

Abstract
Many biosensors, DNA arrays, and next-generation DNA sequencing technologies need common methods for end modification of random DNA sequences generated from a sample of DNA. Surface immobilization of chemically modified DNA is often the first step in creating appropriate sensing platforms. We describe a simple technique for efficient heterobifunctional modification of arbitrary double-stranded DNA fragments with chosen chemical groups. The modification requires the use of short (10–20 base pairs) synthetic adaptors having desired terminal functional groups and installs known sequences, which can be used for hybridization of primers in the sequencing-by-synthesis approaches. The method, based on ligation under optimized conditions, is selective and provides high yields of the target heterobifunctional DNA product. An additional two-step procedure can be applied to select further for the desired bifunctionalized product using PCR amplification with a chemically modified primer. Both functional groups in the modified DNA are chemically active and can be used in surface immobilization of the DNA strands to create the surface of a biosensor or sequencing chip.
Keywords: Biosensors, DNA modification, Next-generation sequencing, DNA attachment, Bioconjugation, Genomic DNA
Introduction
Methods of DNA detection and analysis have advanced at a high pace in the last decade. The completion of the human and many other vertebrate and invertebrate genomes in recent years has revolutionized the course of biological and biomedical research [1, 2]. Development of DNA array technology has accelerated diagnostics and has made possible analysis of gene expressions on thousands of genes simultaneously [3]. Whereas many genomes have been decoded using the state-of-the-art Sanger sequencing technology, ensuring long reads with high accuracy, a number of research groups and biotechnology companies are now developing new, next-generation, sequencing methods to reduce the sequencing costs and increase the throughput by several orders of magnitude [4]. These new approaches are developed with the ultimate goal of achieving full genome sequencing of an individual at a price affordable in a single laboratory or as a clinical test.
One of the key elements in the next-generation sequencing technology (and DNA chip technology, in a broad sense) is the sample preparation of genomic DNA samples. These recent technologies require end modification of the DNA for conjugation of fluorescent labels or reactive groups for subsequent attachment to solid surfaces of flat substrates, microscopic beads, or nanoparticles [4]. Several methods of new generation sequencing rely on a long-term observation of single DNA molecules attached to the surface of an observation fluid cell (DNA chip). Single-molecule sequencing provides the ultimate sensitivity and optimal parallelization with no need for amplification. Therefore, reproducible and robust chemistry for surface immobilization of genomic DNA fragments is critical for rapid progress in this field [5, 6]. Covalent specific chemical attachment has a clear advantage over non-specific adsorption to solid substrates or specific binding based on weak bio-molecular recognition, such as hybridization with a complementary DNA strand, antigen–antibody, or biotin–avidin type interactions. Covalent modification provides attachment that is stable under a variety of denaturing and mildly harsh conditions over long periods of time. Heterobifunctional modification, i.e., different functionality at 3′ and 5′ termini, avoids formation of loops in the surface attachment step.
In addition, most of the demonstrated and proposed next-generation sequencing technologies currently under development use a sequencing-by-synthesis approach, i.e., they use DNA polymerase to read the sequence and require an initial hybridization step of annealing short primers to DNA fragments to ensure efficient polymerase binding to DNA [4, 6]. A general technique for end modification of the genomic DNA fragments, ideally, should install a known sequence needed for hybridization of these short primers in every fragment. For example, the next-generation single molecule sequencing developed by Harris et al. requires introduction of known DNA segments at both ends of the DNA fragment [7]. These authors introduced a 5′-distal sequencing primer recognition site via an adapter ligation and a 3′-poly(dA) tail using terminal deoxynucleotidyl transferase. For other applications, e.g., to track single DNA molecules, it is often favorable to tag one end with a fluorophore, such as Cy3, Cy5, or Alexa dyes. Applications that use direct manipulation of single DNA molecules, such as optical or magnetic tweezers, require two functional groups for attachment to the surfaces of the sample support and force probes (dielectric or magnetic microspheres or a tip of an atomic force microscope) [8].
A modification of synthetic single-stranded DNA (ssDNA) oligomers with biospecific or reactive functional groups, such as biotin, amino, or thiol groups, that enable binding to solid surfaces is readily done using the technology of phosphoramidite-based solid phase DNA synthesis. Both 3′- and 5′-modified oligomers with many different end groups are available commercially. Here, we describe an easy two-step method to introduce distinct functional chemical groups into double-stranded DNA (dsDNA) fragments of a random sequence using short synthetic DNA oligomers carrying desired terminal functional groups. This method can be applied to genomic DNA that has been cut into smaller sized fragments by limited DNAse digest, sonication, or nebulization. We demonstrate the utility of our approach by carrying out a successful controlled chemical attachment of a thiol-modified DNA to gold-coated or silane-modified solid substrates.
