug delivery platform, which offers (a) biomarker-targeted drug delivery, (b) possible delivery of greater therapeutic payloads and (c) possible use of multiple, complementary drug warheads for combination therapy.
Supplementary Material
Experimental Section, 1H NMR spectrum of 13, Histograms of length and height measurements on 50 SWNTs 4 recorded by AFM, UV-Visible spectra of 1 and 2, CMF images of double-stained microtubules, and MTT cytotoxicity assay data plot and graphs for conjugate 3 against L1210FR, L1210, and WI38 cell lines. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 3.
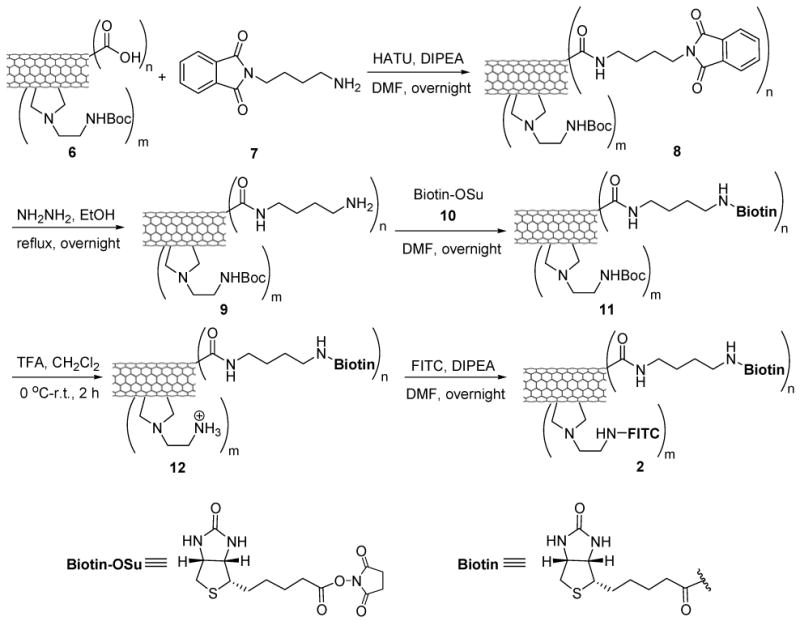
Synthesis of biotin-SWNT-FITC conjugate 2.
Acknowledgments
This research was supported by grants from the National Cancer Institute (CA 103314 to I. O.) and the National Science Foundation (CAREER DMR-0348239 to S.S.W.). SSW also acknowledges the Alfred P. Sloan Foundation for a faculty fellowship (2006-2008). JC thanks the Battelle Memorial Institute for support through CRADA #05-25, administered by Brookhaven National Laboratory. Work at BNL was also supported by the U.S. Department of Energy Office of Basic Energy Sciences under Contract DE-AC-02-98CH10886. The authors acknowledge the technical service and advice provided by Ms. Susan Van Horn for TEM and Dr. Guo-Wei Tian for CFM, performed at the Central Microscopy Imaging Center at Stony Brook. They also thank Ms. Rebecca Rowehl for her valuable help with cell culture preparations at the Cell Culture and Hybridoma facility at Stony Brook.
