

Abstract
Accumulations of β-amyloid (Aβ) contribute to neurological deficits associated with Alzheimer's disease (AD). The effects of Aβ on basal neuronal excitability and learning-related AHP plasticity were examined using whole-cell recordings from hippocampal neurons in the 5XFAD mouse model of AD. A robust increase in Aβ42 (and elevated levels of Aβ38-40) in naïve 5XFAD mice was associated with decreased basal neuronal excitability, evidenced by a select increase in Ca2+-sensitive afterhyperpolarization (AHP). Moreover, trace fear deficits observed in a subset of 5XFAD weak-learner mice were associated with a greater enhancement of the AHP in neurons, as compared to age-matched 5XFAD learner and 5XFAD naïve mice. Importantly, learning-related plasticity of the AHP remained intact in a subset of 5XFAD mice that learned trace fear conditioning to a set criterion. We show that APP-PS1 mutations enhance Aβ and disrupt basal excitability via a Ca2+-dependent enhancement of the AHP, and suggest disruption to learning-related modulation of intrinsic excitability resulted, in part, from altered cholinergic modulation of the AHP in the 5XFAD mouse model of AD.
Keywords: hippocampus, afterhyperpolarization, fear conditioning, Alzheimer's disease
1. Introduction
Alzheimer's disease is a progressive neurodegenerative disease that afflicts the elderly and is characterized by the development of two proteinaceous aggregates, amyloid-rich plaques and tau-rich neurofibrillary tangles (Selkoe, 2001). A large body of evidence suggests that beta-amyloid (Aβ), regardless of the structure (Dodart, et al., 2002,Gong, et al., 2003,Hardy and Selkoe, 2002,Hartley, et al., 1999,Lambert, et al., 1998,Selkoe and Schenk, 2003,Turner, et al., 2003) or location (Tseng, et al., 2004), plays a causal role in the pathogenesis of the disease.
Production of Aβ occurs through the proteolytic processing of amyloid-precursor protein (APP) by λ- and β-secretase (Selkoe, 2001,Sisodia, 1999,Vassar, 2004). Mutations in the genes for APP, presenilin1 (PS1) and presenilin2 (PS2) that cause early-onset familial Alzheimer's disease (FAD) drive the processing of APP toward the amyloidgenic pathway, leading to an overproduction of Aβ (Scheuner, et al., 1996,Younkin, 1994). Moreover, the inheritable FAD mutations preferentially increase the 42 amino acid species of Aβ (Aβ42) that is considered the most toxic variant of Aβ (Hutton, et al., 1998,Sisodia, 1999,Younkin, 1998). Although most cases of AD are idiopathic, the robust association of Aβ42 with FAD suggests a causative role for Aβ42 in the etiology of the disease. Therefore, the 5XFAD mouse was generated in order to drive very high levels of Aβ42 in the brain (Oakley, et al., 2006). This was accomplished by co-expressing five FAD mutations (altering both the APP and PS1 genes).
The onset of Aβ toxicity is closely tied to alterations in Ca2+ homeostasis, making it difficult to determine which event is the proximate cause of cognitive decline and neurodegeneration in AD (LaFerla, 2002). Nevertheless, Aβ peptides (Goodman and Mattson, 1994,Mattson, et al., 1992,Mattson, et al., 1993), and PS1 mutations (Duff, et al., 1996) and APP derivatives (Selkoe, 2001) have been shown to alter cytosolic Ca2+ through multiple, and in some cases concurrent, mechanisms (Arispe, et al., 1994,Bhatia, et al., 2000,Kawahara and Kuroda, 2000). The destabilization of intracellular calcium has been implicated as a cause of cognitive deficits and neurodegeneration (Khachaturian, 1987,Mattson and Chan, 2003,Saito, et al., 1993).
Several lines of evidence support the hypothesis that Aβ-induced synaptic dysfunction contributes to memory deficits observed in AD (Chang, et al., 2006,Jacobsen, et al., 2006,Kamenetz, et al., 2003), in part through the alteration of Ca2+-regulated signaling pathways (Thibault, et al., 2007,Xie, 2004). In addition, Aβ and/or the destabilization of Ca2+ homeostasis has been posited to disrupt the modulation of intrinsic excitability that has also been shown to be important for learning and memory (Disterhoft and Oh, 2006,Zhang and Linden, 2003).
The present study examined the functional consequences of FAD mutations, and resulting Aβ overproduction, on hippocampal neuronal excitability in the Tg6799 mouse model of AD (5XFAD), and mechanisms underlying Aβ's action. Moreover, we explored the effects of performance and retention of trace-fear conditioning on the intrinsic plasticity of CA1 neurons in the 5XFAD mouse.
2. Methods
2.1 Experimental procedures
The experimenters were kept blind to genotype, training status and retention status until after all data were analyzed.
2.2 Animals
Transgenic mice (5XFAD) that co-overexpress the human APP695 with the Swedish (K670N, M671L), Florida (I716V), London (V717I) mutations and human PS1 (M146L and L286V) mutations were generated (Oakley, et al., 2006). Heterozygous 5XFAD were intercrossed with B6/SJL mice (Taconic Farms Inc.) to generate the genotypes of interest. Males were selected and genotyping was performed by PCR analysis of tail DNA. Negative aged-matched littermates were used as wild type controls. Mice were generated and bred at Northwestern University and all animal procedures were approved by the Northwestern University Animal Care and Use Committee.
2.3 Trace-fear conditioning paradigm
The basic protocol for trace fear conditioning has been described previously (Kaczorowski and Disterhoft, 2009; Supplementary Figure 1). Adult male mice (2 and 4 mo) and middle-aged mice (8 mo) were trained in a Plexiglas conditioning chamber with stainless steel floor grid used for shock delivery. After the baseline period (150 s), mice received four pairings of the CS (tone, 3 kHz, 85 dB) and US (shock 1 s, 0.7 mA). The CS and US were separated by a 30-s empty trace interval and the intertrial interval was set at 210 s ± 10 s. The training chamber was wiped with 95% ethyl alcohol, placed in the hood to provide white noise at 75 dB, and illuminated with a 10 W bulb with lights in the experimental room off in order to make it distinct. Retention of the tone CS:US memory was tested 24 hours later in a novel context that contrasted the training chamber (original context) in the following ways: floor was lined with bedding rather than shock grid, chamber was reduced in size, chamber was located across the room, background noise was reduced to 60 dB, room lights remained on and the chamber was rinsed with water. Following a 150 s baseline, mice received 4 presentations of a 15-s tone trial with an intertrial interval set at 210 ± 10 s. Auditory trace fear memory was assessed by scoring freezing behavior with automated procedures (Freeze Frame) during the four 15-s CS and subsequent 30-s trace periods. One hour after testing in the novel context, retention of the contextual CS:US memory was tested by placing mice into the original context. Fear memory was tested for the entire 10 minute session. Mice were trained and tested one at a time. After behavioral tests were completed, mice were killed by decapitation under halothane anesthesia. Brains were removed and bisected down the midline; the right hemibrain was used for electrophysiological experiments and the left for either biochemical or histological assays.
2.4 ELISA quantification of brain Aβ42 levels
Hemibrains were snap-frozen to be used for sandwich ELISA assays of total Aβ42. The hemibrains were homogenized in 1× phosphate buffered saline supplemented with 1× AEBSF (Calbiochem protease inhibitor cocktail) (pH 8.0), and then extracted with 5 m guanidine HCl, and centrifuged at 20 000 g for 20 min at 4 °C to remove insoluble material. Supernata nt fractions were analyzed by sandwich ELISA using well-established human Aβ42 Signal Select TM immunoassay kits (Biosource, Camarillo, CA, USA) to quantify total levels of cerebral Aβ42. Detailed procedures were reported previously (Oakley, et al., 2006).
2.5 Histology
Hemibrains were fixed in paraformaldehyde/PBS and transferred into 30% sucrose/PBS containing 0.01% sodium azide, sectioned sagittally, and stained according to Oakley and colleagues (Oakley et al., 2006). Plaque analyses were obtained on 3-7 sections per animal and normalized to the area of CA1 for each section (Craft, et al., 2004). Plaque counts were performed by manually counting all the plaques in CA1 region on each section by a blinded experimenter, and the area of CA1 was determined by outlining the boundaries of all plaques on each section using Image J Software (NIH public domain software) so that the average density of plaques in CA1 was calculated. No obvious differences were observed in the size of the plaques in age-matched 5XFAD learners versus weak-learners, and therefore we did not measure total plaque area.
2.6 Electrophysiology
Whole-cell current-clamp recordings were made from CA1 pyramidal neurons from mouse dorsal hippocampal slices (300 μm) that were first incubated at 34°C in bubbled artificial cerebral spinal fluid (aCSF, in mM): 125 NaCl, 25 glucose, 25 NaHCO3, 2.5 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2 (pH 7.5, bubbled with 95%O2/5%CO2) for 30 minutes, and then incubated at room temperature in bubbled aCSF for 1-4 hours before use. Electrodes prepared from thin-walled capillary glass were filled with 115 mM potassium methylsulfate based internal solution that contained 20 KCl, 10 Na-PCr, 10 HEPES, 2 MgATP, 0.3 NaGTP and 0.10% Biocytin (pH to 7.3) and had a resistance of 4-5 MΩ. The effects of KMeth internal solution on input resistance in CA1 mouse neurons has been shown to provide a stable recording environment (Kaczorowski, et al., 2007). Unless otherwise stated, chemicals were obtained from Sigma (St. Louis, MO). Potassium methylsulfate was purchased from ICN (New York, NY). Slices were transferred to a recording chamber mounted on a Zeiss Axioskop (Oberkochen, Germany) where they were submerged in oxygenated aCSF at 33-34°C.