Materials and methods
Chemicals and synthetic DNA
Chemicals were ordered from Fluka, Aldrich, EMD, and MP Biomedicals and used without further purification. All of the synthetic DNA oligomers were ordered from IDT (most with HPLC purification) and were dissolved in water to 100 μM concentration upon arrival. λ DNA, enzymes, and buffers for enzymes were supplied by New England Biolabs. Deoxynucleotides (dNTP) were ordered from Promega. Water used in experiments was deionized (DI) with Millipore Simplicity UV and autoclaved before use; 5× TBE stock buffer (Tris–base (0.445 M), boric acid (0.445 M), and EDTA (7.85 mM)) was diluted to 1× before use. All reactions were carried out in autoclaved microcentrifuge and PCR tubes. DNA electrophoresis was run in SureLock minicell (Novex) using VWR power supply at 100 V for 1 h. Precast 6% polyacrylamide gels containing TB buffer (Invitrogen) were used for DNA electrophoresis. The 3 M sodium acetate solution was adjusted to pH of 4.0 with glacial acetic acid; 10× annealing buffer was made from 100 mM Tris–HCl, 1 M NaCl, and 10 mM EDTA and was diluted to 1× for use.
PCR amplification
The 100-mer template (5′-AGCGACTGCTATCATGTCATATCGACGTGCTCACTAGCTCTACATATGCGTGCGTGATCAGATGACGTATCGATACGTACTATAGTCTCGTATGCGAGTG-3′) was diluted to a 1 μM stock solution for storage. The 5′-end phosphorylated primers (5′-AGCGACTGCTATCATGTCATATCG-3′, forward primer; 5′-CACTCGCATACGAGAC TATAGTACG-3′, reverse primer) were diluted to 2.5 μM. PCR was carried out according to the protocol recommended by NEB for Taq DNA polymerase. One microliter of 100-mer template with 2.5 μL of forward and reverse primers were used with 10 μL of 10× Thermopol II buffer and 2 μL 100 mM MgSO4. One microliter Taq DNA polymerase (5 units) was added; the total reaction volume was adjusted to 100 μL with DI water and exposed to 30 cycles of 15 s denaturation at 95 °C, 10 s annealing at 60 °C, and 30 s extension at 72 °C. After amplification, the reaction mixture was purified using a QIAGEN nucleotide removal kit and eluted with 50 μL of water.
dA tailing
To the purified PCR product, 500× dATP was added to produce a single dA overhang to each 3′-end with 1.5 μL of Taq polymerase for 1 h at 70 °C. The mixture was then purified using a QIAGEN MinElute reaction cleanup kit. The DNA was eluted with 10 μL of water.
Adapter annealing
Twenty microliters of 100 μM (in annealing buffer) end-modified oligonucleotides of a complementary sequence with T-overhang (5′-CCTAGTCGAACGATCTGACCT-3′, 5′-thiol-modified; 5′-GGTCAGATCGTTCGACTAGG-3′, 3′-amino modified) were annealed. To ensure high yield of desired adapter, the mixture was placed in a thermocycler (Eppendorf, EPgradient S) and allowed to denature at 95 °C for 2 min, with an annealing and denaturing cycle at 95 °C for 15 s, 40°C at 15 s, 72°C at 1 min repeated five times, and, finally, annealing at 72 °C for 5 min. The adapters were used directly without any further purification.
dA-T ligation
dA-tailed DNA (45 pmol) was mixed with 25 molar excess of adapter to add at both ends, then 7,500 cohesive-end units of T4 ligase (NEB) were added. The volume of the mixture was brought to 75 μL. The reaction mixture was then incubated at 16°C overnight in a thermocycler (unless other conditions are stated). The ligated products were loaded onto a 6% polyacrylamide gel electrophoresis (PAGE) gel, and a band of desired product with both ends ligated was excised, DNA was eluted from the gel with 1× TBE buffer at 37 °C overnight and purified using a QIAGEN MinElute Reaction Cleanup kit into 10 μL of water.
Genomic DNA preparation
λ-Phage DNA (33 μg) was sonicated four times at 5 W for 20 s on ice in 300 μL of buffered solution. Sheared DNA was then loaded on a PAGE gel for size selection. Selected genomic DNA was eluted out from the excised bands in 1× TBE buffer at 37 °C overnight. Eluted DNA was purified and concentrated in 35 μL of water using a QIAGEN nucleotide removal kit. For dA tailing and dA-dT ligation of genomic DNA, the same procedures were used as those described above for the PCR-amplified synthetic DNA.