References
- 1.Leuschner C, Kumar C. In: Nanofabrication Towards Biomedical Application. Kumar CSSR, Jormes J, Leuschner C, editors. Wiley-VCH; 2005. pp. 289–326. [Google Scholar]
- 2.Ferrari M. Nature Rev. 2005;5:161–171. doi: 10.1038/nrc1566. [DOI] [PubMed] [Google Scholar]
- 3.Lacerda L, Bianco A, Prato M, Kostarelos K. Adv Drug Delivery Rev. 2006;58:1460–1470. doi: 10.1016/j.addr.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 4.Dresselhaus M, Dai H. MRS 2004 Carbon Nanotube Special Issue. 2004. [Google Scholar]
- 5.Dai H. Surf Sci. 2002;500:218–241. [Google Scholar]
- 6.Prato M, Kostarelos K, Bianco A. Acc Chem Res. 2008;41:60–68. doi: 10.1021/ar700089b. [DOI] [PubMed] [Google Scholar]
- 7.Kam NWS, Dai H. Physica Status Solidi B: Basic Solid State Phys. 2006;243:3561–3566. [Google Scholar]
- 8.Klumpp C, Kostarelos K, Prato M, Bianco A. Biochim Biophys Acta. 2006;1758:404–412. doi: 10.1016/j.bbamem.2005.10.008. [DOI] [PubMed] [Google Scholar]
- 9.Campidelli S, Klumpp C, Bianco A, Guldi DM, Prato M. J Phys Org Chem. 2006;19:531–539. [Google Scholar]
- 10.Pastorin G, Kostarelos K, Prato M, Bianco A. J Biomed Nanotech. 2005;1:133–142. [Google Scholar]
- 11.Bianco A, Hoebeke J, Kostarelos K, Prato M, Partidos CD. Curr Drug Delivery. 2005;2:253–259. doi: 10.2174/1567201054367959. [DOI] [PubMed] [Google Scholar]
- 12.Bianco A, Kostarelos K, Partidos CD, Prato M. Chem Commun. 2005:571–577. doi: 10.1039/b410943k. [DOI] [PubMed] [Google Scholar]
- 13.Bianco A, Kostarelos K, Prato M. Curr Opin Chem Biol. 2005;9:674–679. doi: 10.1016/j.cbpa.2005.10.005. [DOI] [PubMed] [Google Scholar]
- 14.Bianco A. Exp Opin Drug Delivery. 2004;1:57–65. doi: 10.1517/17425247.1.1.57. [DOI] [PubMed] [Google Scholar]
- 15.Liu Z, Davis C, Cau W, He L, Chen X, Dai H. Proc Nat Acad Sci USA. 2008;105:1410–1415. doi: 10.1073/pnas.0707654105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Z, Cai W, He L, Nakayama N, Chen K, Sun X, Chen X, Dai H. Nature Nanotech. 2007;2:47–52. doi: 10.1038/nnano.2006.170. [DOI] [PubMed] [Google Scholar]
- 17.Singh R, Pantarotto D, Lacerda L, Pastorin G, Klumpp C, Prato M, Bianco A, Kostarelos K. Proc Nat Acad Sci USA. 2006;103:3357–3362. doi: 10.1073/pnas.0509009103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang H, Wang J, Deng X, Sun H, Shi Z, Gu Z, Liu Y, Zhao Y. J Nanosci Nanotechnol. 2004;4:1019–1024. doi: 10.1166/jnn.2004.146. [DOI] [PubMed] [Google Scholar]
- 19.McDevitt MR, Chattopadhyay D, Jaggi JS, Finn RD, Zanzonico PB, Villa C, Rey D, Mendenhall J, Batt CA, Njardarson JT, Scheinberg DA. PLoS ONE. 2007:e907. doi: 10.1371/journal.pone.0000907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang ST, Fernando KAS, Liu JH, Wang J, Sun HF, Liu Y, Chen M, Huang Y, Wang X, Wang H, Sun YP. Small. 2008;4:940–944. doi: 10.1002/smll.200700714. [DOI] [PubMed] [Google Scholar]
- 21.McDevitt MR, Chattopadhyay D, Kappel BJ, Jaggi JS, Schiffman SR, Antczak C, Njardarson JT, Brentjens R, Scheinberg DA. J Nuclear Med. 2007;48:1180–1189. doi: 10.2967/jnumed.106.039131. [DOI] [PubMed] [Google Scholar]