Neurons were visualized with infrared differential video interference microscopy. Cells were evaluated and accepted for use only if they had a resting membrane potential < -58 mV, access resistance (Rs) < 40 MΩ, input resistance (RN) > 30 MΩ, and an action potential amplitude greater than 70 mV relative to action potential threshold. All recordings were done in the current-clamp mode using a Dagan current-clamp amplifier; cells were held at -66 mV by manually adjusting the holding current (< -100 pA). The electrode capacitance and series resistance were monitored, compensated, and recorded. Data were transferred to a computer using an ITC-16 digital-to-analog converter (InstruTech, Port Washington, NY). IgorPro (Wavemetrics, Lake Oswego, OR) software was used for acquisition and analysis. AHPs were triggered by a high-frequency train of action potentials that consisted of 25 action potentials at 50 Hz and were reported as an average of 2-4 sweeps per cell. The somatic current injection was set at +1 nA to ensure faithful generation of action potentials with reproducible timing. The AHP was defined as the membrane potential beginning at the peak negative value (relative to the initial baseline) following the last spike and ending when the voltage had decayed to 95% of that peak value. The integral of the AHP was calculated and shown as the area relative to the initial baseline from the peak (0 s) out +10 s. The AHP was then divided into bins of varying durations. In order to facilitate repeated-measures statistical comparisons of AHPs obtained under different conditions, the fast repolarizing component of the AHP (the first 1000 ms) was divided into bins of varying duration (y, in milliseconds), according to the equation y=i*2(x+1), where i is the sample interval (0.05 ms for 20 Hz sampling rate), 2(x+1) is the number of points being averaged, and x is the bin number (beginning with x=0). This equation afforded us the ability to sample the early component of the AHP at a very high rate, while gradually reducing the sampling rate as the magnitude of the AHP decreased over time. Because the AHP slowly decays after 1 second, the remainder of the AHP (> 1000 ms) values were averaged into bins of 1024 ms. The value of the AHP in each bin was plotted versus time on a log scale beginning at 1 ms after the AHP peak. The Ca2+-sensitve AHP (Ca2+-AHP) was identified based on the sensitivity to divalent Ca2+ blockers (Cd2+ or Ni2+).
Single action potentials were elicited using a brief (2 ms) current step, the amplitude of which was set to be within 20 pA of the threshold current (Ithreshold). The fast afterdepolarization (fADP) amplitude measured following a single action potential was reported as the average membrane potential, relative to the resting membrane potential, in the first 10 ms after the fast repolarization phase of the action potential, as described previously (Metz, et al., 2005). The total fADP was reported as the integral of the fADP beginning after the fast repolarization phase of the action potential and terminating when membrane potential returned to the resting value. Action potential threshold was defined as the voltage when dV/dt first exceeded 28 mV/ms and was confirmed by eye as corresponding to the inflection point at which the action potential began. The action potential amplitude (spike height) was defined as the change in voltage from action potential threshold to the maximum voltage achieved during the action potential, as described previously (Golding, et al., 2001). Membrane potentials were not corrected for the liquid junction potential, which was calculated to be 8 mV.
2.7 Statistical analysis
Statistical tests were performed using SPSS software (SPSS Inc., Chicago, IL). Significance was determined by repeated measures ANOVA or one-way ANOVA with post-hoc Fisher's least significant difference t-tests where appropriate (unless otherwise noted). All results are reported as mean ± SEM.
3. Results
3.1 Age-related increase in Aβ42, more so than plaque density, coincides with spatial memory deficits in the 5XFAD mouse
We first set out to characterize the behavioral phenotype, and measure Aβ42 levels in male 5XFAD mice from 2, 4 and 8 mo of age. Mice used for behavioral experiments were 70 males; WT, 2 mo (n = 6), 4 mo (n = 8) and 8 mo (n = 25); 5XFAD, 2 mo (n = 5), 4 mo (n = 9) and 8 mo (n = 17). To assess hippocampal function, mice were trained on a trace fear conditioning task followed by retention tests of the tone CS and contextual CS memory.
During training on trace fear conditioning, no effect of the transgene was observed on measures of baseline freezing (F (1, 64) = 0.01, p = 0.9), the expression of freezing during tone CS (F (1, 64) = 0.3, p = 0.6), or post-shock freezing (F (1, 64) = 2.5, p = 0.1) suggesting that measures of anxiolysis or expression of behavioral freezing (measured index of fear) did not differ between WT and 5XFAD mice (Figure A1, B1, C1). Retention of the tone CS:US memory was tested 24 hours later in a novel context (see Methods). Data from two mice were excluded due to video malfunction. Following a 150 s baseline, mice received 4 presentations of the tone CS alone (in the absence of foot shock). Neither baseline freezing level (F (1, 62) = 0.5, p = 0.5), conditional freezing in response to the tone CS (F (1, 62) = 0.002, p = 0.95) or trace CS (F (1, 62) = 0.004, p = 0.95) differed between WT and 5XFAD mice (Figure A2, B2, C2). Thus, retention of the tone CS:US memory following trace fear conditioning was intact in 5XFAD compared to WT mice across all age groups.
In contrast, 5XFAD mice showed significantly less freezing (F (1, 62) = 4.5, p < 0.05) compared to WTs when retention of the contextual CS:US memory was tested (Figure 2A). Post-hoc analysis revealed middle-aged (8 mo) 5XFAD mice froze significantly less than age-matched WT mice (p < 0.01). Behavioral impairments reported here in a subset of 5XFAD mice, similar to those reported elsewhere (Oakley, et al., 2006,Ohno, et al., 2006a), mapped onto an increase in Aβ42 with age (F (2, 18) = 25.0, p < 0.001, Figure 2B) and such impairments were correlated with Aβ42 in a subset of 8 mo learner and weak-learner 5XFAD mice (R2 = 0.61, p < 0.05). Although some previous reports have shown a correlation between hippocampus-dependent memory and plaque burden (Chen, et al., 2000,Hsiao, et al., 1996), we found only a weak correlation between plaque density and degree of contextual fear (R2 = 0.23, p = 0.10) in a subset of 8 mo learner and weak-learner 5XFAD mice (Figure 2C, inset). Thus, our data are more consistent with reports showing memory deficits independent of Aβ plaque burden in mouse models of AD (Holcomb, et al., 1999,Jacobsen, et al., 2006,Koistinaho, et al., 2001) and in human AD patients (Neary, et al., 1986,Terry, et al., 1991). Note that we observed an increase in the heterogeneity in the performance of 5XFAD mice with increasing Aβ42 across the age groups (Figure 2C).
Figure 2. Age-related increase in Aβ42 coincides with deficits on retention of contextual fear in the 5XFAD Alzheimer's disease mouse model.
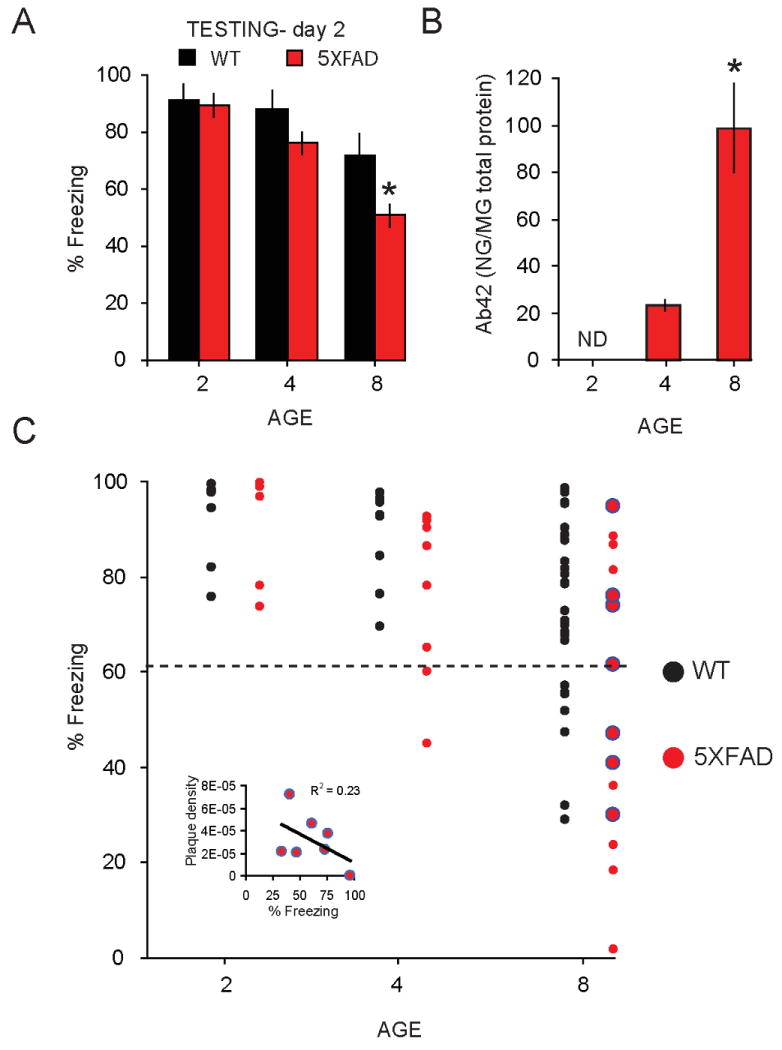
A, Significant decrease in mean % freezing during contextual fear retention test in 8 mo old 5XFAD (red) relative to WT mice (black) (* = p < 0.05). B, Age-related enhancement of Aβ42 in 5XFAD mice. A significant elevation in Aβ42 was observed at 4 mo, and continued to at 8 mo of age (p < 0.05). No detectable levels (ND) of Aβ42 were observed in brains of 2 mo old 5XFAD mice. C, Distribution of mice relative to their mean % freezing during contextual fear retention testing, age (2, 4, and 8 mo) in WT and 5XFAD mice. Points below black dashed line represent weak-learners, defined as mice with freezing levels < 3 SD from the mean freezing in 2 mo WT mice (61%).
