

Abstract
The wild-type cholecystokinin type 2 (CCK2) receptor is expressed in many gastrointestinal and lung tumours. A splice variant of the CCK2 receptor with retention of intron 4 (CCK2Ri4sv) showing constitutive activity associated with increased tumour growth was described in few colorectal, pancreatic and gastric cancers. Given the potential functional and clinical importance of this spliceoform, its occurrence was quantitatively characterized in a broad collection of 81 gastrointestinal and lung tumours, including insulinomas, ileal carcinoids, gastrointestinal stromal tumours (GIST), gastric, colorectal and pancreatic ductal adenocarcinomas, cholangiocellular and hepatocellular carcinomas, small cell lung cancers (SCLC), non-SCLC (nSCLC) and bronchopulmonary carcinoids, as well as 21 samples of corresponding normal tissues. These samples were assessed for transcript expression of total CCK2 receptor, wild-type CCK2 receptor and CCK2Ri4sv with end-point and real-time RT-PCR, and for total CCK2 receptor protein expression on the basis of receptor binding with in vitro receptor autoradiography. Wild-type CCK2 receptor transcripts were found in the vast majority of tumours and normal tissues. CCK2Ri4sv mRNA expression was present predominantly in insulinomas (incidence 100%), GIST (100%) and SCLC (67%), but rarely in pancreatic, colorectal and gastric carcinomas and nSCLC. It was not found in wild-type CCK2 receptor negative tumours or any normal tissues tested. CCK2Ri4sv transcript levels in individual tumours were low, ranging from 0.02% to 0.14% of total CCK2 receptor transcripts. In conclusion, the CCK2Ri4sv is a marker of specific gastrointestinal and lung tumours. With its high selectivity for and high incidence in SCLC and GIST, it may represent an attractive clinical target.
Keywords: CCK2 receptor intron 4 splice variant, RT-PCR, receptor autoradiography, gastrointestinal stromal tumour, small cell lung cancer
Introduction
The cholecystokinin type 2 (CCK2) receptor is a G protein-coupled, 7 transmembrane domain receptor. Physiologically, it is expressed mainly in the brain and gastrointestinal tract where it mediates the effects of gastrin on gastric acid secretion and epithelial and endocrine cell growth [1]. The CCK2 receptor is also a cancer biomarker of increasing clinical importance. It is expressed by a wide variety of tumours in and outside the gastrointestinal tract. It is present in especially high levels in medullary thyroid carcinomas, small cell lung cancer (SCLC), gastrointestinal stromal tumours (GIST) and insulinomas [2]. This has led to the successful development of clinical applications using radioactive CCK2 receptor ligands for imaging and targeted radiotherapy of some of these tumours [3]. Conversely, the effects of the CCK2 receptor on proliferation particularly of gastric and pancreatic cancer could, unfortunately, not yet be translated into established therapeutic strategies [4, 5].
Tumoral CCK2 receptors have been found to correspond not only to the wild-type form. A CCK2 receptor splice variant that has the fourth intron inappropriately retained (CCK2Ri4sv) has been identified in cancer [6, 7]. The molecular basis for this missplicing event was shown to be a weak 3′ splice site of intron 4 of the CCK2 receptor in association with reduced splice factor levels [7]. Intron 4 retention leads to insertion of an additional 69 amino acids in the third intracellular loop of the CCK2 receptor protein, a domain important for signal transduction. Correspondingly, the splice variant was found to differ from the wild-type CCK2 receptor with respect to activation of intracellular signalling pathways, whereas ligand binding characteristics were unchanged [8]: In specific cell types, the CCK2Ri4sv was constitutively active, leading to increased basal calcium mobilization and MAP kinase and Src kinase phosphorylation as compared with cells expressing wild-type CCK2 receptor [6, 9, 10]. Expression of constitutively active CCK2Ri4sv was associated with increased basal cell proliferation, and activation of the CCK2Ri4sv by ligand binding resulted in enhanced cell growth compared with the wild-type receptor [6, 8]. A second aberrant CCK2 receptor form that was identified in cancer, namely in a single case of human colorectal carcinoma, was a mutant with a single point mutation in the third intracellular loop [11]. This receptor form also led to increased cell proliferation, was, however, not associated with a tumorigenic potential as observed for experimental, not naturally occurring CCK2 receptor mutants [12].
Splice variants with specific expression and functions in cancer such as the CCK2Ri4sv have been receiving increasing attention because of their possible role in tumour biology and potential use for diagnostic and/or therapeutic applications [13, 14]. One important step towards a better understanding of their functional or clinical relevance in cancer is the validation of their expression in human tumour tissues with respect to, for instance, their tumour selectivity, frequency and quantity [15]. As for the CCK2Ri4sv, only selected tumours have been investigated for this missplicing event so far. Although it has been found in colorectal, pancreatic and gastric adenocarcinomas as well as in Barrett’s mucosa, a cancer precursor lesion [6, 7, 16–18], it has not been quantified in all of these tissues. Moreover, tumours known for their particularly high CCK2 receptor levels have not been considered for their CCK2Ri4sv expression. In fact, the molecular mechanisms leading to intron 4 retention may not be restricted to the tumour types investigated so far, but could occur universally in cancers expressing the CCK2 receptor.
Therefore, the aim of the present study was to quantitatively characterize the total CCK2 receptor, wild-type CCK2 receptor and CCK2Ri4sv expression at the transcriptional and protein level in a broader spectrum of different tumour types using end-point and real-time RT-PCR and in vitro receptor autoradiography. It was furthermore intended to assess the cancer selectivity of the CCK2Ri4sv by including normal tissues of tumour origin in the study. The primary focus was laid on gastrointestinal and lung tumours, as the CCK2 receptor is a marker for the major tumours of these organ systems [2].
Materials and methods
Tissues
Human tumour and normal tissue samples were obtained from surgical resection specimens, immediately frozen and stored at –80°C. A total of 81 tumour samples was investigated, including 4 insulinomas, 8 ileal carcinoid tumours, 4 GIST, 8 gastric adenocarcinomas, 13 colorectal adenocarcinomas, 7 pancreatic ductal adenocarcinomas, 3 cholangiocellular carcinomas, 5 hepatocellular carcinomas, 9 SCLC, 12 non-SCLC (nSCLC) and 8 bronchopulmonary carcinoid tumours. Furthermore, three samples of each normal gastric mucosa, gastric muscularis layer, ileal mucosa, colorectal mucosa, pancreas, liver and lung were assessed. The tissues originated either from samples investigated previously for peptide receptors [19–25] collected in accordance with the required international ethical guidelines or from samples collected prospectively at the Institute of Pathology of the University of Berne in agreement with the World Medical Association Declaration of Helsinki, including informed consent and approval by the Institutional Review Board.