Mass spectrometry [9]
The DNA samples were prepared by ethanol precipitation followed by dissolution in DI water. Microflex (Bruker Daltonics, Germany) was used to obtain MALDI-TOF mass spectra of the DNA oligomers. A solution of 0.3 M 3-hydroxypicolinic acid, 0.5 M picolinic acid, and 0.3 M ammonium fluoride (9:1:1 M ratio) was used to form a matrix on the ground steel plate (Bruker Daltonics, Germany). The matrix solution (1 μL) was plated and dried, and then 1 μL of DNA solution was placed on top of the matrix and dried. Spectra were taken under Linear Positive mode using a 20-kV laser. Since the molecular weight of the DNA fragments analyzed was fairly high (up to 90 kDa), laser intensity was higher than the typical intensity required for detection of small proteins. The laser attenuator range was set to 45%, and the full dynamic range of the laser intensity within the attenuator range was used.
DNA attachment to gold surfaces
Gold-coated coverslips were prepared by e-beam deposition of 12 nm Au with 1.5 nm Ti adhesion layer on piranha-cleaned 24 mm× 60 mm coverslips (Superslips, VWR). Coverslips were cut into 1 cm×1 cm pieces. Prior to surface modification, goldcoated coverslips were cleaned by nitrogen-oxygen plasma (Harrick Plasma, NJ, USA) on high power for 1 min, rinsed with ethanol, and dried with nitrogen.
The thiol group of the heterobifunctionally labeled DNA (size selected genomic DNA or control PCR product) was deprotected with tris(2-carboxyethyl)phosphine (5 mM in 6× saline-sodium citrate buffer containing 100 nM of citric acid, and 1 M NaCl). The DNA and the deprotecting solution were mixed in 1:10 volume ratio and incubated at room temperature (RT) for 1 h. The deprotected DNA was purified using a QIAGEN MinElute Reaction Cleanup kit into 10 μL of water. The purified DNA was reacted with Au-coated substrates in PBS buffer containing (1) 1 M NaCl for attachment of double-stranded DNA or (2) 8 M urea, 4 M guanidine·HCl (Gu·HCl), or 0.5 M NaOH for attachment of single-stranded DNA. The final concentration of DNA in the 15-μL reaction volume was 50 nM double stranded or 100 nM single stranded.
Fifteen microliters of solution was deposited onto a plasma-cleaned gold-coated coverslip and incubated in a humid chamber at RT for 1 h. The coverslip was then rinsed with DI water, soaked three times for 15 min in 1 M NaCl PBS buffer, rinsed with DI water, and dried with a stream of nitrogen. The dried samples were incubated in a vial containing 15 mL of 3 mM mercaptohexanoic acid (MHA) in 1 M NaCl PBS buffer overnight at RT to block the remaining Au surface and to remove non-specifically bound DNA. The samples were then washed as described above after reaction with DNA. For dsDNA, two different samples were made: (1) one sample was analyzed with X-ray photoelectron spectroscopy (XPS) as is; (2) another sample was incubated for 45 min in 4 M Gu·HCl solution in order to denature the immobilized double-stranded DNA into single strands prior to XPS analysis. As a control, Au surfaces were exposed to a 100-mer-long PCR product in 4 M Gu·HCl in the same manner. High-resolution XPS spectra (pass energy of 300 eV, 60° tilt angle with respect to surface normal) of Au, N, P, S, C, and O regions for each sample were taken (Scienta ESCA) and quantified using a multiple peak fitting routine of IGOR Pro 6.0 (WaveMetrics, OR, USA).
Silicon surface modification with maleimide silane
3-Maleimidopropyltriethoxy silane (MPTES) was synthesized following a method adapted from Shaltout et al. [10] Briefly, a 2.2 M solution of maleic anhydride in anhydrous toluene was prepared, and then a 3.3 M solution of 3-aminopropyltriethoxysilane in anhydrous toluene was added dropwise. The mixture was stirred for 2 h at RT, and ZnCl2 powder (1:1 in mole ratio with maleic anhydride) was added. After stirring for 30 min, a 3.3 M solution of hexamethyldisilazane (HMDS) in anhydrous toluene was added dropwise. The reaction mixture was refluxed for 2 h after complete addition of HMDS. Solvent was removed using a rotary evaporator following removal of excess ZnCl2 by vacuum filtration. Using vacuum distillation (~300 mTorr), excess maleic anhydride and byproducts were distilled at 65 and 96 °C, respectively. The final product (MPTES) was collected at 112 °C. 1H NMR (CDCl3): δ=6.67 (s, 2H), 3.78 (q, 6H), 3.48 (t, 2H), 1.67 (m, 2H), 1.19 (t, 9H), 0.56 (m, 2H).