The degree of impairment of contextual fear memory in each mouse was determined by comparison to a reference group of young WT mice tested concurrently (figure 2C). Dashed line indicates the criterion used to distinguish between learners and weak-learners, as shown previously (Kaczorowski and Disterhoft, 2009). Weak-learners were characterized as those mice whose performance on the contextual fear memory test fell below 3 SD of the mean (91 ± 10%) of our control group, the young 2 mo old mice. Based on this criterion, contextual memory deficits were first evident in a subset of 5XFAD 4 mo weak-learner mice. An examination of retention of the contextual fear memory indicated that 2 of 8 (25%) 4 mo old 5XFAD were weak-learner mice, whereas all 4 mo old WT mice met the criterion for learner status. An increase in the percentage of WT and 5XFAD weak-learners was observed at 8 mo, where a subset of both WT and 5XFAD mice performed below criterion (Figure 2C). Note that the percentage of 8 mo 5XFAD mice that were impaired on contextual fear memory, and termed weak-learners, differed significantly from that of the WT controls (WT, 30%; 5XFAD, 68%; Pearson chi-square χ2(1, N = 46) = 5.10, p < .05).
3.2 Intrinsic excitability reduced in neurons from 5XFAD mice
In order to examine the relationship between FAD mutations, memory deficits, and neuronal excitability in hippocampal neurons, we selected two age groups for biophysical characterization. The 2 mo group was selected because 5XFAD mice at this age were unimpaired on trace fear conditioning, and are reported to have no detectable levels of Aβ40 and Aβ42 in the hippocampus (Oakley et al., 2006). We replicated this finding and showed that no detectable levels of Aβ42 were observed in brains from 2 mo 5XFAD mice (Figure 2B); thus 2 mo 5XFAD mice served as our controls. The 8 mo group was selected because a subset of 5XFAD mice (67%) at this age showed a significant impairment of contextual fear memory, which corresponded to increased levels of Aβ42 reported herein (Figure 2B), and elsewhere (Oakley et al., 2006).
Whole-cell current-clamp recordings were performed in dorsal CA1 pyramidal neurons from naive 5XFAD and WT mice. The post-burst afterhyperpolarization (AHP) was elicited by twenty-five action potentials at a frequency of 50 Hz (Figure 3A), a stimulus shown to reliably evoke an AHP (Figure 3 C,D) of sizable - but not maximal - amplitude from hippocampal neurons of mice (Kaczorowski and Disterhoft, 2009,Ohno, et al., 2006b). Measurements of the AHP were used to assess neuronal excitability because AHPs have been shown to be inversely related to increased firing in pyramidal neurons (Lancaster and Nicoll, 1987,Madison and Nicoll, 1984,Sah, 1996,Storm, 1990). Neuronal excitability was significantly reduced in CA1 neurons from naïve 8 mo old 5XFAD mice (F (1, 60) = 5.3, p < 0.05) as evidenced by a increase in the mean amplitude of the AHP peak (AHP(peak) : 5XFAD, -4.4 ± 0.3 mV, n = 27cells; WT, -3.7 ± 0.2 mV, n = 35; p < 0.01) lasting 300 ms (AHP(300 ms) : 5XFAD, -1.6 ± 0.2 mV, n = 27cells; WT, -1.2 ± 0.1 mV, n = 35; p < 0.01) in neurons from 8 mo naïve 5XFAD mice compared to age-matched WTs (Figure 3). No differences were observed in the amplitude of the AHP measured from 300 ms to 10 s in neurons from 8 mo naïve 5XFAD compared to WT mice (p = 0.8). Note that no changes in basic membrane properties were observed in neurons from 5XFAD compared to WT 8 mo mice (Table 1).
Figure 3. Neuronal excitability is decreased in CA1 hippocampal neurons from 8 mo old 5XFAD mice.
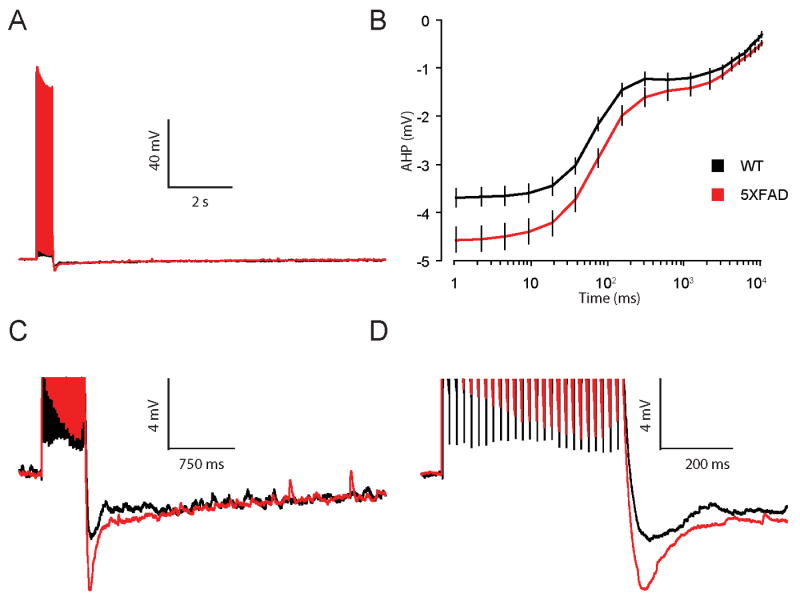
A, Representative traces of the AHP measured in whole-cell current clamp mode from neurons of 8 mo old 5XFAD (red) and WT (black) mice. AHPs were elicited by 25 brief current injections to elicit 25 action potentials at 50 Hz. B, Summary plot of the AHP versus time on a log scale for 8 mo old 5XFAD (red) and WT (black) mice. The AHP was averaged into bins to give mean and SEM at multiple time points (see Methods). A significant increase was observed in the mean AHP measured from the AHP peak (1 ms) out to 300 ms, but not at any time point > 600 ms, in neurons from 5XFAD compared to WT mice (* p < 0.05). C,D, AHP on an expanded scale.
Table 1. Basic membrane properties in neurons from naïve 8 mo 5XFAD and WT mice.
|
Vm (mV) |
Threshold (mV) |
Spike height (mV) |
Half-width (ms) |
fADP (mV) |
|
|---|---|---|---|---|---|
| 5XFAD (30) | -67.2 ± 0.1 | -53.6 ± 0.7 | 113 ± 1 | 0.99 ± .05 | 12.8 ± 0.9 |
| WT (35) | -68.0 ± 0.1 | -54.1 ± 0.4 | 116 ± 0.7 | 1.0 ± 0.01 | 12.3 ± 0.5 |
Numbers of cells per group are indicated in parentheses.
In contrast to neurons from 8 mo naïve 5XFAD mice, the AHP from neurons of 2 mo naïve 5XFAD mice were not significantly different from age-matched WT controls measured from 1 ms to 10 s (F(1, 13) = 0.6, p = 0.4, Figure 4 A-D). Taken together, these data suggest that FAD mutations coincident with elevated Aβ42 and/or plaque deposition (8 mo 5XFAD mice), corresponded to decreased neuronal excitability in neurons from 5XFAD mice evidenced by an increase in the AHP (300 ms).
Figure 4. Neuronal excitability is comparable in CA1 hippocampal neurons from 2 mo old WT and 5XFAD mice.
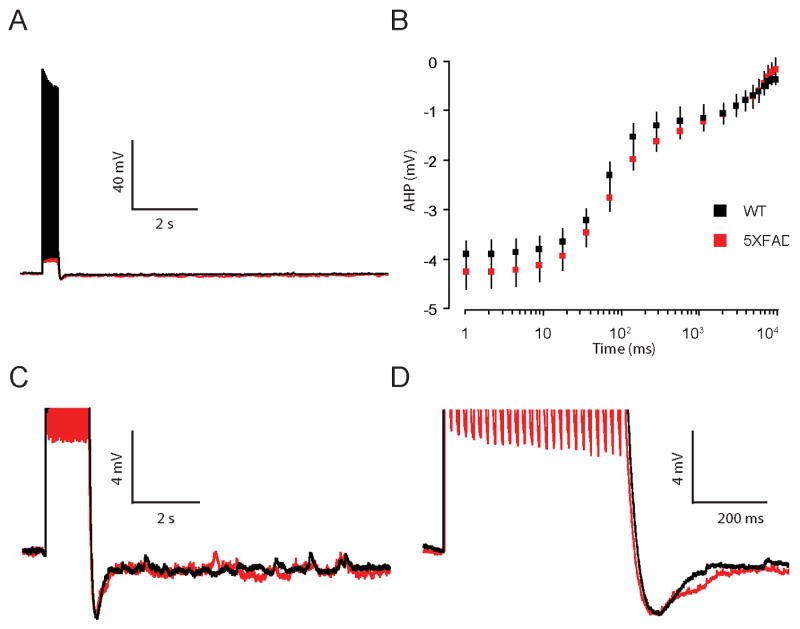
A, Representative traces of the AHP measured in whole-cell current clamp mode from neurons of 2 mo old 5XFAD (red) and WT (black) mice. AHPs were elicited by 25 brief current injections to elicit 25 action potentials at 50 Hz. B, Summary plot of the AHP versus time on a log scale for 2 mo old 5XFAD (red) and WT (black) mice. The AHP was averaged into bins to give mean and SEM at multiple time points (see Methods). No significant differences in the mean AHP were observed in 5XFAD compared to WT mice at 2 mo of age (p = 0.4). C,D, AHP on an expanded scale.
3.3 Selective enhancement of the Ca2+-sensitive AHP in neurons from 5XFAD mice
Based on the known effects FAD mutations, and Aβ peptides, on resting and evoked intracellular Ca2+ levels (Goodman and Mattson, 1994,Mattson, et al., 1992,Mattson, et al., 1993), we hypothesized that the enhancement of the AHP in neurons from 8 mo 5XFAD mice resulted from an increase in the Ca2+-sensitive component of the AHP. In line with our hypothesis, the AHP peak, AHP(300 ms), in CA1 neurons from 8 mo 5XFAD mice was reduced by 73 ± 19% with bath application of either the divalent Ca2+-channel blocker Ni2+ (750 μM) or Cd2+ (200 μM), (Figure 5A). Divalent Ca2+-channel blockers (CCB) were also effective in reducing the AHP in CA1 neurons from WT (Figure 5B) mice, albeit to a lesser extent (51 ± 13%). As a control measure, the effects of bath application of Cd2+ on suppression of EPSPs were monitored in a subset of neurons to verify efficacy of Ca2+-channel blockers (data not shown, n = 5). Comparison of AHPs in neurons from 5XFAD and WT mice in the presence of Ca2+-channel blockers showed that these AHPs were indistinguishable, and no differences between groups were observed. Overall, blockade of Ca2+ channels reduced the peak AHP (collectively driven by IAHP and sIAHP) in neurons from 5XFAD to a greater extent than in WT mice (t (1,31) = 1.8, p < 0.05, one-tailed, Figure 5C, inset), thus restoring the level of excitability in 5XFAD neurons to that of WT mice (Figure 5C). These data confirmed that the mechanism underlying the effects of FAD mutations on enhancement of the AHP, in concert with over production of Aβ, is a Ca2+-dependent one that has been shown previously to relate to learning and memory abilities (Disterhoft and Oh, 2006).