In vitro receptor autoradiography for binding sites of CCK2 receptor protein
In vitro receptor autoradiography to assess the expression of CCK2 receptor binding sites in tumour tissues was performed as described previously [19]. Briefly, 20-μm-thick tissue sections mounted on glass slides were incubated with 2000 Ci/mmol of the radioligand 125I-D-Tyr-Gly-[(Nle28,31)CCK-26–33] (125I-CCK; Anawa, Wangen, Switzerland) either alone or in competition with 50 nM cold sulphated CCK-8 (Bachem, Bubendorf, Switzerland) or 50 nM cold gastrin (Bachem). Thus, CCK2 receptors can be distinguished from CCK1 receptors, as the former show a high affinity for both CCK-8 and gastrin, whereas the latter bind only CCK-8 with high affinity. The slides were then exposed to a radiation-sensitive film. The density of the CCK2 receptor-specific signal on the film was quantified in the tumour region that was later excised from the original tumour sample for PCR analysis. This was achieved with a computer-assisted image processing system (Interfocus, Mering, Germany) and radioactive tissue standards (Autoradiographic [14C] microscales, GE Healthcare, Little Chalfont, UK) containing known amounts of isotope, cross-calibrated to tissue-equivalent ligand concentrations [26, 27].
RNA extraction and reverse transcription
A tumour region either positive or negative for CCK2 receptor binding sites by autoradiography was selected and cut out of the frozen tissue block with a sterile scalpel blade. Care was taken to choose an area of tumour tissue without admixed normal tissue with the help of a matching haematoxylin and eosin stained tissue section. Similarly, aliquots of normal tissues were obtained. The dissected tissue samples (≤5 mg) were disrupted and homogenized using the TissueLyser (Qiagen, Hilden, Germany), and total RNA was isolated with the RNeasy Micro Kit (Qiagen), with inclusion of a DNase I digestion step. The total RNA concentration was measured by spectrophotometry, and the RNA quality was checked with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA). Reverse transcription to cDNA was performed with SuperScript III (Invitrogen, Carlsbad, CA, USA) using oligo(dT)s or random hexamers. For end-point PCR, 500 ng total RNA was converted per 20 μl volume reaction.
End-point PCR for wild-type CCK2 receptor and CCK2Ri4sv transcripts
CCK2 receptor transcripts were identified with a nested PCR, where the first round reaction amplified total CCK2 receptor cDNA, and the second round reaction either wild-type CCK2 receptor or CCK2Ri4sv. The primer characteristics are shown in Table 1. PCR was carried out on 2 μl of cDNA or first round PCR product with 5 U/ml Platinum Taq Polymerase (Invitrogen), 2 mM MgCl2 (Invitrogen), 0.2 mM dNTPs (Stratagene, La Jolla, CA, USA) and 0.2 μM of both sense and antisense primers in a 50 μl reaction volume. The first round reaction was cycled 35 times for 30 sec. at 94°C, 45 sec. at 57°C and 3 min. at 72°C. The second round reaction included 37 cycles of 30 sec. at 94°C, 45 sec. at 57°C for the wild-type receptor or 50 sec. at 55°C for the splice variant and 45 sec. at 72°C. Reactions were run at least in duplicate. As positive controls, the previously described CHO cell lines stably expressing wild-type CCK2 receptor (CHO-CCK2R cells) or CCK2Ri4sv (CHO-CCK2R-i4 cells) [8] were used, and for negative control no template was added. Furthermore, β-actin control PCR reactions were run for each sample with 32 cycles comprising 30 sec. at 94°C, 45 sec. at 59°C and 1 min. at 72°C (Table 1). Fifteen microlitre fractions of final PCR products were resolved on a 1.5% agarose/TAE gel along with a 100 bp DNA Ladder (New England Biolabs, Ipswich, MA, USA) as molecular weight marker and visualized by ethidium bromide staining. Representative bands were excised and sequenced to confirm their identities.
Table 1.
Primers and TaqMan MGB probe used for end-point and real-time PCR: sequences, complementary gene regions and amplicon sizes
| Target | Primer/probe | Sequence (5′-3′) | Gene region | Amplicon size | |
|---|---|---|---|---|---|
| End-point PCR | Total CCK2 receptor* | Sense primer | AATCGCAGCGTGAGCAGGTG | Exon 1 | 2032 nt‡ |
| Antisense primer | AGCAATGGAGGGAGTGGGAG | Exon 5 | |||
| Wild-type CCK2 receptor† | Sense primer | CGCGTGATTGTAGCCACGTG | Exon 3 | 311 nt | |
| Antisense primer | TTCTGGTGAACAGCCCCTGG | Junction exons 4/5 | |||
| CCK2Ri4sv† | Sense primer | CTCGCGTGATTGTAGCCAC | Exon 3 | 399 nt | |
| Antisense primer | CTTCCTTCTCACCCTCACC | Intron 4 | |||
| β-actin | Sense primer | CCAGCTCACCATGGATGATGATATCG | Junction exons 1/2 | 853 nt | |
| Antisense primer | GGAGTTGAAGGTAGTTTCGTGGATGC | Exon 5 | |||
| Real-time PCR | Total CCK2 receptor | Sense primer | CGCCAGACCTGGTCCGTACT | Junction exons 3/4 | 164 nt |
| Antisense primer | GCCCGCCTTGGTTTCG | Exon 4 | |||
| Probe | 6FAM-TCGCTGTCACTGTCGC | Exon 4 | |||
| CCK2Ri4sv | Sense primer | CGCCAGACCTGGTCCGTACT | Junction exons 3/4 | 227 nt | |
| Antisense primer | CCCGTCGCCCTCCAAA | Intron 4 | |||
| Probe | 6FAM-TCGCTGTCACTGTCGC | Exon 4 |
Real-time PCR for total CCK2 receptor and CCK2Ri4sv transcripts
Of 52 of the 63 tumours positive for CCK2 receptor transcripts by end-point PCR, sufficient RNA was available for further real-time PCR analysis. Two different real-time PCR reactions were established to amplify either total CCK2 receptor or CCK2Ri4sv cDNA. Primers and the TaqMan MGB probe (Table 1) were designed using the Primer Express software version 3.0 (Applied Biosystems, Foster City, CA, USA) and synthesized by Applied Biosystems. The optimal primer and probe concentrations yielding the lowest Ct and highest ΔRn values for a given RNA concentration were found to be 900 nM for all primers and 250 nM for the probe. Using these concentrations, cDNA corresponding to 100 ng total RNA was amplified in a 20 μl reaction volume with the TaqMan Universal PCR Master Mix (Applied Biosystems) on a 7900HT Fast Real-time PCR System (Applied Biosystems) as follows: 2 min. at 50°C, 10 min. at 95°C, followed by 50 cycles of 15 sec. at 95°C and 1 min. at 60°C. The endogenous control genes 18S rRNA, GAPDH and human β-actin were amplified in each sample using pre-designed primer and probe sets by Applied Biosystems. Standard curves for all genes were constructed using serial cDNA dilutions of CHO-CCK2R or CHO-CCK2R-i4 cells or the pancreatic cancer cells CAPAN2 or BxPC3 (American Type Culture Collection, Manassas, VA, USA). On each plate, the same CCK2 receptor expressing tumour was included as calibrator, and no template reactions as negative controls. All reactions were run at least in triplicate.