A silicon wafer was cut into 1 cm×2.5 cm pieces and cleaned in piranha solution (60% sulfuric acid with 40% hydrogen peroxide) for 1 h (Warning: piranha solution reacts violently with organics and must be handled with extreme care). The pieces were washed with water and dried with N2 for ellipsometry measurements. In a round-bottomed flask, MPTES was added to 30 mL of anhydrous toluene to make 0.1% (v/v) solution. The pieces of Si wafer were submerged in the solution and secured using a Teflon comb. The solution was refluxed for 1 h. After cooling to RT, the pieces of Si wafer were washed with toluene (HPLC grade) and ethanol, dried with N2, and stored in scintillation vials. The formation of 3-maleimidopropyltriethoxy silane monolayer was confirmed by ellipsometry and contact angle measurements.
DNA attachment to the MPTES-modified Si surface
The reactivity of the MPTES monolayer was tested with 5′-TAMRA and 3′-thiol-modified synthetic oligonucleotide (5′-HS-T15TCATCGCACATCGTAGCACAAGACTAMRA-3′). Fifteen microliters of the 100 nM solution of ssDNA (deprotected and purified as described above) in 1 M NaCl PBS was deposited onto the surface of the Si wafer, and the sample was incubated in a humid chamber at RT for 1 h. The surface was then washed with water and soaked three times for 45 min total in 1 M NaCl PBS buffer. After washing, the wafers were incubated in 3 mM MHA (in 1 M NaCl PBS buffer) overnight to block the surface, washed as described after DNA reaction, dried, and stored in a vial prior to characterization. A control to test the surface reactivity was done with an MPTES surface blocked by MHA. The same procedure (deposition, washing, and blocking steps) was followed using (1) 5′-TAMRA and 3′-thiol-modified ssDNA oligo; (2) 5′-FAM-modified ssDNA without 3′-thiol group (5′-FAM-TTTGTCTTGTGCTACGATGTGCGATGA-3′); (3) 150-mer heterobifunctionally labeled DNA. For the 150-mer, instead of 1 M NaCl PBS buffer, a denaturing 4 M Gu·HCl buffer was used in a surface reaction to generate ssDNA at a final concentration of 100 nM.
FITC attachment to the amine end of the surface-attached DNA
Fluorescein isothiocyanate (FITC; 17 mM) solution was made by dissolving FITC powder in 1 M Na2CO3-NaHCO3 buffer (pH 9), DMF, and water (5:2:8 vol. ratio). After reaction with DNA, the Si surfaces were covered with the FITC solution and incubated overnight (16 h) at RT. The wafers were washed twice with dimethylsulfoxide then four times with water, and dried with N2. The characterization of the wafers was done with a fluorescent microscope (Olympus, IX71).
PCR amplification using end-modified primers
One microliter of end-modified 150-mer DNA (337 nM in water), 5 μL of 10 μM 5′-thiol-modified oligo (5′-HS-CCTAGTCGAACGATCTGACCT-3′), 1 μL of 25 μM dNTP mix, 10 μL of 10× Taq DNA polymerase buffer, 1 μL Taq DNA polymerase, and 82 μL of DI water were mixed together for the PCR reaction. After the amplification reaction, the mixture was purified with a QIAGEN Nucleotide Removal kit into 50 μL of DI water.
Terminal deoxynucleotidyl transferase (TdT) reaction with ddATP-NH2
A 100 mM stock solution of 3′-amino-2′, 3′-dideoxyadenosine-5′-triphosphate (ddATP-NH2) was ordered from TriLink Bio Technologies (San Diego, CA) and was diluted to 25 μM before use. TdT was ordered from New England Biolabs. All reaction mixtures contained 5 μL of PCR-amplified 150-mer DNA with a 5′-thiol group, 1 μL of 25 μM ddATP-NH2, 5 μL of 10× TdT buffer, and 5 μL of 10× CoCl2 buffer. Four different TdT concentrations were used to test the efficiency of the enzyme: 10, 15, 20, and 40 units of TdT were added to each tube and the volume was brought up to 50 μL. The tubes were incubated at 37 °C for 20 min, and then TdT was heat-inactivated at 72 °C for 20 min. The reaction mixture was then purified using a QIAGEN MinElute Reaction Cleanup kit into 10 μL of water.