Figure 5. FAD mutations selectively increase Ca2+-sensitive AHP in CA1 hippocampal neurons from 8 mo old 5XFAD mice.
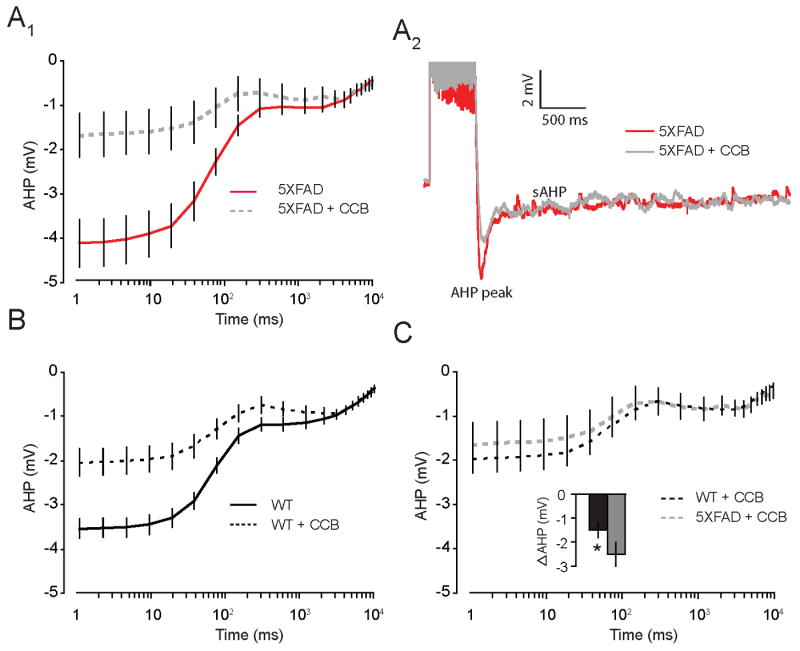
A1, Plot shows mean AHP versus time on a log scale in neurons from 8 mo 5XFAD mice. AHPs (1 ms – 300 ms) measured in artificial cerebral spinal fluid (aCSF) were significantly reduced after bath application of the divalent Ca2+ channel blockers Cd2+ (200 μM) or Ni2+ (750 μM). A2, Representative traces of the AHP measured in whole-cell current clamp mode from neurons of 8 mo old 5XFAD in ACSF (red) and following bath application of calcium channel blockers (CCB; Cd2+ (200 μM) or Ni2+ (750 μM); grey). B, Plot shows mean AHP versus time on a log scale in neurons from 8 mo WT mice. AHPs (1 ms – 300 ms) measured in artificial cerebral spinal fluid (aCSF), which were significantly reduced after bath application of the CCBs. C, Plot shows AHPs in presence of CCBs were equivalent in neurons from WT and 5XFAD mice. Inset, Plot shows amplitude of peak AHP from 5XFAD mice was significantly reduced compared to WTs (p < 0.05).
3.4 Learning-related plasticity of the AHP is impaired in 5XFAD mice
Work from our laboratory, and others, have demonstrated reductions in the AHP in neurons from animals that learn hippocampal-dependent spatial watermaze and trace eyeblink conditioning (Disterhoft and Oh, 2006). Recently, our laboratory has demonstrated a similar reduction in the AHP in CA1 neurons from mice (Kaczorowski, 2006; Kaczorowski and Disterhoft, 2009) and rats (McKay, et al., 2009) that learned trace fear conditioning. We also demonstrated that, during aging, animals that are impaired in hippocampal-dependent learning tasks exhibit an increased AHP in pyramidal neurons. Given that the AHP is enhanced in neurons of 8 mo naive 5XFAD mice, we hypothesized learning-related modulation of the AHP (AHP plasticity) - shown previously in WT learner mice (Kaczorowski and Disterhoft, 2009) - would be impaired in neurons from 5XFAD weak-learner mice.
In order to examine this hypothesis, whole-cell current clamp recordings were performed on CA1 neurons from a subset of 8 mo 5XFAD mice trained on trace fear conditioning. The AHPs from trained 5XFAD mice were compared based on a criterion set for contextual fear memory. Comparison of learner and weak-learner 5XFAD mice during acquisition of trace-fear, as well as auditory and contextual fear memory tests were performed. Examination of freezing behavior revealed no significant differences in measures of baseline freezing (F (1, 14) = 1.15, p = 0.3), the expression of freezing during tone CS (F (1, 14) = 0.7, p = 0.4), trace CS (F (1, 14) = 0.3, p = 0.6), or post-shock freezing (F (1, 14) = 0.3, p = 0.6) during training, and suggest that measures of anxiolysis or expression of behavioral freezing (measured index of fear) did not differ in 5XFAD learners compared to weak-learners (Figure 6A). Similarly, during testing of fear memory for the auditory and trace CS, no difference in baseline freezing was observed (F (1, 14) = 0.1, p = 0.8). A meaningful difference in mean % freezing to the tone CS (F (1, 14) = 6.3, p < 0.05), auditory trace CS (F (1, 14) = 6.4, p < 0.05), and contextual CS (F (1, 14) = 55.0, p < 0.001) was observed with a significant decrease in mean percent freezing exhibited by 5XFAD weak-learners compared to 5XFAD learners (Figure 6B,C).
Figure 6. Fear memory deficits in subset of 8 mo old 5XFAD mice.
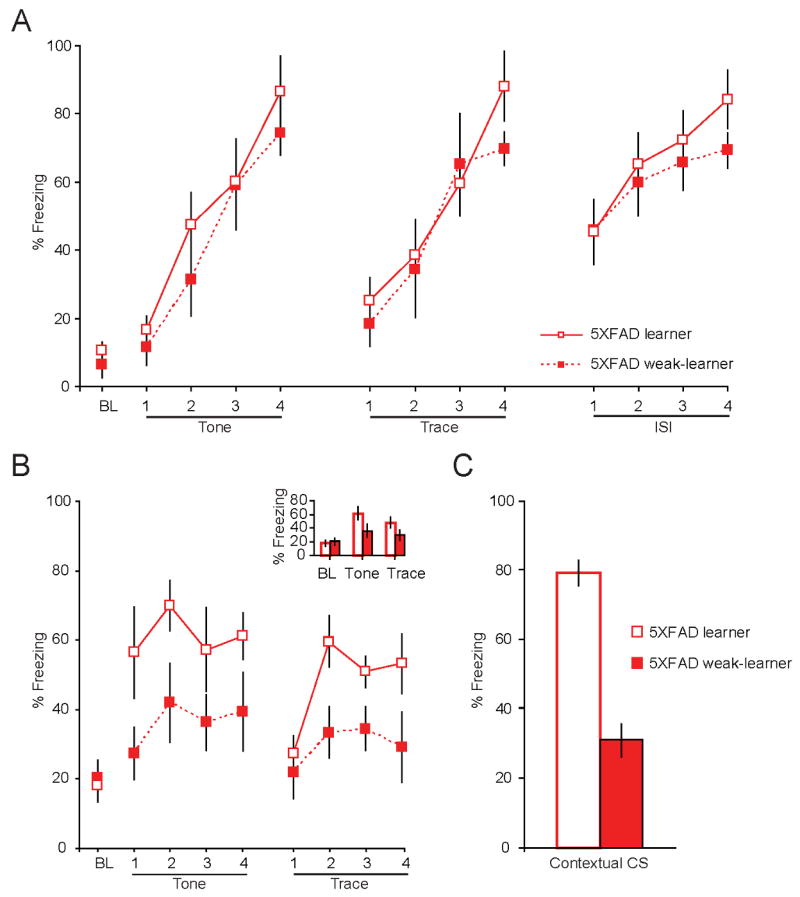
A, Mean % freezing during the baseline (BL), tone CS, trace CS and post-shock period (ISI) were comparable between 8 mo 5XFAD learners and weak-learners during training on trace fear conditioning. B, Retention of fear –as indexed by behavioral freezing – to the auditory tone CS (p < 0.05) and trace CS (p < 0.05) was significantly reduced in 8 mo old 5XFAD weak-learner compared to learner mice. Inset shows mean freezing, averaged across 4 trails, was reduced in response to tone CS presentations and trace CS in 5XFAD weak-learners compared to learners. B, Retention of the contextual CS memory was similarly impaired in 8 mo 5XFAD weak-learners compared to learners, as evidenced by a significant reduction in mean % freezing during a 10 minute presentation to the original training chamber (p < 0.001).