- 22.Liu Z, Chen K, Davis C, Sherlock S, Cao Q, Chen X, Dai H. Cancer Res. 2008;68:6652–6660. doi: 10.1158/0008-5472.CAN-08-1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu Z, Winters M, Holodniy M, Dai H. Angew Chem Int Ed. 2007;46:2023–2027. doi: 10.1002/anie.200604295. [DOI] [PubMed] [Google Scholar]
- 24.Gao L, Nie L, Wang T, Qin Y, Guo Z, Yang D, Yan X. Chem Bio Chem. 2006;7:239–242. doi: 10.1002/cbic.200500227. [DOI] [PubMed] [Google Scholar]
- 25.Kam NWS, Liu Z, Dai H. Angew Chem Int Ed. 2006;45:577–581. doi: 10.1002/anie.200503389. [DOI] [PubMed] [Google Scholar]
- 26.Kam NWS, Liu Z, Dai H. J Am Chem Soc. 2005;127:12492–12493. doi: 10.1021/ja053962k. [DOI] [PubMed] [Google Scholar]
- 27.Kam NWS, O'Connell M, Wisdom JA, Dai H. Proc Nat Acad Sci USA. 2005;102:11600–11605. doi: 10.1073/pnas.0502680102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kam NWS, Dai H. J Am Chem Soc. 2005;127:6021–6026. doi: 10.1021/ja050062v. [DOI] [PubMed] [Google Scholar]
- 29.Kam NWS, Jessop TC, Wender PA, Dai H. J Am Chem Soc. 2004;126:6850–6851. doi: 10.1021/ja0486059. [DOI] [PubMed] [Google Scholar]
- 30.Georgakilas V, Tagmatarchis N, Pantarotto D, Bianco A, Briand JP, Prato M. Chem Commun. 2002:3050–3051. doi: 10.1039/b209843a. [DOI] [PubMed] [Google Scholar]
- 31.Pantarotto D, Briand JP, Prato M, Bianco A. Chem Commun. 2004:16–17. doi: 10.1039/b311254c. [DOI] [PubMed] [Google Scholar]
- 32.Pantarotto D, Partidos CD, Graff R, Hoebeke J, Briand JP, Prato M, Bianco A. J Am Chem Soc. 2003;125:6160–6164. doi: 10.1021/ja034342r. [DOI] [PubMed] [Google Scholar]
- 33.Pantarotto D, Partidos CD, Hoebeke J, Brown F, Kramer E, Briand JP, Muller S, Prato M, Bianco A. Chem Biol. 2003;10:961–966. doi: 10.1016/j.chembiol.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 34.Lacerda L, Bianco A, Prato M, Kostarelos K. J Mater Chem. 2008;18:17–22. [Google Scholar]
- 35.Singh R, Pantarotto D, McCarthy D, Chaloin O, Hoebeke J, Partidos CD, Briand JP, Prato M, Bianco A, Kostarelos K. J Am Chem Soc. 2005;127:4388–4396. doi: 10.1021/ja0441561. [DOI] [PubMed] [Google Scholar]
- 36.Bianco A, Hoebeke J, Godefroy S, Chaloin O, Pantarotto D, Briand JP, Muller S, Prato M, Partidos CD. J Am Chem Soc. 2005;127:58–59. doi: 10.1021/ja044293y. [DOI] [PubMed] [Google Scholar]
- 37.Pantarotto D, Singh R, McCarthy D, Erhardt M, Briand JP, Prato M, Kostarelos K, Bianco A. Angew Chem Int Ed. 2004;43:5242–5246. doi: 10.1002/anie.200460437. [DOI] [PubMed] [Google Scholar]
- 38.Wu W, Wieckowski S, Pastorin G, Benincasa M, Klumpp C, Briand JP, Gennaro R, Prato M, Bianco A. Angew Chem Int Ed. 2005;44:6358–6362. doi: 10.1002/anie.200501613. [DOI] [PubMed] [Google Scholar]
- 39.Kostarelos K, Lacerda L, Pastorin G, Wu W, Wieckowski S, Luangsivilay J, Godefroy S, Pantarotto D, Briand JP, Muller S, Prato M, Bianco A. Nature Nanotech. 2007;2:108–113. doi: 10.1038/nnano.2006.209. [DOI] [PubMed] [Google Scholar]
- 40.Feazell RP, Nakayama-Ratchford N, Dai H, Lippard SJ. J Am Chem Soc. 2007;129:8438–8439. doi: 10.1021/ja073231f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ali-Boucetta H, Al-Jamal KT, McCarthy D, Prato M, Bianco A, Kostarelos K. Chem Commun. 2008:459–461. doi: 10.1039/b712350g. [DOI] [PubMed] [Google Scholar]