FITC attachment to amine end of the amplified heterobifunctional DNA
Following TdT treatment, 10 μL of purified heterobifunctional DNA was mixed with 100 μL of 17 mM FITC solution and 90 μL of 1 M Na2CO3-NaHCO3 buffer (pH 9) in a microcentrifuge tube. The mixture was incubated at RT overnight. To remove unreacted FITC, DNA from the reaction mixture was purified using a QIAGEN MinElute Reaction Cleanup kit into 10 μL of DI water. Purified FITC-reacted DNA solution was dropped onto a low-fluorescence coverslip (sonicated in 10% Alconox solution for 20 min, water for 5 min, acetone for 15 min, and 1 M KOH for 20 min; rinsed with DI water; and flash-flamed to remove residual organics). After air drying the DNA solution, fluorescence was quantified using a CCD camera mounted on a fluorescence microscope (Olympus, IX71).
Results and discussion
Strategy for efficient DNA modification
Figure 1 schematically outlines the steps in our approach to DNA end modification. We rely on ligation of synthetic oligomers of known sequence (hereafter called adaptors) bearing desired chemical functional groups to DNA fragments generated by shearing the genomic DNA. Our strategy to achieve high yields of desired ligation products was to introduce single-base long, complementary overhangs: a 3′-A-tail in the DNA fragments and a 5′-T-tail in the synthetic adaptors. A-tailing of the dsDNA genomic fragment prevents self-ligation of the DNA, whereas the T-overhang in the synthetic adapter prevents adapter dimerization in the presence of ligase, hence, guiding the reaction towards targeted DNA-adapter ligation (Fig. 1). An A-tailing modification of dsDNA is readily achieved using the A-tailing activity of Taq DNA polymerase, whereas a T-tail can be easily added during the solid-phase synthesis of one of the strands forming dsDNA adapters. At the same time, 5′-phosphorylation in both adaptors and fragments ensure the formation of covalent phosphodiester bonds between the 3′-hydroxl- and 5′-phosphate groups. The adaptors are formed by annealing two complementary sequences: one carrying a 3′-modification and another carrying a 5′-modification [11]. Successful ligation at the two ends of a genomic dsDNA fragment introduces chemical groups at both ends of each complimentary strand of the genomic DNA.
Fig. 1.
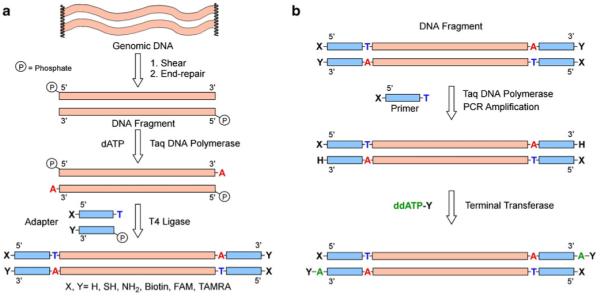
a Adapter annealing method. This method can be applied to any dsDNA fragment that requires the introduction of heterobifunctional reactive groups on both ends of the DNA by synthetic adapters. b Amplification of modified DNA. Using ssDNA with a 5′-functional group as a primer, modified DNA can be PCR-amplified, following terminal deoxynucleotidyl transferase addition of ddATP with another functional group to produce a large amount of heterobifunctional dsDNA
This approach has several advantages in terms of its specificity and flexibility in achieving the desired modification: (1) neither genomic DNA nor adaptors are consumed in the side reactions of self-ligation due to base mismatch, a clear improvement over any blunt-end ligation strategy; (2) the ligation is self-limiting, i.e., the ligation is terminated after addition of a single adaptor at each end due to presence of the chemical groups; (3) the approach can be used to incorporate either single chemical functionality at one end only (3′ or 5′ terminus) or a different chemical functionality at each; and (4) a known sequence is introduced at both 3′- and 5′-ends that can be used in further steps, e.g., for amplification (Fig. 1b), hybridization of primers for polymerase binding, or annealing of labeled oligonucleotides to identify the location of the strands attached to the surface of the DNA chip.
Chemical modification of model DNA
For proof of principle, we performed initial studies using a double-stranded 100 base pair (bp) long PCR fragment derived from a synthetic oligonucleotide of known sequence (5′-AGCGACTGCTATCATGTCATATCGACGTGCTCACTAGCTCTACATATGCGTGCGTGATCAGATGACGTATCGATACGTACTATAGTCTCGTATGCGAGTG-3′). Ligation was carried out by mixing 6 pmol of DNA with a 25 molar excess of adapter in 10 μL volume in the presence of 1,000 units of ligase overnight at 16 °C (as recommended by the manufacturer—NEB). Controls comprised dsDNA PCR product or adapter only in the absence or presence of ligase, as well as DNA product and adapter together in the absence of ligase. The resulting reaction products were separated by electrophoresis on a 6% PAGE gels using TBE buffer. Figure 2 displays an image of the gel separating reaction products obtained under optimized conditions as well as the products from several control experiments.
Fig. 2.