Analysis of electrophysiological recordings from neurons in the CA1 pyramidal layer in neurons from 5XFAD learners, 5XFAD weak-learners, and naïve 5XFAD mice revealed that AHPs from 5XFAD learners were significantly reduced compared to both 5XFAD weak-learners and 5XFAD naïve mice (F(2, 62) = 9.8, p < 0.001). Post-hoc students’ test revealed that the membrane potential from the peak AHP lasting for 10 s -AHP(10 s) was significantly reduced in neurons from 5XFAD learners compared to 5XFAD weak-learners (Figure 7A). AHPs from 5XFAD learner mice were similarly reduced compared to recordings from naïve 5XFAD mice (peak lasting 150 ms; 2 s – 10 s). The mean membrane potential of the AHP from naive 5XFAD mice fell between that of 5XFAD learners and 5XFAD weak-learners (Supplementary Figure 2). A similar learning-related reduction in the AHP was observed in neurons from WT learners compared to WT weak-learners (Kaczorowski and Disterhoft, 2009). Interestingly, the AHPs from WT learners and 5XFAD learners were virtually indistinguishable with no significant difference in the membrane potential of the AHP at all time points (Figure 7B). Thus, although the amplitude of the AHP in neurons from naïve 5XFAD mice is larger compared to naïve WTs, the learning-related reduction of the AHP in neurons from both 5XFAD and WT learner mice reaches the same amplitude. Taken together, these data suggest that FAD mutations – and increased Aβ - disrupt learning-related modulation of neuronal excitability. The AHP is greatest in neurons from weak-learners in our mouse model of AD. Compared to age-matched WTs, the 8 mo 5XFAD mice demonstrate more robust deficits on retention of spatial (contextual) and temporal (trace) associative memory following trace fear conditioning. The degree of AHP reduction in 5XFAD learners was greater compared to WT mice, thereby restoring the AHP in 5XFAD mice to an equivalent level of excitability found in 8 mo WT learners. Application of the mAChR agonist carbachol (10 μM) blocked the slow component of AHP in neurons from 5XFAD mice (Supplementary figure 3), suggesting the learning-related reduction of the AHP is sensitive to cholinergic modulation, as shown previously (Kronforst-Collins, et al., 1997a,Ohno, et al., 2004,Weiss, et al., 2000).
Figure 7. Learning-related modulation of the AHP was impaired in CA1 hippocampal neurons from 5XFAD weak-learner mice.
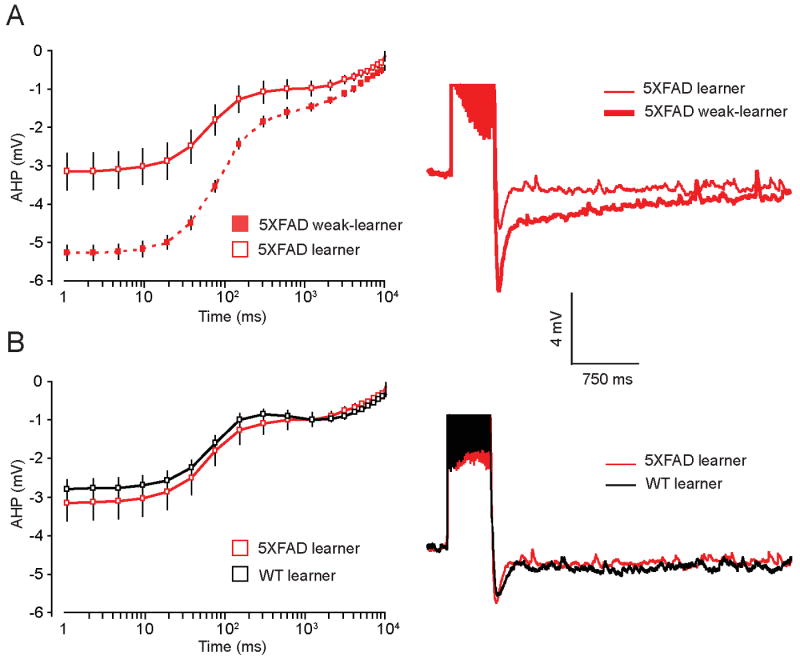
A, Plot shows mean AHP versus time on a log scale in neurons from 5XFAD learners and weak-learners. A significant increase in the mean amplitude of the late AHP (1 ms – 10 s), was observed in neurons from weak-learner 5XFAD mice compared to learner 5XFAD mice (p < 0.001). Right, representative traces of the AHP measured in neurons of 8 mo old 5XFAD learners (thin red) and weak-learners (thick red). B, Plot shows mean AHP versus time on a log scale, which is comparable in 5XFAD learners (red) and WT learners (black). Right, representative traces of the AHP measured in neurons of 8 mo old 5XFAD learners (red) and WT learners (black).
4. Discussion
Our combined use of behavioral, electrophysiological and molecular assays to examine the functional consequences of FAD mutations demonstrate that an increase in the Ca2+-dependent AHP observed in naïve 8 mo 5XFAD mice corresponded to elevated Aβ42 levels, and the onset of clear deficits in contextual fear memory. Thus, therapeutics aimed at restoring intracellular Ca2+ dynamics and/or reducing Ca2+-activated K+ currents (IAHP and sIAHP) that underlie the AHP may be efficacious in the treatment of AD. Second, the degree of trace and contextual fear conditioning deficits in 8 mo 5XFAD mice relates to the neuronal excitability of hippocampal neurons – the AHPs of neurons from 5XFAD weak-learner mice were larger than that of 5XFAD learners, as well as the average of 5XFAD naïve mice. This is the first demonstration of decreased excitability of hippocampal neurons associated with impaired trace fear conditioning – evidenced by larger AHP in 5XFAD weak-learners - in a mouse model of AD. This result may be driven by either an active enhancement of the AHP, occurring during, or as a result of neural activity evoked by fear conditioning procedures (albeit unsuccessful). Alternatively, this result may reflect the extreme end of the heterogeneity in the AHP in 5XFAD mice where a subset of 5XFAD mice have larger basal AHPs compared to the “average” naïve 5XFAD mice – such that mice with largest basal AHP show highest degree of impairment on the trace fear task. The latter is a less likely possibility given that we did not observe high degree of variation in the AHP amplitude in neurons from naïve 5XFAD mice that would be expected if the basal AHP in naïve 5XFAD mice were an average of two extremes.
Previous reports from our laboratory have shown that trace fear conditioning is a useful assay to examine mechanisms underlying Aβ-dependent deficits in temporal associative memory (Ohno, et al., 2006a). Although this early work describes deficits on both contextual and auditory trace fear memory in the 5XFAD mice as early as 4 mo of age, the present report did not observe a global deficit in auditory trace fear memory of 5XFAD mice compared to age-matched WTs at multiple age groups (2 mo, 4 mo, and 8 mo). This is likely due to the degree of heterogeneity in retention of auditory trace-fear in our 4 mo and 8 mo old 5XFAD mice. In support of this suggestion, deficits in both trace fear memory and contextual fear memory were exposed in a subset of 8 mo 5XFAD mice (weak-learners) whose performance did not reach established criterion for retention of conditioned fear. Additionally, the inclusion of female 5XFAD mice in early reports, compared to our exclusive use of males, may account for these differences as female 5XFAD mice show accelerated onset and increased magnitude of Aβ42 expression compared to male 5XFAD mice from 2-6 mo of age (Oakley, et al., 2006). Because the retention of contextual fear (spatial associative learning) is more sensitive to hippocampal dysfunction than retention of auditory trace fear (Chowdhury, et al., 2005,Kaczorowski, 2006,Kaczorowski and Disterhoft, 2009,Moyer and Brown, 2006), the 5XFAD mouse is a good model of AD as it recapitulates progressive, age-sensitive deficits in hippocampal-dependent memory impairment that accompany an increase in Aβ and a decrease in neuronal excitability. Importantly, the expression of freezing observed during training was unaltered in the 5XFAD compared to WT mice – indicative of a functional amygdala in 2-8 mo old 5XFAD mice. With further aging, we expect more profound retention deficits, and eventually acquisition deficits as the production of Aβ increases in the hippocampus, cortex and amygdala - regions required for several variants of fear conditioning.
Age-related reductions in intrinsic excitability, evidenced by an increase in the AHP in neurons from rat (Landfield and Pitler, 1984,Matthews, et al., 2009,Pitler and Landfield, 1990) and rabbit (Moyer, et al., 1992) hippocampal pyramidal neurons, correlate with learning and memory deficits (Moyer, et al., 2000,Power, et al., 2002,Tombaugh, et al., 2005). Moreover, AD-related enhancement of AHPs have been observed in response to acute application of Aβ-peptides in granule cells (Yun, et al., 2006), as well as in CA3 neurons from mice with PS1 mutations (Barrow, et al., 2000). However, the Ca2+ dependence of these AHPs was not explored. Here we show APP/PS1 FAD mutations result in a similar reduction in excitability that is evidenced by a selective increase in the Ca2+-dependent AHP in neurons from naïve 8 mo 5XFAD mice. Previous work from our laboratory failed to detect changes in the amplitude of the AHP in an alternative mouse model, the Tg2576 mouse (Ohno, et al., 2004). Thus, FAD mutations restricted to the APP transgene (e.g. Tg2576 model), in the absence of PS1 mutations, may not produce an enhancement of the AHP. This contention is supported by a recent report showing PS1 mutations significantly enhance calcium signals in neurons compared to mouse models that incorporate only APP mutations (Stutzmann, et al., 2006). Taken together, our work parallels early work in aging studies. We observed increased Ca2+-dependent AHPs in neurons from middle-aged (8 mo) 5XFAD mice that show clear hippocampal-dependent memory deficits.
Moreover, we show AHPs from 5XFAD mice are sensitive to the application of the cholinergic agonist, carbachol. This finding is significant because the selective muscarinic acetylcholine receptor1 (mAChR1) agonist, AF267B, restores cognitive function in the 3x-Tg mouse model of AD (Caccamo, et al., 2006). Thus, AF267B may facilitate learning and memory through enhancement of neuronal excitability by reducing the Ca2+-dependent AHP. Reductions in the AHP using the mAChR1 agonist CI-1017 (Weiss, et al., 2000) or metrifonate (Oh, et al., 1999,Power, et al., 2001), a cholinesterase inhibitor, have been shown previously to coincide with restored learning in aged rabbits (Kronforst-Collins, et al., 1997a,Kronforst-Collins, et al., 1997b,Weiss, et al., 2000). It is important to note that although we did not observe differences in the AHP lasting longer than 300 ms in naïve 5XFAD compared to WTs, cholinergic function may still be disrupted in 5XFAD mice. In fact, our laboratory has shown that although the AHP in naïve Tg2576 and WTs were comparable, the ability of Tg2576 neurons to be modulated by low levels of mAChR agonists was significantly impaired (Ohno, et al., 2004).