CCK2 receptor gene expression was relatively quantified using the efficiency (E)-corrected ΔCt method. Total CCK2 receptor transcript levels were expressed relative to 18S rRNA and a calibrator sample using the equation:
E (total CCK2R) ΔCt (total CCK2R) (calibrator – sample)/E (18S rRNA) ΔCt (18S rRNA) (calibrator – sample)[28].
CCK2Ri4sv transcript expression was reported relative to total CCK2 receptor transcript expression as follows:
E (CCK2Ri4sv) −Ct (CCK2Ri4sv)/E (total CCK2R) −Ct (total CCK2R)[29, 30].
Immunohistochemistry for the proliferation marker MIB-1
Immunohistochemistry for the proliferation marker MIB-1 was performed on nine SCLC, four nSCLC and four pancreatic adenocarcinomas. Cryostat sections (10 μm thick) were post-fixed in formalin. The primary antibody (DAKO, Zug, Switzerland) was added in a 1:50 dilution. The secondary antibody was a biotinylated goat antimouse immunoglobulin (DAKO). Antibody binding was visualized using the VECTASTAIN Elite ABC kit (Vector, Burlingame, CA, USA). Staining was carried out with DAB, and counterstaining with hemalum.
Statistical analysis
Statistical analysis was performed with the GraphPad Prism software version 4.0 (La Jolla, CA, USA), using the unpaired t-test, Mann-Whitney test, linear regression analysis and Spearman correlation. P < 0.05 was considered statistically significant.
Results
Real-time PCR: specificity, dynamic range, normalization
Several lines of evidence of the specificities of the real-time PCR reactions for total CCK2 receptor and the CCK2Ri4sv, respectively, were obtained. By separating amplification products on an agarose gel, each reaction yielded a single band migrating with the expected molecular size (Fig. 1A). Moreover, the reaction for CCK2Ri4sv produced no amplification signal in CHO-CCK2R cells which express only wild-type, but not splice variant CCK2 receptor. Finally, in samples where the reverse transcriptase had been omitted, no amplification signal was obtained.
Fig 1.
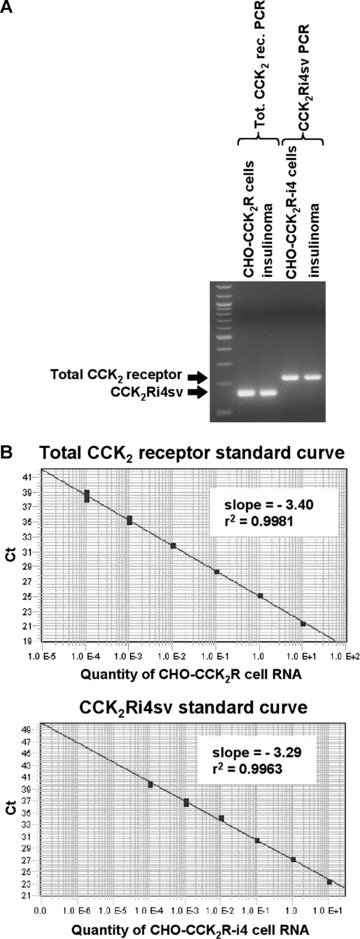
Real-time PCR: specificities (A) and dynamic ranges (B) of total CCK2 receptor and CCK2Ri4sv assays. (A) Visualization of real-time PCR products obtained for total CCK2 receptor and CCK2Ri4sv in CHO-CCK2R and CHO-CCK2R-i4 cells, respectively, as well as in an insulinoma expressing both wild-type and splice variant CCK2 receptor in an agarose gel. A 100 bp DNA ladder was included as molecular weight marker. Both PCR assays yield a single product migrating with the expected size. (B) Standard curves of total CCK2 receptor and CCK2Ri4sv real-time PCR reactions. Both curves show linearity over a 6-log RNA concentration range.
The real-time PCR assays for total and splice variant CCK2 receptors were tested on serial cDNA dilutions of CHO-CCK2R and CHO-CCK2R-i4 cells, respectively. In both assays, the relationship between the Ct values, which spanned the Ct values anticipated for the samples, and the logarithm of the starting RNA amount remained linear over the tested 6-log concentration range (Fig. 1B). Similar plots were created for 18S rRNA, GAPDH and β-actin, yielding slopes of –3.40, –3.39 and –3.50, respectively, and r2 > 0.99 for all three genes. These plots were then generated in each real-time PCR run, to calculate the efficiencies used for relative quantification.
The endogenous control genes 18S rRNA, GADPH and β-actin were evaluated for their suitability for normalization, although a larger number of candidate genes could not be tested because of the limited availability of sample RNA. The expression levels of these genes were determined in each sample using the formula E−Ct. 18S rRNA expression was fairly constant among the various tumour groups, whereas expression of GAPDH and β-actin was much more variable. We concluded that 18S rRNA was the most suitable endogenous control in the studied tumour population and, therefore, used it for relative quantification of total CCK2 receptor transcript levels.
Wild-type CCK2 receptor transcript expression
The wild-type CCK2 receptor transcript expression was assessed with end-point PCR. It was found to be very common (Table 2). It was detected in every tumour type studied, except for cholangiocellular carcinoma. Moreover, the frequency of the wild-type CCK2 receptor transcript expression in the individual tumour types was mostly high. Furthermore, all investigated normal tissues were positive for the wild-type CCK2 receptor except for the lung. Typical end-point PCR results for the wild-type CCK2 receptor in tumours and normal tissues are shown in Fig. 2(A) in the top gel.
Table 2.