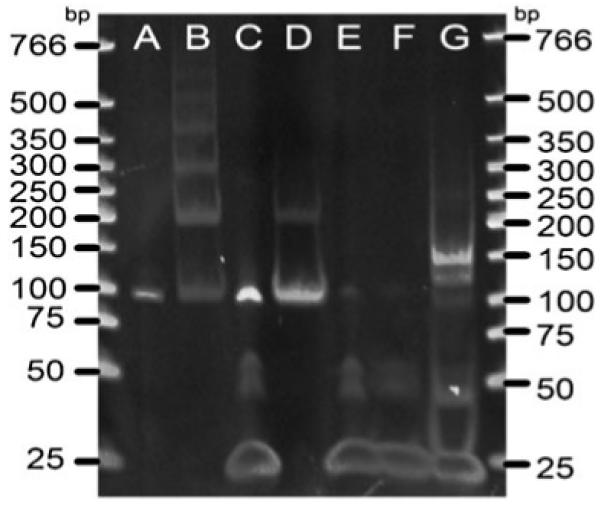
PAGE gel demonstrating the generation of a 150-mer fragment after successful adapter ligation. Lanes A 100 bp without ligase, B 100-mer with 1,000 units of T4 ligase, C 100-mer and 25 molar excess adapters with T4 ligase, D dA-tailed 100-mer with T4 ligase, E adapter without ligase, F adapter with T4 ligase, G dA-tailed 100-mer and 25 M excess adapters with 1,000 units of T4 ligase
As shown in Fig. 2, A-tailing of the DNA fragments (lane D) almost completely prevented self-ligation in the presence of ligase, whereas blunt-ended phosphorylated DNA product self-ligated and formed a ladder of higher molecular weight reaction products (lane B). The 25-bp-long adapter carrying T-overhang contained small amounts of impurities (lane E) around higher molecular weight (around 50-mer and 100-mer), as supplied. The 25-mer adaptor did not polymerize in the presence of ligase (compare lane E and F).
In contrast, the ligation reaction containing dA-tailed DNA fragment and T-overhang adapter in a 1:25 molar ratio resulted in two distinct products (lane G) that migrated at 125-mer and 150-mer sizes. Under these conditions, the 100-mer fragment has mostly disappeared, suggesting almost complete reaction of the original dA-tailed PCR product with adapter molecules. According to observed relative intensity, the 150-mer DNA oligomers, corresponding to the addition of adaptors at both ends of the model dsDNA, was the dominant product.
To corroborate this interpretation, we carried out several additional experiments characterizing the nature of these ligation products [12-14]. Analysis by MALDI mass spectrometry revealed that 150-bp and 125-bp-long products were indeed prevalent in the ligated mixture (Fig. 3). Signal for the original 100-mer PCR product was not detected in mass spectra, most likely due to its much lower concentration than that of the ligation reaction products. Relatively high amounts of singly (M+) and doubly charged (M2+) molecular ions of 150-mer ligated product were detected around m/z of 90,000 and 45,000, respectively. As expected from the gel analysis, a significant amount of 125-mer product was present and detected as M2+ approximately at 38,000. In addition, an excess amount of adapter was detected around m/z 13,700. The spectra, along with the PAGE analysis, indicated that ligation efficiency towards the 150-mer product was high.
Fig. 3.

Characterization of reaction products by MALDI-TOF mass spectrometry. a Mass spectrum of 100-mer PCR product. The peak around m/z of 64,000 represents singly charged molecular ion (M+), and the peak around 32,000 represents a doubly charged molecular ion (M2+ ). The broad peaks may have resulted from Na ion adducts and/or wide isotope distributions. b Mass spectrum of annealed adapter. The peak at m/z around 13,600 shows M+ of the adapter and M2+ appears at around 6,800. M4+ is also detected around 3,400. Other peaks (m/z around 10,000 and in between 4,000 and 6,000) are non-specifically bound products during annealing process. c–e Mass spectra of ligated product without gel selection. Strong signal of unreacted adapters was present (c), and M+ around m/z 90,000 showed the dominant presence of 150-mer-ligated product (e). M2+ of 150-mer product appeared around m/z 45,000, and only M2+ of 125-mer one-end-ligated product was observed around m/z 38,000 (d). No significant peak around 64,000 or 32,000 was detected, confirming that the initial 100-mer PCR product was mostly converted to 125-mer- or 150-mer-ligated products
Next, we confirmed that the fusion point between the model DNA and adaptors occurred at the expected location in the sequence. We separated the 125-mer and 150-mer reaction products on a preparative PAGE and eluted the bands overnight at 37 °C into TBE running buffer followed by concentration with a spin column. These purified reaction products were then characterized by DNA sequencing (Fig. 4). Sequencing primers (20-mers) were selected so that they would prime to the fusion site between the adapter and 3′-end of the PCR product. Extension of the sequencing reactions demonstrated that the 125-mer was comprised of the 3′ part of the adapter sequence (otherwise sequencing primer hybridization would not have taken place), the known PCR template ending in an A-base (Fig. 4, top trace) showing that the in vitro A-tailing reaction was also successful. In contrast, the 150-mer sequence extended after the A-base into the ligated adapter sequence (Fig. 4, bottom trace), demonstrating a successful ligation of the adapter to the 3′-end of the 100-mer PCR product. Our ability to use known sequence incorporated into a model (generally, unknown) DNA sequence for hybridization of primers and successful polymerase reaction demonstrates the utility of our approach for adding both known chemical groups and priming sites to genomic DNA.