Previous work from our laboratory and others has shown that increased hippocampal excitability, evidenced as a reduction in the AHP, is a cellular consequence of learning and is acetylcholine dependent (Saar, et al., 2001,Weiss, et al., 2000). Learning a hippocampus-dependent task in young and aged rabbits (Moyer, et al., 2000), rats (Kuo, 2004,Oh, et al., 2003,Tombaugh, et al., 2005) and mice (Ohno, et al., 2006b) results in a reduction in the AHP in CA1 pyramidal neurons. Here we found a similar and significant learning-related reduction in the Ca2+-sensitive AHP in neurons from a subset of middle-aged 5XFAD learners as compared to 5XFAD weak-learners and naïve 5XFAD mice. The amplitude of the AHP in neurons from 5XFAD learners was comparable to that of age-matched WT learners.
In conclusion, our data demonstrate that FAD mutations, and downstream targets, alter the intrinsic excitability of CA1 hippocampal pyramidal neurons via two distinct mechanisms. First, a FAD specific enhancement in the Ca2+-dependent AHP (1 – 300 ms) was observed in neurons from naïve 8 mo 5XFAD mice, which correspond to the age of onset of memory impairments. These data suggest that a general decrease in basal neuronal excitability observed in neurons from naïve 8 mo 5XFAD mice contribute to learning and memory impairments that emerge during middle-age. Second, the late AHP (1 - 10 s), shown to be carbachol-sensitive, in neurons from 5XFAD weak-learner mice was increased in both amplitude and duration compared to that observed in neurons from 5XFAD learners, but not 5XFAD naïve mice. Thus, neurons from 5XFAD weak-learners fail to express/undergo learning-induced modulation of the late, carbachol-sensitive, AHP seen in neurons from 5XFAD learners. Importantly, such altered modulation of the carbachol-sensitive AHP reported herein has recently been reported in neurons from middle-aged WT weak-learner mice (Kaczorowski and Disterhoft, 2009).
Deficits in hippocampal synaptic transmission and long-term potentiation (LTP) have been well characterized in a number of mouse models of AD (Fitzjohn, et al., 2001,Hsia, et al., 1999,Jacobsen, et al., 2006,Nalbantoglu, et al., 1997). Given that changes in intrinsic excitability can alter synaptic throughput (Zhang and Linden, 2003), the changes we see here likely contribute to changes at the level of the synapse. In support of this idea, manipulations that alter the AHP have also been shown to affect EPSP temporal summation (Lancaster, et al., 2001) and LTP (Kramar, et al., 2004,Kumar and Foster, 2004,Le Ray, et al., 2004,Murphy, et al., 2004,Sourdet, et al., 2003,Thibault, et al., 2001). Thus, our results, in combination with previous work, suggest that FAD mutations and overproduction of Aβ disrupt basal and learning-related changes in neuronal excitability, which underlie hippocampal-learning and memory deficits observed in Alzheimer's disease.
Supplementary Material
A, Training: Day 1, schematic representation of the temporal relationship between the tone and the footshock for trace-fear conditioning training procedures. During training, freezing is a direct index of fear. Testing: Day 2, schematic representation of procedures used to determine retention of the auditory and contextual CS-US memory 24 hours following trace-fear conditioning. Tests of tone CS:US memories occur in a novel context where freezing, an index of fear memory, is measured during presentation of the tone. Tests of contextual CS:US fear measure freezing during a10 minute exposure to the original context, the location where the animal was shocked the day before.
A, Plot shows mean AHP versus time on a log scale in neurons from 5XFAD naïve (grey), learners (red, solid) and weak-learners (red, dashed). A significant reduction in the mean amplitude of the AHP (1 ms – 150 ms; 2 – 10 s), was observed in neurons from 5XFAD learners compared to 5XFAD naïve mice (p < 0.001). Right, representative traces of the AHP measured in neurons of 8 mo old 5XFAD naïve (grey), 5XFAD learners (thin red) and 5XFAD weak-learners (thick red).
A1, The slow component of the AHP in neurons from 8-month old 5XFAD mice was completely blocked by the mAChR agonist carbachol (10 μM) (n = 7 cells/3 mice). Similar results were obtained in WT neurons and reported elsewhere (Kaczorowski, 2006). A2, Representative traces of the AHP measured in neurons of 8 mo old 5XFAD naïve in ACSF (red) and carbachol (grey).
Figure 1. Acquisition and retention of auditory trace fear conditioning is comparable in wild-type mice and the 5XFAD mouse model of Alzheimer's disease.
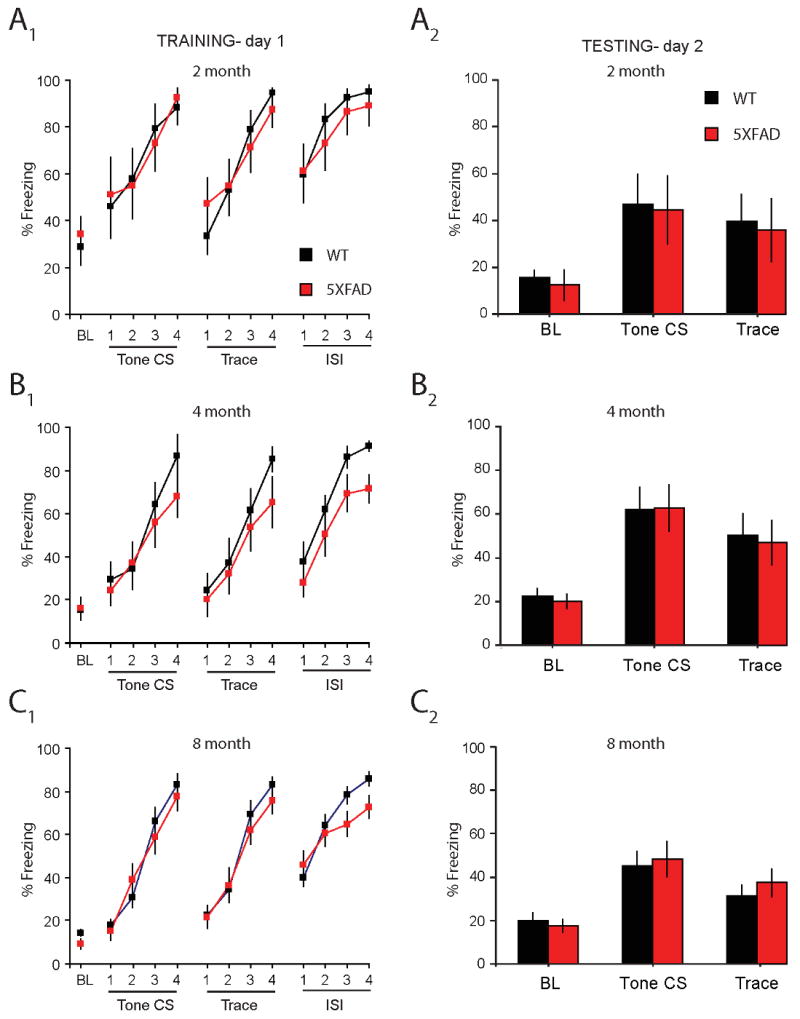
A1, B1, C1, Baseline (BL) freezing, tone CS freezing, trace CS freezing, and post-shock freezing (ISI) during training on trace fear conditioning was similar between young (2 mo and 4 mo) and middle-aged (8 mo) WT and 5XFAD mice. A2, B2, C2, Baseline (BL) freezing, retention of the tone CS memory (average of tone CS 1-4), and retention of the trace CS memory (average of trace CS intervals1-4) was comparable in WT and 5XFAD young (2 mo and 4 mo) and middle-aged (8 mo) mice.
Acknowledgments
The authors wish to thank Caroline Cook who provided advice on the illustrations. This work was supported by the National Institutes of Mental Health grant F31 MH-067445 awarded to C.C.K., and National Institutes of Health grant R37 AG-08796 and T32 AG20506 awarded to J.F.D.