Expression of wild-type CCK2 receptor and CCK2Ri4sv transcripts and total CCK2R binding sites in the same gastrointestinal and lung tumour tissues
| Wild-type CCK2 receptor transcripts | CCK2Ri4sv transcripts | Total CCK2 receptor binding sites | |||
|---|---|---|---|---|---|
| Incidence: positive / total cases (%) | Incidence: positive / total cases (%) | CCK2Ri4sv/total CCK2 receptor transcripts: mean ± S.E.M. | Incidence: positive / total cases (%) | Density: mean ± S.E.M. (dpm/mg) | |
| Insulinomas | 4 / 4 (100%) | 4 / 4 (100%) | 0.56 ± 0.08‰ | 4 / 4 (100%) | 4076 ± 1760 |
| Ileal carcinoid tumours | 8 / 8 (100%) | 0 / 8 (0%) | 7 / 8 (88%) | 629.2 ± 217 | |
| GIST | 4 / 4 (100%) | 4 / 4 (100%) | 0.21 ± 0.02‰ | 4 / 4 (100%) | 6590.5 ± 545 |
| Gastric adenocarcinomas | 6 / 8 (75%) | 0 / 8 (0%) | 2 / 8 (25%) | 1745 ± 806 | |
| Colorectal adenocarcinomas | 9 / 13 (69%) | 0 / 13 (0%) | 0 / 13 (0%) | ||
| Pancreatic adenocarcinomas | 4 / 7 (57%) | 1/ 7 (14%) | NT* | 0 / 7 (0%) | |
| Cholangiocarcinomas | 0 / 3 (0%) | 0 / 3 (0%) | 0 / 1 (0%) | ||
| Hepatocellular carcinomas | 4 / 5 (80%) | 0 / 5 (0%) | 0 / 5 (0%) | ||
| SCLC | 8 / 9 (89%) | 6 / 9 (67%) | 1.05 ± 0.09‰ | 7 / 9 (78%) | 2054.8 ± 702 |
| nSCLC | 9 / 12 (75%) | 1 / 12 (8%) | 1.37‰ | 1 / 12 (8%) | 1975 |
| Lung carcinoid tumours | 7 / 8 (88%) | 0 / 8 (0%) | 4 / 8 (50%) | 1272 ± 635 | |
NT = not tested because of limited availability of sample.
Fig 2.
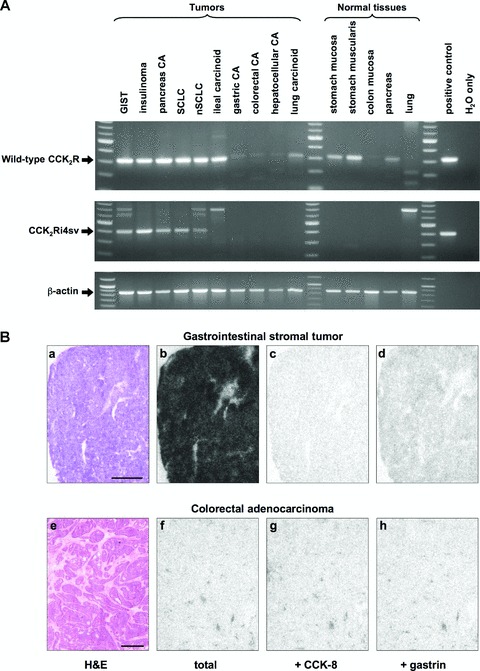
Expression of CCK2 receptor transcripts (A) and binding sites (B) in tissues. (A) Agarose gels showing representative end-point RT-PCR products for wild-type CCK2 receptor (top gel), CCK2Ri4sv (middle gel) and β-actin (bottom gel) in tumours and normal tissues. Positive control reactions were performed on cDNA of CHO-CCK2R and CHO-CCK2R-i4 cells for the wild-type and splice variant CCK2 receptor, respectively. For negative control no template was added (H2O only). A 100 bp DNA ladder served as molecular weight marker. Wild-type CCK2 receptor transcripts are amplified in all displayed tumours and normal tissues except for normal lung. Note the weak bands obtained in gastric, colorectal and hepatocellular carcinomas. In the colon mucosa, the band is barely visible. CCK2Ri4sv transcripts are detected in the GIST, insulinoma, pancreatic adenocarcinoma, SCLC and nSCLC, but not in the remaining tumours or normal tissues. Additional bands migrating with higher molecular weights correspond to amplified genomic CCK2 receptor or are non-specific, as confirmed by sequencing. (B) In vitro receptor autoradiography on serial tissue sections to assess CCK receptor binding sites in a GIST (a–d) and a colorectal adenocarcinoma (e–h). a, e: Haematoxylin and eosin stained tissue sections showing the tumour tissues (bars = 1 mm). b, f: Autoradiograms showing total 125I-CCK binding to the tumour tissues. Very strong 125I-CCK binding to the GIST, but hardly any labelling of the colorectal carcinoma. c, g: Autoradiograms showing 125I-CCK binding in the presence of 50 nM cold CCK-8 and e, h: autoradiograms showing 125I-CCK binding in the presence of 50 nM cold gastrin. In the GIST, 125I-CCK is completely displaced by CCK-8 and almost to the same extent by gastrin, proving that the vast majority of the 125I-CCK binding sites correspond to CCK2 receptors. (The small fraction of total 125I-CCK binding that is displaced by CCK-8 but not gastrin corresponds to concomitantly expressed CCK1 receptors.) Conversely, in the colorectal carcinoma, the traces of 125I-CCK binding are not displaced by either CCK-8 or gastrin, proving that they reflect non-specific 125I-CCK binding to the tissue. This tumour has no detectable CCK receptor binding sites.
CCK2Ri4sv transcript expression
Using end-point PCR, CCK2Ri4sv transcripts were found in a subset of tumours expressing wild-type CCK2 receptor mRNA, whereas they were not detected in tumours without wild-type receptors. Unlike the wild-type form, the splice variant was restricted to specific tumour types (Table 2, Fig. 2A). It was commonly expressed in insulinomas, GIST and SCLC. Indeed, in the former two tumour types it was present in every case studied. By contrast, the CCK2Ri4sv expression was rare in pancreatic adenocarcinomas and nSCLC, where only single cases were found to be positive. Splice variant transcripts were not identified in the remaining tumours. Likewise, they were not detected in any of the normal tissue samples.
In the tumours that expressed the CCK2Ri4sv by end-point PCR, the transcript levels were quantified relative to total CCK2 receptor levels with real-time PCR. Splice variant transcripts were found to account for a very small fraction of all CCK2 receptor transcripts. The mean expression levels ranged from 0.21‰ of total CCK2 receptors in GIST to 1.05‰ in SCLC (Table 2). The expression levels in the individual cases are shown in Fig. 3(A). This figure also demonstrates how the relative splice variant amounts varied between the different tumour types. In SCLC, a significantly higher proportion of all CCK2 receptor transcripts corresponded to the splice variant than in GIST (P= 0.0003) or insulinomas (P= 0.0045).