Fig. 4.
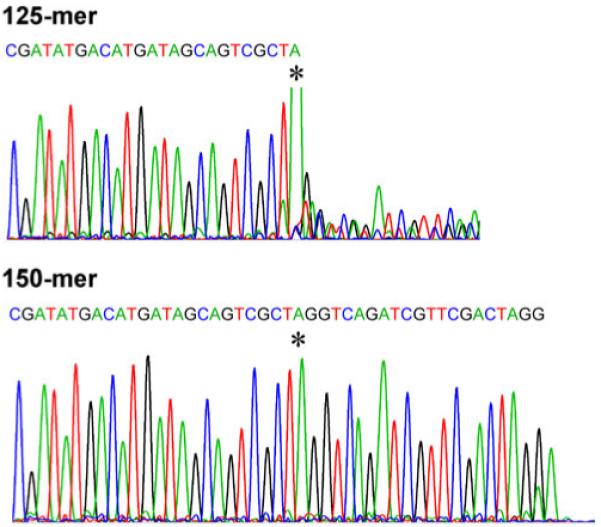
Ending sequences of the eluted 125-mer and 150-mer bands from PAGE. The asterisks mark the A added by Taq polymerase at the end of the model DNA sequence
We observed that, under conditions suggested by the suppliers of the T4 ligase (NEB and Promega), the predominant product was persistently a single-addition product (~100% 125-mer versus ~0% of 150-mer DNA), whereas the yield of conversion was relatively low (>70% of original DNA remained unmodified in spite of the 25-fold molar excess of adaptors). We investigated a range of conditions to enable efficient and complete ligation of a model DNA with chemically modified adaptors at both ends. We varied reaction time, reaction temperature, and concentration of ligase or DNA and quantified the composition of resulting reaction mixtures using intensity of peaks corresponding to 100-mer-, 125-mer-, and 150-mer-long DNA fragments (Fig. 5a).
Fig. 5.
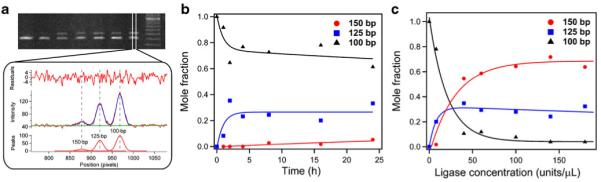
Analysis of PAGE data. a Image intensity profiles are averaged over 20–40 pixels in a given lane and then fitted to multiple Gaussian peaks (using IGOR Pro, Wavemetrics) representing 100-mer-, 125-mer-, and 150-mer-long DNA fragments. Peak areas (normalized by the total fragment length) are used as a quantitative measure of the molar amount of given oligomers. b Time course of changes in the molar fraction of the dA-tailed unmodified DNA (100-mer) and DNA ligated at one end (125-mer) or both ends (150-mer). Six picomoles of 100-mer DNA with 25 molar excess adapters and 400 units of T4 ligase in a total volume of 50 μL were incubated at 16 °C. c Dependence of the conversion of the dA-tailed DNA into end-modified DNA on the concentration of the enzyme for 6-h-long reactions. Six picomoles of 100-mer DNA and 25 molar excess of adapters in a total volume of 10 μL were incubated at 16 °C. Curves in b and c are fits to exponential or double-exponential functions and serve as guides to the eye
Increasing reaction time did not affect the yields of the end-modified products significantly, and after 4 h, no substantial change was observed (Fig. 5b), possibly due to the loss of enzyme activity over long times. Changes in temperature (reactions at 4, 16, and 25 °C) or concentration of the DNA (doubling the concentration) did not result in significant changes in the outcome of the reaction. Concentration of ligase in the reaction mixture, on the other hand, played a major role in defining the yield of end-modified products: with the total concentration exceeding 50 units/μL, more than 90% of original dsDNA was converted into double- and single-addition ligation products in approximately 2:1 ratio (Fig. 5c).