Footnotes
Disclosure Statement. The authors have no actual or potential conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Arispe N, Pollard HB, Rojas E. beta-Amyloid Ca(2+)-channel hypothesis for neuronal death in Alzheimer disease. Mol Cell Biochem. 1994;140(2):119–25. doi: 10.1007/BF00926750. [DOI] [PubMed] [Google Scholar]
- Barrow PA, Empson RM, Gladwell SJ, Anderson CM, Killick R, Yu X, Jefferys JG, Duff K. Functional phenotype in transgenic mice expressing mutant human presenilin-1. Neurobiol Dis. 2000;7(2):119–26. doi: 10.1006/nbdi.1999.0276. [DOI] [PubMed] [Google Scholar]
- Bhatia R, Lin H, Lal R. Fresh and globular amyloid beta protein (1-42) induces rapid cellular degeneration: evidence for AbetaP channel-mediated cellular toxicity. Faseb J. 2000;14(9):1233–43. doi: 10.1096/fasebj.14.9.1233. [DOI] [PubMed] [Google Scholar]
- Caccamo A, Oddo S, Billings LM, Green KN, Martinez-Coria H, Fisher A, LaFerla FM. M1 receptors play a central role in modulating AD-like pathology in transgenic mice. Neuron. 2006;49(5):671–82. doi: 10.1016/j.neuron.2006.01.020. [DOI] [PubMed] [Google Scholar]
- Chang EH, Savage MJ, Flood DG, Thomas JM, Levy RB, Mahadomrongkul V, Shirao T, Aoki C, Huerta PT. AMPA receptor downscaling at the onset of Alzheimer's disease pathology in double knockin mice. Proc Natl Acad Sci U S A. 2006;103(9):3410–5. doi: 10.1073/pnas.0507313103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Chen KS, Knox J, Inglis J, Bernard A, Martin SJ, Justice A, McConlogue L, Games D, Freedman SB, Morris RG. A learning deficit related to age and beta-amyloid plaques in a mouse model of Alzheimer's disease. Nature. 2000;408(6815):975–9. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- Chowdhury N, Quinn JJ, Fanselow MS. Dorsal hippocampus involvement in trace fear conditioning with long, but not short, trace intervals in mice. Behav Neurosci. 2005;119(5):1396–402. doi: 10.1037/0735-7044.119.5.1396. [DOI] [PubMed] [Google Scholar]
- Craft JM, Van Eldik LJ, Zasadzki M, Hu W, Watterson DM. Aminopyridazines attenuate hippocampus-dependent behavioral deficits induced by human beta-amyloid in a murine model of neuroinflammation. J Mol Neurosci. 2004;24(1):115–22. doi: 10.1385/JMN:24:1:115. [DOI] [PubMed] [Google Scholar]
- Disterhoft JF, Oh MM. Pharmacological and molecular enhancement of learning in aging and Alzheimer's disease. J Physiol Paris. 2006;99(2-3):180–92. doi: 10.1016/j.jphysparis.2005.12.079. [DOI] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Abeta burden in Alzheimer's disease model. Nat Neurosci. 2002;5(5):452–7. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Duff K, Eckman C, Zehr C, Yu X, Prada CM, Perez-tur J, Hutton M, Buee L, Harigaya Y, Yager D, Morgan D, Gordon MN, Holcomb L, Refolo L, Zenk B, Hardy J, Younkin S. Increased amyloid-beta42(43) in brains of mice expressing mutant presenilin 1. Nature. 1996;383(6602):710–3. doi: 10.1038/383710a0. [DOI] [PubMed] [Google Scholar]
- Fitzjohn SM, Morton RA, Kuenzi F, Rosahl TW, Shearman M, Lewis H, Smith D, Reynolds DS, Davies CH, Collingridge GL, Seabrook GR. Age-related impairment of synaptic transmission but normal long-term potentiation in transgenic mice that overexpress the human APP695SWE mutant form of amyloid precursor protein. J Neurosci. 2001;21(13):4691–8. doi: 10.1523/JNEUROSCI.21-13-04691.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golding NL, Kath WL, Spruston N. Dichotomy of action-potential backpropagation in CA1 pyramidal neuron dendrites. J Neurophysiol. 2001;86(6):2998–3010. doi: 10.1152/jn.2001.86.6.2998. [DOI] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100(18):10417–22. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman Y, Mattson MP. Secreted forms of beta-amyloid precursor protein protect hippocampal neurons against amyloid beta-peptide-induced oxidative injury. Exp Neurol. 1994;128(1):1–12. doi: 10.1006/exnr.1994.1107. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353–6. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19(20):8876–84. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcomb LA, Gordon MN, Jantzen P, Hsiao K, Duff K, Morgan D. Behavioral changes in transgenic mice expressing both amyloid precursor protein and presenilin-1 mutations: lack of association with amyloid deposits. Behav Genet. 1999;29(3):177–85. doi: 10.1023/a:1021691918517. [DOI] [PubMed] [Google Scholar]
- Hsia AY, Masliah E, McConlogue L, Yu GQ, Tatsuno G, Hu K, Kholodenko D, Malenka RC, Nicoll RA, Mucke L. Plaque-independent disruption of neural circuits in Alzheimer's disease mouse models. Proc Natl Acad Sci U S A. 1999;96(6):3228–33. doi: 10.1073/pnas.96.6.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274(5284):99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hutton M, Perez-Tur J, Hardy J. Genetics of Alzheimer's disease. Essays Biochem. 1998;33:117–31. doi: 10.1042/bse0330117. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Wu CC, Redwine JM, Comery TA, Arias R, Bowlby M, Martone R, Morrison JH, Pangalos MN, Reinhart PH, Bloom FE. Early-onset behavioral and synaptic deficits in a mouse model of Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103(13):5161–6. doi: 10.1073/pnas.0600948103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski CC. Dissertation. 2006. Intrinsic plasticity in CA1 pyramidal neurons: Role of calcium(2+)- and sodium(+)-activated potassium currents in aging and Alzheimer's disease; pp. 1–291. [Google Scholar]
- Kaczorowski CC, Disterhoft J, Spruston N. Stability and plasticity of intrinsic membrane properties in hippocampal CA1 pyramidal neurons: effects of internal anions. J Physiol. 2007;578(Pt 3):799–818. doi: 10.1113/jphysiol.2006.124586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczorowski CC, Disterhoft JF. Memory deficits are associated with impaired ability to modulate neuronal excitability in middle-aged mice Learning & Memory. 2009 doi: 10.1101/lm.1365609. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37(6):925–37. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kawahara M, Kuroda Y. Molecular mechanism of neurodegeneration induced by Alzheimer's beta-amyloid protein: channel formation and disruption of calcium homeostasis. Brain Res Bull. 2000;53(4):389–97. doi: 10.1016/s0361-9230(00)00370-1. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Hypothesis on the regulation of cytosol calcium concentration and the aging brain. Neurobiol Aging. 1987;8(4):345–6. doi: 10.1016/0197-4580(87)90073-x. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Ort M, Cimadevilla JM, Vondrous R, Cordell B, Koistinaho J, Bures J, Higgins LS. Specific spatial learning deficits become severe with age in beta -amyloid precursor protein transgenic mice that harbor diffuse beta -amyloid deposits but do not form plaques. Proc Natl Acad Sci U S A. 2001;98(25):14675–80. doi: 10.1073/pnas.261562998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramar EA, Lin B, Lin CY, Arai AC, Gall CM, Lynch G. A novel mechanism for the facilitation of theta-induced long-term potentiation by brain-derived neurotrophic factor. J Neurosci. 2004;24(22):5151–61. doi: 10.1523/JNEUROSCI.0800-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronforst-Collins MA, Moriearty PL, Ralph M, Becker RE, Schmidt B, Thompson LT, Disterhoft JF. Metrifonate treatment enhances acquisition of eyeblink conditioning in aging rabbits. Pharmacol Biochem Behav. 1997a;56(1):103–10. doi: 10.1016/S0091-3057(96)00164-5. [DOI] [PubMed] [Google Scholar]
- Kronforst-Collins MA, Moriearty PL, Schmidt B, Disterhoft JF. Metrifonate improves associative learning and retention in aging rabbits. Behav Neurosci. 1997b;111(5):1031–40. doi: 10.1037//0735-7044.111.5.1031. [DOI] [PubMed] [Google Scholar]
- Kumar A, Foster TC. Enhanced long-term potentiation during aging is masked by processes involving intracellular calcium stores. J Neurophysiol. 2004;91(6):2437–44. doi: 10.1152/jn.01148.2003. [DOI] [PubMed] [Google Scholar]
- Kuo AG, Lee G, Disterhoft JF. Alteration of sIAHP underlies reduction of AHP in rat CA1 pyramidal neurons after trace eyebink conditioning. Society for Neuroscience Abstracts 741.13 2004 [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3(11):862–72. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95(11):6448–53. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Hu H, Ramakers GM, Storm JF. Interaction between synaptic excitation and slow afterhyperpolarization current in rat hippocampal pyramidal cells. J Physiol. 2001;536(Pt 3):809–23. doi: 10.1111/j.1469-7793.2001.00809.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Nicoll RA. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J Physiol. 1987;389:187–203. doi: 10.1113/jphysiol.1987.sp016653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landfield PW, Pitler TA. Prolonged Ca2+-dependent afterhyperpolarizations in hippocampal neurons of aged rats. Science. 1984;226(4678):1089–92. doi: 10.1126/science.6494926. [DOI] [PubMed] [Google Scholar]
- Le Ray D, Fernandez De Sevilla D, Belen Porto A, Fuenzalida M, Buno W. Heterosynaptic metaplastic regulation of synaptic efficacy in CA1 pyramidal neurons of rat hippocampus. Hippocampus. 2004;14(8):1011–25. doi: 10.1002/hipo.20021. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Control of the repetitive discharge of rat CA 1 pyramidal neurones in vitro. J Physiol. 1984;354:319–31. doi: 10.1113/jphysiol.1984.sp015378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews E, Linardakis JM, Disterhoft JF. The fast AHP and the slow AHP are differentially modulated in hippocampal neurons by aging and by learning. Journal of Neuroscience. 2009 doi: 10.1523/JNEUROSCI.0384-09.2009. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Chan SL. Neuronal and glial calcium signaling in Alzheimer's disease. Cell Calcium. 2003;34(4-5):385–97. doi: 10.1016/s0143-4160(03)00128-3. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12(2):376–89. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP, Tomaselli KJ, Rydel RE. Calcium-destabilizing and neurodegenerative effects of aggregated beta-amyloid peptide are attenuated by basic FGF. Brain Res. 1993;621(1):35–49. doi: 10.1016/0006-8993(93)90295-x. [DOI] [PubMed] [Google Scholar]