Fig 3.
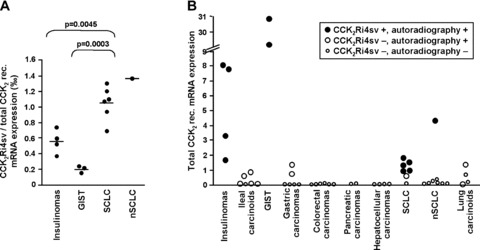
CCK2Ri4sv transcript expression in tumours. (A) Real-time PCR quantification of CCK2Ri4sv transcripts relative to total CCK2 receptor transcripts. Each point represents an individual tumour; scale bars = mean values. In all tumours, the CCK2Ri4sv transcripts account for a small fraction in the per mill range of total CCK2 receptor transcripts. SCLC and nSCLC express significantly higher relative amounts of CCK2Ri4sv transcripts than insulinomas or GIST. (B) Total CCK2 receptor transcripts levels relative to 18S rRNA levels in tumours with CCK2Ri4sv expression (black points) and without CCK2Ri4sv expression (white points), as well as in autoradiographically positive tumours (large points) and autoradiographically negative tumours (small points). Total CCK2 receptors could not be quantified in a GIST and a SCLC because 18S rRNA levels strongly deviated from expected values in the former and not enough RNA was available for endogenous control gene PCR in the latter. The CCK2Ri4sv is identified only in tumours with relative total CCK2 receptor levels above 1, whereas almost all tumours without CCK2Ri4sv expression show total receptor transcript levels below 1. Furthermore, most of the tumours without autoradiographically measurable CCK2 receptor binding sites exhibit very low total receptor transcript levels, whereas the tumours with identifiable receptor binding sites express higher transcript levels.
Real-time PCR analysis furthermore revealed that the identification of the CCK2Ri4sv was associated with the amount of total CCK2 receptor mRNA. Tumours with splice variant expression showed significantly higher total receptor transcript levels than tumours without (P < 0.0001). This is shown in Fig. 3(B) for the individual cases. The splice variant was identified only in tumours with total CCK2 receptor mRNA levels above a certain level. Conversely, in only 2 of the 37 tumours without splice variant expression, the total CCK2 receptor mRNA amounts equalled those of tumours expressing the splice variant, whereas the remaining tumours showed lower total receptor transcripts levels.
Immunohistochemistry with the proliferation marker MIB-1 was performed in selected tumour samples to assess the relation between CCK2Ri4sv expression and the proliferative index. In SCLC, the mean proportion of tumour cells in the proliferative phase, as visualized by nuclear staining for MIB-1, was larger in tumours with than in tumours without CCK2Ri4sv expression (64.8% MIB-1 positive tumour cells in SCLC with CCK2Ri4sv expression versus 56.5% in SCLC without CCK2Ri4sv expression). This difference was, however, not statistically significant (P= 0.2774). Conversely, in nSCLC and pancreatic adenocarcinomas, the single cases expressing CCK2Ri4sv exhibited a lower proliferative index than the tumours without splice variant expression (nSCLC: 30.5%versus mean 37.7% MIB-1 positive tumour cells; pancreatic adenocarcinomas: 9%versus mean 30.1%).
Total CCK2 receptor binding site expression
Total CCK2 receptor protein expression in tumour tissues was assessed on the basis of CCK2 receptor binding with in vitro receptor autoradiography [19]. With this method, the binding sites of both wild-type CCK2 receptors and CCK2Ri4sv are simultaneously identified, as these two receptor forms cannot pharmacologically be differentiated [8]. The autoradiography results were comparable with published data (Table 2) [2]: The CCK2 receptor binding site expression with respect to both incidence and density was particularly high in insulinomas, GIST and SCLC, but virtually absent in gastric, colorectal and pancreatic carcinomas, liver tumours and nSCLC. This is illustrated with two representative cases in Fig. 2(B). The GIST in the first row displays strong 125I-CCK binding, corresponding to a high number of CCK receptor binding sites; displacement of 125I-CCK by both CCK-8 and gastrin provides proof of specific CCK2 receptor identification. Conversely, in the colorectal carcinoma in the second row there are no CCK receptor binding sites identifiable, as this tumour shows no specific 125I-CCK binding.
Correlation of CCK2 receptor binding site and transcript expression in tumour tissues
Receptor binding sites were not identified in over half of the tumours showing CCK2 receptor transcripts (Table 2). Real-time PCR analysis revealed that these autoradiographically negative tumours showed particularly low total CCK2 receptor transcript levels, whereas tumours with detectable receptor binding sites displayed significantly higher receptor mRNA amounts (P < 0.0001). This is demonstrated for the individual tumours in Fig. 3(B). Furthermore, it was found that in autoradiographically positive tumours the actual binding site density deviated considerably from that predicted from the transcript levels in a linear regression model in the majority of cases, as can be seen in Fig. 4(A) and (B). Taking into account all autoradiographically positive and negative cases, the density levels of CCK2 receptor binding sites correlated statistically fairly well with total CCK2 receptor transcript levels (r2= 0.6579, P= 0.0002).
Fig 4.
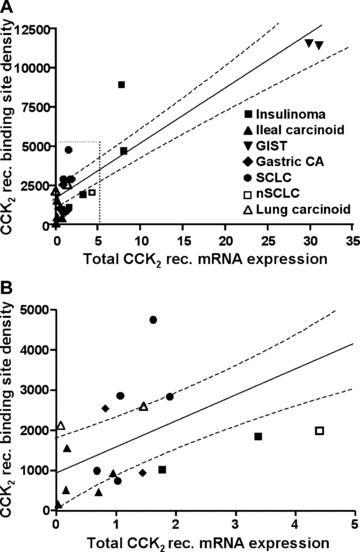
Correlation of the CCK2 receptor binding site density measured with autoradiography with receptor transcript levels relative to 18S rRNA levels assessed by real-time PCR in autoradiographically positive tumours. (B) is a higher magnification of the area indicated with dotted lines in (A). The straight line represents the line of best fit obtained by linear regression analysis (dashed lines = 95% confidence intervals).