- 42.Liu Z, Sun X, Nakayama-Ratchford N, Dai H. ACS Nano. 2007;1:50–56. doi: 10.1021/nn700040t. [DOI] [PubMed] [Google Scholar]
- 43.Ojima I. Acc Chem Res. 2008;41:108–119. doi: 10.1021/ar700093f. [DOI] [PubMed] [Google Scholar]
- 44.Jaracz S, Chen J, Kuznetsova LV, Ojima I. Bioorg Med Chem. 2005;13:5043–5054. doi: 10.1016/j.bmc.2005.04.084. [DOI] [PubMed] [Google Scholar]
- 45.Kuznetsova L, Chen J, Sun L, Wu X, Pepe A, Weith JM, Pera P, Bernachi RJ, Ojima I. Bioorg Med Chem Lett. 2006;16:974–977. doi: 10.1016/j.bmcl.2005.10.089. [DOI] [PubMed] [Google Scholar]
- 46.Chen J, Jaracz S, Zhao X, Chen S, Ojima I. Exp Opin Drug Delivery. 2005;2:873–890. doi: 10.1517/17425247.2.5.873. [DOI] [PubMed] [Google Scholar]
- 47.Miller ML, Roller EE, Wu X, Leece BA, Goldmacher VS, Chari RVJ, Ojima I. Bioorg Med Chem Lett. 2004;14:4079–4082. doi: 10.1016/j.bmcl.2004.05.027. [DOI] [PubMed] [Google Scholar]
- 48.Ojima I. Chem Bio Chem. 2004;5:628–635. doi: 10.1002/cbic.200300844. [DOI] [PubMed] [Google Scholar]
- 49.Saito G, Swansonb JA, Lee KD. Adv Drug Deliv Rev. 2003;55:199–215. doi: 10.1016/s0169-409x(02)00179-5. [DOI] [PubMed] [Google Scholar]
- 50.West KR, Otto S. Cur Drug Discov Tech. 2005;2:123–160. doi: 10.2174/1570163054866882. [DOI] [PubMed] [Google Scholar]
- 51.Sanderson RJ, Hering MA, James SF, Sun MMC, Doronina SO, Siadak AW, Senter PD, Wahl AF. Clin Cancer Res. 2005;11:843–852. [PubMed] [Google Scholar]
- 52.Vlahov IR, Santhapuram HKR, Kleindl PJ, Howard SJ, Stanford KM, Leamon CP. Bioorg Med Chem Lett. 2006;16:5093–5096. doi: 10.1016/j.bmcl.2006.07.030. [DOI] [PubMed] [Google Scholar]
- 53.Leamon CP, Reddy JA, Vlahov IR, Westrick E, Dawson A, Dorton R, Vetzel M, Santhapuram HK, Wang Y. Mol Pharm. 2007;4:659–667. doi: 10.1021/mp070049c. [DOI] [PubMed] [Google Scholar]
- 54.Satyam A. Bioorg Med Chem Lett. 2008;18:3196–3199. doi: 10.1016/j.bmcl.2008.04.063. [DOI] [PubMed] [Google Scholar]
- 55.Cuchelkar V, Kopeckova P, Kopecek J. Macromol Biosci. 2008;8:375–383. doi: 10.1002/mabi.200700240. [DOI] [PubMed] [Google Scholar]
- 56.Austin CD, Wen X, Gazzard L, Nelson C, Scheller RH, Scales SJ. Proc Nat Acad Sci. 2005;102 doi: 10.1073/pnas.0509035102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Duncan R. Nature Rev Drug Discovery. 2003;2:347–360. doi: 10.1038/nrd1088. [DOI] [PubMed] [Google Scholar]
- 58.Bottini M, Cerignoli F, Dawson MI, Magrini A, Rosato N, Mustelin T. Biomacromolecules. 2006;7:2259–2263. doi: 10.1021/bm0602031. [DOI] [PubMed] [Google Scholar]
- 59.Russel-Jones G, McTavish K, McEwan J, Rice J, Nowotnik D. J Inorg Biochem. 2004;98:1625–1633. doi: 10.1016/j.jinorgbio.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 60.Russel-Jones G, Mcewan J. US 2006/0127310 A1 US Pat Appl Pub. 2006
- 61.Leamon CP, Reddy JA. Adv Drug Deliv Rev. 2004;56:1127–1141. doi: 10.1016/j.addr.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 62.Meister A. Metabolism and transport of glutathione and other γ-glutamyl compounds. Raven Press; New York: 1983. [Google Scholar]