- McKay BM, Matthews EA, Oliveira FA, Disterhoft JF. Intrinsic neuronal excitability is reversibly altered by a single experience in fear conditioning. J Neurophysiology. 2009 doi: 10.1152/jn.00347.2009. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz AE, Jarsky T, Martina M, Spruston N. R-type calcium channels contribute to afterdepolarization and bursting in hippocampal CA1 pyramidal neurons. J Neurosci. 2005;25(24):5763–73. doi: 10.1523/JNEUROSCI.0624-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JR, Jr, Brown TH. Impaired trace and contextual fear conditioning in aged rats. Behav Neurosci. 2006;120(3):612–24. doi: 10.1037/0735-7044.120.3.612. [DOI] [PubMed] [Google Scholar]
- Moyer JR, Jr, Power JM, Thompson LT, Disterhoft JF. Increased excitability of aged rabbit CA1 neurons after trace eyeblink conditioning. J Neurosci. 2000;20(14):5476–82. doi: 10.1523/JNEUROSCI.20-14-05476.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer JR, Jr, Thompson LT, Black JP, Disterhoft JF. Nimodipine increases excitability of rabbit CA1 pyramidal neurons in an age- and concentration-dependent manner. J Neurophysiol. 1992;68(6):2100–9. doi: 10.1152/jn.1992.68.6.2100. [DOI] [PubMed] [Google Scholar]
- Murphy GG, Fedorov NB, Giese KP, Ohno M, Friedman E, Chen R, Silva AJ. Increased neuronal excitability, synaptic plasticity, and learning in aged Kvbeta1.1 knockout mice. Curr Biol. 2004;14(21):1907–15. doi: 10.1016/j.cub.2004.10.021. [DOI] [PubMed] [Google Scholar]
- Nalbantoglu J, Tirado-Santiago G, Lahsaini A, Poirier J, Goncalves O, Verge G, Momoli F, Welner SA, Massicotte G, Julien JP, Shapiro ML. Impaired learning and LTP in mice expressing the carboxy terminus of the Alzheimer amyloid precursor protein. Nature. 1997;387(6632):500–5. doi: 10.1038/387500a0. [DOI] [PubMed] [Google Scholar]
- Neary D, Snowden JS, Mann DM, Bowen DM, Sims NR, Northen B, Yates PO, Davison AN. Alzheimer's disease: a correlative study. J Neurol Neurosurg Psychiatry. 1986;49(3):229–37. doi: 10.1136/jnnp.49.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26(40):10129–40. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh MM, Kuo AG, Wu WW, Sametsky EA, Disterhoft JF. Watermaze learning enhances excitability of CA1 pyramidal neurons. J Neurophysiol. 2003;90(4):2171–9. doi: 10.1152/jn.01177.2002. [DOI] [PubMed] [Google Scholar]
- Oh MM, Power JM, Thompson LT, Moriearty PL, Disterhoft JF. Metrifonate increases neuronal excitability in CA1 pyramidal neurons from both young and aging rabbit hippocampus. J Neurosci. 1999;19(5):1814–23. doi: 10.1523/JNEUROSCI.19-05-01814.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M, Chang L, Tseng W, Oakley H, Citron M, Klein WL, Vassar R, Disterhoft JF. Temporal memory deficits in Alzheimer's mouse models: rescue by genetic deletion of BACE1. Eur J Neurosci. 2006a;23(1):251–60. doi: 10.1111/j.1460-9568.2005.04551.x. [DOI] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Silva AJ, Disterhoft JF. Differential effects of alphaCaMKII mutation on hippocampal learning and changes in intrinsic neuronal excitability. Eur J Neurosci. 2006b;23(8):2235–40. doi: 10.1111/j.1460-9568.2006.04746.x. [DOI] [PubMed] [Google Scholar]
- Ohno M, Sametsky EA, Younkin LH, Oakley H, Younkin SG, Citron M, Vassar R, Disterhoft JF. BACE1 deficiency rescues memory deficits and cholinergic dysfunction in a mouse model of Alzheimer's disease. Neuron. 2004;41(1):27–33. doi: 10.1016/s0896-6273(03)00810-9. [DOI] [PubMed] [Google Scholar]
- Pitler TA, Landfield PW. Aging-related prolongation of calcium spike duration in rat hippocampal slice neurons. Brain Res. 1990;508(1):1–6. doi: 10.1016/0006-8993(90)91109-t. [DOI] [PubMed] [Google Scholar]
- Power JM, Oh MM, Disterhoft JF. Metrifonate decreases sI(AHP) in CA1 pyramidal neurons in vitro. J Neurophysiol. 2001;85(1):319–22. doi: 10.1152/jn.2001.85.1.319. [DOI] [PubMed] [Google Scholar]
- Power JM, Wu WW, Sametsky E, Oh MM, Disterhoft JF. Age-related enhancement of the slow outward calcium-activated potassium current in hippocampal CA1 pyramidal neurons in vitro. J Neurosci. 2002;22(16):7234–43. doi: 10.1523/JNEUROSCI.22-16-07234.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saar D, Grossman Y, Barkai E. Long-lasting cholinergic modulation underlies rule learning in rats. J Neurosci. 2001;21(4):1385–92. doi: 10.1523/JNEUROSCI.21-04-01385.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P. Ca(2+)-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 1996;19(4):150–4. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- Saito K, Elce JS, Hamos JE, Nixon RA. Widespread activation of calcium-activated neutral proteinase (calpain) in the brain in Alzheimer disease: a potential molecular basis for neuronal degeneration. Proc Natl Acad Sci U S A. 1993;90(7):2628–32. doi: 10.1073/pnas.90.7.2628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D, Eckman C, Jensen M, Song X, Citron M, Suzuki N, Bird TD, Hardy J, Hutton M, Kukull W, Larson E, Levy-Lahad E, Viitanen M, Peskind E, Poorkaj P, Schellenberg G, Tanzi R, Wasco W, Lannfelt L, Selkoe D, Younkin S. Secreted amyloid beta-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat Med. 1996;2(8):864–70. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81(2):741–66. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, Schenk D. Alzheimer's disease: molecular understanding predicts amyloid-based therapeutics. Annu Rev Pharmacol Toxicol. 2003;43:545–84. doi: 10.1146/annurev.pharmtox.43.100901.140248. [DOI] [PubMed] [Google Scholar]
- Sisodia SS. Alzheimer's disease: perspectives for the new millennium. J Clin Invest. 1999;104(9):1169–70. doi: 10.1172/JCI8508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sourdet V, Russier M, Daoudal G, Ankri N, Debanne D. Long-term enhancement of neuronal excitability and temporal fidelity mediated by metabotropic glutamate receptor subtype 5. J Neurosci. 2003;23(32):10238–48. doi: 10.1523/JNEUROSCI.23-32-10238.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–87. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE, Smith I, Caccamo A, Oddo S, Laferla FM, Parker I. Enhanced ryanodine receptor recruitment contributes to Ca2+ disruptions in young, adult, and aged Alzheimer's disease mice. J Neurosci. 2006;26(19):5180–9. doi: 10.1523/JNEUROSCI.0739-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry RD, Masliah E, Salmon DP, Butters N, DeTeresa R, Hill R, Hansen LA, Katzman R. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572–80. doi: 10.1002/ana.410300410. [DOI] [PubMed] [Google Scholar]
- Thibault O, Gant JC, Landfield PW. Expansion of the calcium hypothesis of brain aging and Alzheimer's disease: minding the store. Aging Cell. 2007;6(3):307–17. doi: 10.1111/j.1474-9726.2007.00295.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault O, Hadley R, Landfield PW. Elevated postsynaptic [Ca2+]i and L-type calcium channel activity in aged hippocampal neurons: relationship to impaired synaptic plasticity. J Neurosci. 2001;21(24):9744–56. doi: 10.1523/JNEUROSCI.21-24-09744.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombaugh GC, Rowe WB, Rose GM. The slow afterhyperpolarization in hippocampal CA1 neurons covaries with spatial learning ability in aged Fisher 344 rats. J Neurosci. 2005;25(10):2609–16. doi: 10.1523/JNEUROSCI.5023-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng BP, Kitazawa M, LaFerla FM. Amyloid beta-peptide: the inside story. Curr Alzheimer Res. 2004;1(4):231–9. doi: 10.2174/1567205043332045. [DOI] [PubMed] [Google Scholar]
- Turner PR, O'Connor K, Tate WP, Abraham WC. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog Neurobiol. 2003;70(1):1–32. doi: 10.1016/s0301-0082(03)00089-3. [DOI] [PubMed] [Google Scholar]
- Vassar R. BACE1: the beta-secretase enzyme in Alzheimer's disease. J Mol Neurosci. 2004;23(1-2):105–14. doi: 10.1385/JMN:23:1-2:105. [DOI] [PubMed] [Google Scholar]
- Weiss C, Preston AR, Oh MM, Schwarz RD, Welty D, Disterhoft JF. The M1 muscarinic agonist CI-1017 facilitates trace eyeblink conditioning in aging rabbits and increases the excitability of CA1 pyramidal neurons. J Neurosci. 2000;20(2):783–90. doi: 10.1523/JNEUROSCI.20-02-00783.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie CW. Calcium-regulated signaling pathways: role in amyloid beta-induced synaptic dysfunction. Neuromolecular Med. 2004;6(1):53–64. doi: 10.1385/NMM:6:1:053. [DOI] [PubMed] [Google Scholar]
- Younkin SG. The amyloid beta protein precursor mutations linked to familial Alzheimer's disease alter processing in a way that fosters amyloid deposition. Tohoku J Exp Med. 1994;174(3):217–23. doi: 10.1620/tjem.174.217. [DOI] [PubMed] [Google Scholar]
- Younkin SG. The role of A beta 42 in Alzheimer's disease. J Physiol Paris. 1998;92(3-4):289–92. doi: 10.1016/s0928-4257(98)80035-1. [DOI] [PubMed] [Google Scholar]
- Yun SH, Gamkrelidze G, Stine WB, Sullivan PM, Pasternak JF, Ladu MJ, Trommer BL. Amyloid-beta(1-42) reduces neuronal excitability in mouse dentate gyrus. Neurosci Lett. 2006;403(1-2):162–5. doi: 10.1016/j.neulet.2006.04.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Linden DJ. The other side of the engram: experience-driven changes in neuronal intrinsic excitability. Nat Rev Neurosci. 2003;4(11):885–900. doi: 10.1038/nrn1248. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A, Training: Day 1, schematic representation of the temporal relationship between the tone and the footshock for trace-fear conditioning training procedures. During training, freezing is a direct index of fear. Testing: Day 2, schematic representation of procedures used to determine retention of the auditory and contextual CS-US memory 24 hours following trace-fear conditioning. Tests of tone CS:US memories occur in a novel context where freezing, an index of fear memory, is measured during presentation of the tone. Tests of contextual CS:US fear measure freezing during a10 minute exposure to the original context, the location where the animal was shocked the day before.
A, Plot shows mean AHP versus time on a log scale in neurons from 5XFAD naïve (grey), learners (red, solid) and weak-learners (red, dashed). A significant reduction in the mean amplitude of the AHP (1 ms – 150 ms; 2 – 10 s), was observed in neurons from 5XFAD learners compared to 5XFAD naïve mice (p < 0.001). Right, representative traces of the AHP measured in neurons of 8 mo old 5XFAD naïve (grey), 5XFAD learners (thin red) and 5XFAD weak-learners (thick red).
A1, The slow component of the AHP in neurons from 8-month old 5XFAD mice was completely blocked by the mAChR agonist carbachol (10 μM) (n = 7 cells/3 mice). Similar results were obtained in WT neurons and reported elsewhere (Kaczorowski, 2006). A2, Representative traces of the AHP measured in neurons of 8 mo old 5XFAD naïve in ACSF (red) and carbachol (grey).