Discussion
The CCK2 receptor splice variant with intron 4 retention was previously described in a limited number of colorectal, pancreatic and gastric adenocarcinomas [6, 7, 16, 17]. It was considered to be important in these particular tumours, because of its special functional characteristics, namely constitutive activity associated with increased proliferation [6]. By quantitatively assessing the CCK2Ri4sv expression in a larger number of gastrointestinal and lung tumours, the present study reveals that this splice variant is in fact a marker of a different spectrum of tumours. It was found in several tumour types that up to now have not been considered for this splicing event, including such diverse tumours as insulinomas, GIST, SCLC and nSCLC. In most of these tumours the CCK2Ri4sv was expressed in high frequency, whereas it was rare or not detected in colorectal, pancreatic and gastric adenocarcinomas. The CCK2Ri4sv was never the only CCK2 receptor form present in the tumours. Indeed, it accounted for only a small fraction of all CCK2 receptor transcripts. The occurrence of the CCK2Ri4sv was strongly associated with elevated total CCK2 receptor mRNA levels. This is suggestive of an intrinsic small likelihood of intron 4 not to be recognized by the splicing machinery because of its weak 3′ splice site [7], which reaches detection levels when enough CCK2 receptor is transcribed. Conversely, the significant differences in the relative CCK2Ri4sv expression levels observed between the various tumour types indicate additional tumour-specific aberrant molecular processes involved in missplicing of the CCK2 receptor. The tumoral CCK2Ri4sv expression was not significantly associated with an increased proliferation rate.
Existing data indicate a high selectivity of the CCK2Ri4sv for tumours over non-neoplastic tissues, which is in fact much higher than that of the wild-type CCK2 receptor [6, 7, 16, 17, 31]. In the present series, the CCK2Ri4sv was not identified in a variety of normal gastrointestinal and lung tissues. It was in particular not detected in normal tissues of origin of tumours that frequently express it, such as gastric muscularis and lung. This extends previous findings of the absence of CCK2Ri4sv in normal colon and pancreas [6, 7, 16]. Of note, the CCK2Ri4sv may not absolutely be restricted to neoplasia, as it has been described in peripheral blood mononuclear cells of a minority of healthy individuals and in normal gastric mucosa [17, 31].
For some tumour and normal tissues, discrepancies exist between various reports concerning the presence of CCK2Ri4sv mRNA. Colorectal cancer was originally reported to express CCK2Ri4sv, but not wild-type CCK2 receptor transcripts [6]. However, Schmitz et al. recognized only the wild-type, but not the splice variant receptor in as many as 79 colorectal carcinomas [11], in full agreement with our present findings. Furthermore, we could not detect the CCK2Ri4sv in gastric cancer or normal gastric mucosa where it had been described before [17]. Finally, the single cases of CCK2Ri4sv-expressing pancreatic adenocarcinomas reported in the literature suggested frequent CCK2 receptor missplicing in these tumours [7, 16]. However, in this study the splice variant could be identified in only one out of eight investigated cases. This variable identification of CCK2Ri4sv transcripts may at least in part be because of the very low expression levels. The amount of RNA employed for RT-PCR could become critical. Zhou et al. used 10 times more RNA than we were able to utilize [17]. Furthermore, primers may underperform in amplification of low abundant transcripts when located in the vicinity of AG-rich stretches [32] which are present in intron 4 of the CCK2 receptor. On the other hand, it has to be considered that primers not able to distinguish between amplified cDNA and genomic DNA [6, 16] may be responsible for a false high incidence of splice variants with intron retention.
Divergent data has also accumulated over the last years on the CCK2 receptor protein and transcript expression in gastrointestinal tumours, in particular in colorectal, pancreatic and gastric adenocarcinomas [5]. In these tumours, the CCK2 receptor incidence was considerably lower when assessed with receptor binding studies compared with RT-PCR. In the present study, both methods were for the first time applied in parallel on the same tumour specimens, and the quantitative expression of total tumoral CCK2 receptor binding sites was correlated with that of total CCK2 receptor mRNA. This revealed that the cases with divergent results, i.e. negative autoradiography but positive RT-PCR, consistently expressed very low amounts of total CCK2 receptor transcripts. This may reflect a lower sensitivity of CCK2 receptor autoradiography compared with RT-PCR. The correlation furthermore demonstrated that CCK2 receptor transcript levels could not reliably predict the amounts of CCK2 receptor binding sites in a substantial number of cases. Possible explanations for this non-linear relationship between CCK2 receptor mRNA and protein levels are for instance variations in the protein translation rate or in the compartmentalization of the receptor protein in intracellular pools [33] between individual tumours.
The protein expression of the CCK2Ri4sv cannot specifically be assessed in tissues, as selective ligands or immunohistochemical antibodies are not available. In transfected cell lines, it could, however, be shown that the CCK2Ri4sv translated into protein to a similar extent as the wild-type receptor [8]. If this also holds true for original tumours can only be speculated at present. In this case, the density levels of total CCK2 receptor binding sites, which most probably correspond largely to wild-type receptors, would suggest very low CCK2Ri4sv protein amounts in tumours. It has to be considered, though, that the splice variant protein may not show the same stability or be expressed to the same extent in the plasma membrane as the wild-type receptor. Indeed, in transfected cell lines the CCK2Ri4sv was found to be located mainly in the intracellular compartment, whereas the wild-type receptor was expressed predominantly in the cell membrane [34].
The CCK2Ri4sv has been shown to stimulate cell proliferation in vitro[6, 7, 9, 35]. In the present study, however, CCK2Ri4sv-expressing tumours did not show a significantly increased proliferation rate compared with tumours without the splice variant, although the number of investigated cases may be too small to permit definite conclusions. At present, it remains unclear in how far the functional effects of CCK2Ri4sv observed in vitro may be extrapolated to original tumours. It has in particular to be considered that the CCK2Ri4sv expression in its natural environment may show significant differences to that in experimental models. Indeed, transfected cell lines used to explore CCK2Ri4sv functionality express the splice variant abundantly and selectively [6, 7, 35], whereas original tumour tissues express CCK2Ri4sv transcripts at very low levels and always together with high levels of wild-type CCK2 receptor and sometimes even CCK1 receptors [2]. In vivo, the CCK2Ri4sv may well dimerize with either of these receptors, similar to the wild-type CCK2 receptor [36]. Would such heterodimers behave in terms of receptor binding and signalling more like wild-type CCK2 or CCK1 receptors than like the CCK2Ri4sv, or would they elicit even new characteristics? Moreover, as CCK2Ri4sv is expressed at very low levels in original tumours, it may not contribute to cell proliferation to the same extent as in the cell line models. Finally, the constitutive activity of the CCK2Ri4sv, which is largely responsible for the increased proliferation, may not be present in all original tumours. In fact, it was found to be restricted to certain cell types and to be in particular absent in epithelial tumour cells [7, 9]. A possible strategy to further elucidate CCK2Ri4sv-mediated biologic effects may be to study the association between CCK2Ri4sv expression and proliferative index or activation of down-stream signalling pathways in an extended number of original tumours. Unfortunately, the use of cancer cell lines as an alternative to transfected cell lines for similar studies is limited, because cancer cell lines exhibit splicing patterns that are significantly different from those occurring in vivo and have often lost splice variant expression [9, 15, 18, 37] (personal observations).