- 63.Zheng ZB, Zhu G, Tak H, Joseph E, Eiseman JL, Creighton DJ. Bioconjugate Chem. 2005;16:598–607. doi: 10.1021/bc0499634. [DOI] [PubMed] [Google Scholar]
- 64.Jordan MA, Ojima I, Rosas F, Distefano M, Wilson L, Scambia G, Ferlini C. Chem Biol. 2002;9:93–101. doi: 10.1016/s1074-5521(01)00097-7. [DOI] [PubMed] [Google Scholar]
- 65.Nogales E, Wolf SG, Khan IA, Luduena RF, Downing KH. Nature. 1995;375:424–427. doi: 10.1038/375424a0. [DOI] [PubMed] [Google Scholar]
- 66.Liu J, Rinzler AG, Dai H, Hafner JH, Bradley RK, Boul PJ, Lu A, Iverson T, Shelimov K, Huffman CB, Rodriguez-Macias F, Shon YS, Lee TR, Colbert DT, Smalley RE. Science. 1998;280:1253–1256. doi: 10.1126/science.280.5367.1253. [DOI] [PubMed] [Google Scholar]
- 67.Hamon MA, Hu H, Bhowmik P, Niyogi S, Zhao B, Itkis ME, Haddon RC. Chem Phys Lett. 2001;347:8–12. [Google Scholar]
- 68.Hamon MA, Hui H, Bhowmik P, Itkis HME, Haddon RC. Appl Phys A. 2002;74:333–338. [Google Scholar]
- 69.Banerjee S, Hemraj-Benny T, Wong SS. Adv Mater. 2005;17:17–29. [Google Scholar]
- 70.Pastorin G, Wu W, Wieckowski S, Briand JP, Kostarelos K, Prato M, Bianco A. Chem Commun. 2006:1182–1184. doi: 10.1039/b516309a. [DOI] [PubMed] [Google Scholar]
- 71.Sarin VK, Kent SBH, Tam JP, Merrifield RB. Anal Biochem. 1981;117:147–157. doi: 10.1016/0003-2697(81)90704-1. [DOI] [PubMed] [Google Scholar]
- 72.Zhu Y, Peng AT, Carpenter K, Maguire JA, Hosmane NS, Takagaki M. J Am Chem Soc. 2005;127:9875–9880. doi: 10.1021/ja0517116. [DOI] [PubMed] [Google Scholar]
- 73.Mukherjee S, Ghosh RN, Masfield FR. Physiol Rev. 1997;77:759–803. doi: 10.1152/physrev.1997.77.3.759. [DOI] [PubMed] [Google Scholar]
- 74.Silverstein SC, Steinman RM, Cohn ZA. Ann Rev Biochem. 1977;46:669–722. doi: 10.1146/annurev.bi.46.070177.003321. [DOI] [PubMed] [Google Scholar]
- 75.Schmid SL, Carter LL. J Cell Biol. 1990;111:2307–2318. doi: 10.1083/jcb.111.6.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Balamurugan K, Vaziri ND, Said HM. Am J Physiol Renal Physiol. 2005;288:F823–F831. doi: 10.1152/ajprenal.00375.2004. [DOI] [PubMed] [Google Scholar]
- 77.Becker JM, Wilchek M, Katchalski E. Proc Nat Acad Sci USA. 1971;68:2604–2607. doi: 10.1073/pnas.68.10.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ojima I, Bounaud PY, Takeuchi C, Pera P, Bernacki RJ. Bioorg Med Chem Lett. 1998;8:189–194. doi: 10.1016/s0960-894x(97)10218-9. [DOI] [PubMed] [Google Scholar]
- 79.Ojima I, Slater JC, Michaud E, Kuduk SD, Bounaud PY, Vrignaud P, Bissery MC, Veith JM, Pera P, Bernacki RJ. J Med Chem. 1996;39:3889–3896. doi: 10.1021/jm9604080. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental Section, 1H NMR spectrum of 13, Histograms of length and height measurements on 50 SWNTs 4 recorded by AFM, UV-Visible spectra of 1 and 2, CMF images of double-stained microtubules, and MTT cytotoxicity assay data plot and graphs for conjugate 3 against L1210FR, L1210, and WI38 cell lines. This material is available free of charge via the Internet at http://pubs.acs.org.