Given its high specificity for and high incidence in clinically important tumours like SCLC and GIST, the CCK2Ri4sv may represent an attractive target for potential clinical applications [13, 14]. The low tumoral CCK2Ri4sv expression levels imply that only diagnostic tests with high sensitivity, such as PCR, may be clinically useful. Indeed, it has been demonstrated with several examples that PCR for splice variants performed for instance on pleural or peritoneal fluid can assist in tumour diagnosis, despite the very high sensitivity of the method and not exclusive expression of splice variants in cancer cells [38–40]. Conversely, targeting CCK2Ri4sv with a radioactive ligand, as suggested recently [41], may be a less promising approach because accumulation of sufficient radioactivity within the tumour area for imaging or therapy may not be achieved because of the low tumoral CCK2Ri4sv expression levels.
Acknowledgments
The study was supported by a postdoctoral fellowship from the Swiss Foundation for Medical-Biological Fellowships (No. 1267)/Novartis to M.K., by a grant from the National Institute of Health (DK32878) to L.J.M. and by a grant from the Swiss National Science Foundation (No. 3200–105726) to J.C.R.
References
- 1.Rozengurt E, Walsh JH. Gastrin, CCK, signaling, and cancer. Annu Rev Physiol. 2001;63:49–76. doi: 10.1146/annurev.physiol.63.1.49. [DOI] [PubMed] [Google Scholar]
- 2.Reubi JC. Targeting CCK receptors in human cancers. Curr Top Med Chem. 2007;7:1239–42. doi: 10.2174/156802607780960546. [DOI] [PubMed] [Google Scholar]
- 3.Behr TM, Bèhè MP. Cholecystokinin-B/Gastrin receptor-targeting peptides for staging and therapy of medullary thyroid cancer and other cholecystokinin-B receptor-expressing malignancies. Semin Nucl Med. 2002;32:97–109. doi: 10.1053/snuc.2002.31028. [DOI] [PubMed] [Google Scholar]
- 4.Jensen RT. Involvement of cholecystokinin/gastrin-related peptides and their receptors in clinical gastrointestinal disorders. Pharmacol Toxicol. 2002;91:333–350. doi: 10.1034/j.1600-0773.2002.910611.x. [DOI] [PubMed] [Google Scholar]
- 5.Baldwin GS, Shulkes A. CCK receptors and cancer. Curr Top Med Chem. 2007;7:1232–8. doi: 10.2174/156802607780960492. [DOI] [PubMed] [Google Scholar]
- 6.Hellmich MR, Rui XL, Hellmich HL, et al. Human colorectal cancers express a constitutively active cholecystokinin-B/gastrin receptor that stimulates cell growth. J Biol Chem. 2000;275:32122–8. doi: 10.1074/jbc.M005754200. [DOI] [PubMed] [Google Scholar]
- 7.Ding WQ, Kuntz SM, Miller LJ. A misspliced form of the cholecystokinin-B/gastrin receptor in pancreatic carcinoma: role of reduced sellular U2AF35 and a suboptimal 3’-splicing site leading to retention of the fourth intron. Cancer Res. 2002;62:947–52. [PubMed] [Google Scholar]
- 8.Cheng ZJ, Harikumar KG, Ding WQ, et al. Analysis of the cellular and molecular mechanisms of trophic action of a misspliced form of the type B cholecystokinin receptor present in colon and pancreatic cancer. Cancer Lett. 2005;222:95–105. doi: 10.1016/j.canlet.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 9.Chao C, Tallman ML, Ives KL, et al. Gastrointestinal hormone receptors in primary human colorectal carcinomas. J Surg Res. 2005;129:313–21. doi: 10.1016/j.jss.2005.04.038. [DOI] [PubMed] [Google Scholar]
- 10.Olszewska-Pazdrak B, Townsend CM, Hellmich MR. Agonist-independent activation of Src tyrosine kinase by a cholecystokinin-2 (CCK2) receptor splice variant. J Biol Chem. 2004;279:40400–4. doi: 10.1074/jbc.C400208200. [DOI] [PubMed] [Google Scholar]
- 11.Schmitz F, Otte JM, Stechele HU, et al. CCK-B/gastrin receptors in human colorectal cancer. Eur J Clin Invest. 2001;31:812–20. doi: 10.1046/j.1365-2362.2001.00870.x. [DOI] [PubMed] [Google Scholar]
- 12.Gales C, Sanchez D, Poirot M, et al. High tumorigenic potential of a constitutively active mutant of the cholecystokinin 2 receptor. Oncogene. 2003;22:6081–9. doi: 10.1038/sj.onc.1206823. [DOI] [PubMed] [Google Scholar]
- 13.Venables JP. Aberrant and alternative splicing in cancer. Cancer Res. 2004;64:7647–54. doi: 10.1158/0008-5472.CAN-04-1910. [DOI] [PubMed] [Google Scholar]
- 14.Xing Y. Genomic analysis of RNA alternative splicing in cancers. Front Biosci. 2007;12:4034–41. doi: 10.2741/2369. [DOI] [PubMed] [Google Scholar]
- 15.Skotheim RI, Nees M. Alternative splicing in cancer: noise, functional, or systematic? Int J Biochem Cell Biol. 2007;39:1432–49. doi: 10.1016/j.biocel.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 16.Smith JP, Verderame MF, McLaughlin P, et al. Characterization of the CCK-C (cancer) receptor in human pancreatic cancer. Int J Mol Med. 2002;10:689–94. [PubMed] [Google Scholar]
- 17.Zhou J, Chen M, Zhang Q, et al. Human gastric tissues simultaneously express the classical and alternative splicing cholecystokinin-B/gastrin receptors. Receptors Channels. 2004;10:185–8. doi: 10.3109/10606820490926179. [DOI] [PubMed] [Google Scholar]
- 18.Harris JC, Clarke PA, Awan A, et al. An antiapoptotic role for gastrin and the gastrin/CCK-2 receptor in Barrett’s esophagus. Cancer Res. 2004;64:1915–9. doi: 10.1158/0008-5472.can-03-2713. [DOI] [PubMed] [Google Scholar]
- 19.Reubi JC, Schaer JC, Waser B. Cholecystokinin(CCK)-A and CCK-B/gastrin receptors in human tumors. Cancer Res. 1997;57:1377–86. [PubMed] [Google Scholar]
- 20.Reubi JC, Waser B, Schmassmann A, et al. Receptor autoradiographic evaluation of cholecystokinin, neurotensin, somatostatin and vasoactive intestinal peptide receptors in gastro-intestinal adenocarcinoma samples: where are they really located? Int J Cancer. 1999;81:376–86. doi: 10.1002/(sici)1097-0215(19990505)81:3<376::aid-ijc11>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 21.Reubi JC, Zimmermann A, Jonas S, et al. Regulatory peptide receptors in human hepatocellular carcinomas. Gut. 1999;45:766–74. doi: 10.1136/gut.45.5.766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Reubi JC, Waser B. Concomitant expression of several peptide receptors in neuroendocrine tumours: molecular basis for in vivo multireceptor tumour targeting. Eur J Nucl Med Mol Imaging. 2003;30:781–93. doi: 10.1007/s00259-003-1184-3. [DOI] [PubMed] [Google Scholar]
- 23.Reubi JC, Waser B, Gugger M, et al. Distribution of CCK1 and CCK2 receptors in normal and diseased human pancreatic tissue. Gastroenterology. 2003;125:98–106. doi: 10.1016/s0016-5085(03)00697-8. [DOI] [PubMed] [Google Scholar]
- 24.Reubi JC, Körner M, Waser B, et al. High expression of peptide receptors as a novel target in gastrointestinal stromal tumours. Eur J Nucl Med Mol Imaging. 2004;31:803–10. doi: 10.1007/s00259-004-1476-2. [DOI] [PubMed] [Google Scholar]
- 25.Körner M, Hayes GM, Rehmann R, et al. Secretin receptors in the human liver: Expression in biliary tract and cholangiocarcinoma, but not in hepatocytes or hepatocellular carcinoma. J Hepatol. 2006;45:825–35. doi: 10.1016/j.jhep.2006.06.016. [DOI] [PubMed] [Google Scholar]
- 26.Miller JA, Zahniser NR. The use of 14C-labeled tissue paste standards for the calibration of 125I-labeled ligands in quantitative autoradiography. Neurosci Lett. 1987;81:345–50. doi: 10.1016/0304-3940(87)90408-3. [DOI] [PubMed] [Google Scholar]
- 27.Baskin DG, Wimpy TH. Calibration of [14C]plastic standards for quantitative autoradiography of [125I]labeled ligands with Amersham Hyperfilm beta-max. Neurosci Lett. 1989;104:171–7. doi: 10.1016/0304-3940(89)90350-9. [DOI] [PubMed] [Google Scholar]
- 28.Pfaffl MW. Quantification strategies in real-time PCR. In: Bustin SA, editor. A-Z of quantitative PCR. LaJolla: IUL Biotechnology Series; 2004. pp. 87–120. [Google Scholar]
- 29.Bustin SA. Absolute quantification of mRNA using real-time reverse transcription polymerase chain reaction assays. J Mol Endocrinol. 2000;25:169–93. doi: 10.1677/jme.0.0250169. [DOI] [PubMed] [Google Scholar]
- 30.Schmittgen TD, Teske S, Vessella RL, et al. Expression of prostate specific membrane antigen and three alternatively spliced variants of PSMA in prostate cancer patients. Int J Cancer. 2003;107:323–9. doi: 10.1002/ijc.11402. [DOI] [PubMed] [Google Scholar]
- 31.Schmitz F, Schrader H, Otte J, et al. Identification of CCK-B/gastrin receptor splice variants in human peripheral blood mononuclear cells. Regul Pept. 2001;101:25–33. doi: 10.1016/s0167-0115(01)00281-6. [DOI] [PubMed] [Google Scholar]
- 32.Galindo J, Jones N, Powell GL, et al. Advanced qRT-PCR technology allows detection of the cholecystokinin 1 receptor (CCK1R) expression in human pancreas. Pancreas. 2005;31:325–31. doi: 10.1097/01.mpa.0000181487.50269.dc. [DOI] [PubMed] [Google Scholar]
- 33.Miller LJ, Dong M, Harikumar KG, et al. Biochemical and cell biological mechanisms of cholecystokinin receptor regulation. Curr Top Med Chem. 2007;12:1166–72. doi: 10.2174/156802607780960474. [DOI] [PubMed] [Google Scholar]
- 34.Chao C, Ives KL, Goluszko E, et al. SRC regulates constitutive internalization and rapid resensitization of a cholecystokinin 2 receptor splice variant. J Biol Chem. 2005;280:33368–73. doi: 10.1074/jbc.M506337200. [DOI] [PubMed] [Google Scholar]
- 35.Chao C, Goluszko E, Lee YT, et al. Constitutively active CCK2 receptor splice variant increases Src-dependent HIF-1 alpha expression and tumor growth. Oncogene. 2007;26:1013–9. doi: 10.1038/sj.onc.1209862. [DOI] [PubMed] [Google Scholar]
- 36.Cheng ZJ, Harikumar KG, Holicky EL, et al. Heterodimerization of type A and B cholecystokinin receptors enhance signaling and promote cell growth. J Biol Chem. 2003;278:52972–9. doi: 10.1074/jbc.M310090200. [DOI] [PubMed] [Google Scholar]
- 37.Hayes GM, Carrigan PE, Dong M, et al. A novel secretin receptor splice variant potentially useful for early diagnosis of pancreatic carcinoma. Gastroenterology. 2007;133:8–861. doi: 10.1053/j.gastro.2007.06.013. [DOI] [PubMed] [Google Scholar]
- 38.Okamoto I, Morisaki T, Sasaki J, et al. Molecular detection of cancer cells by competitive reverse transcription-polymerase chain reaction analysis of specific CD44 variant RNAs. J Natl Cancer Inst. 1998;90:307–15. doi: 10.1093/jnci/90.4.307. [DOI] [PubMed] [Google Scholar]
- 39.Miyake H, Eto H, Arakawa S, et al. Over expression of CD44V8–10 in urinary exfoliated cells as an independent prognostic predictor in patients with urothelial cancer. J Urol. 2002;167:1282–7. [PubMed] [Google Scholar]
- 40.Dong WG, Sun XM, Yu BP, et al. Role of VEGF and CD44v6 in differentiating benign from malignant ascites. World J Gastroenterol. 2003;9:2596–600. doi: 10.3748/wjg.v9.i11.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laverman P, Roosenburg S, Gotthardt M, et al. Targeting of a CCK(2) receptor splice variant with (111)In-labelled cholecystokinin-8 (CCK8) and (111)In-labelled minigastrin. Eur J Nucl Med Mol Imaging. 2008;35:386–92. doi: 10.1007/s00259-007-0604-1. [DOI] [PubMed] [Google Scholar]