Modification of genomic DNA
We carried out similar modification reactions on a sample of sheared and end-repaired λ-phage DNA using conditions optimized for the model 100-mer DNA (Fig. 6). By shearing DNA with ultrasound, we generated a relatively broad selection of molecular weights (100-mers to 600-mers) and selected a small fraction with sizes around 200-mer by excision from the preparatory scale gel. The ligation reaction did not produce well-separated bands, but resulted in a noticeable shift in the size of the genomic DNA ligation products compared to original DNA. By fitting intensity profiles to three Gaussian peaks, the products could be well-described as a mixture of initial dsDNA and double- and single-addition products (1:3 ratio) with a total yield of conversion around 70%.
Fig. 6.
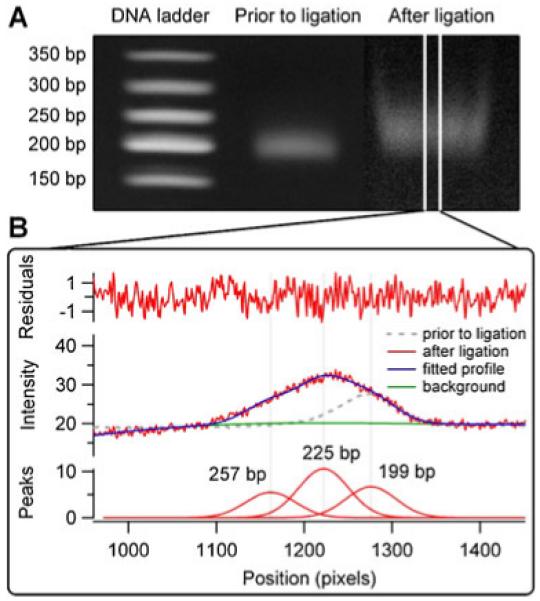
Analysis of PAGE data for modified genomic DNA. a Image intensity profiles are averaged over 40 pixels and b fitted to multiple Gaussian peaks representing bands around 200, 225, and 250 base pair long DNA fragments
Functionalization of model DNA using PCR with modified primers and terminal deoxynucleotidyl transferase with modified ddNTP
We used PCR amplification to select model DNA that was successfully ligated on both ends by the 25-mer adapters. A PCR reaction with a primer having 5′-thiol modification resulted in a 150-mer-long product (as confirmed by PAGE) with 5′-modification, but lost the modification of the 3′-end (Fig. 1b). The single-addition 125-mer product was not amplified because it does not end in the sequence complementary to the primer. Heterobifunctional 150-mer DNA, however, can be readily obtained from the PCR product by modifying the 3′-end with TdT using chemically modified ddATP as a substrate (ddATP-NH2 in this case).
We demonstrated the effectiveness of TdT in addition of the 3′-modification by carrying out a subsequent reaction of the primary amine group in the expected 150-mer DNA product with a fluorescent dye (FITC). We compared four different enzyme concentrations and observed that the fluorescence of the products formed in the reaction between TdT-modified DNA and FITC was consistent with the incorporation of the ddATP-NH2 in the DNA sequence. Without enzyme treatment, no fluorescence was detected. Fluorescence was observed only when TdT was present and increased when higher TdT concentrations were used (Fig. 7), signifying the presence of the amine group in the DNA. The level of fluorescence appeared to saturate after the TdT concentration reached 0.4 units/μL, signifying that, under these conditions, ddATP-NH2 addition by TdT is complete.
Fig. 7.
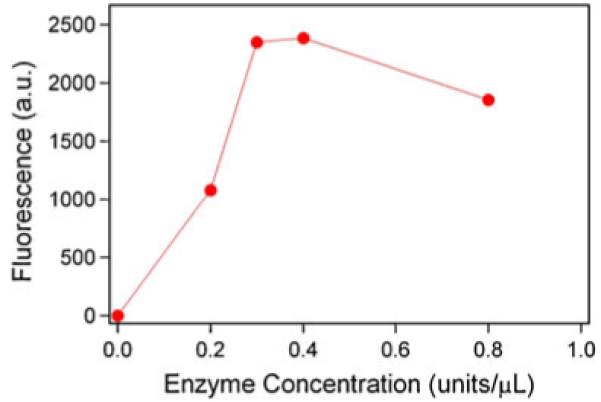
Fluorescent signal versus terminal deoxynucleotidyl transferase concentration in a reaction that adds ddATP-NH2 to the 3′-end of the PCR-amplified model DNA sequence. Experimental error estimated as a width of the peak in the pixel intensity histogram was 10–20% of the average intensity value
Chemical reactivity of functionalized DNA
Chemical functionality of the modified DNAs was tested by attachment of the DNA to Au-coated [15, 16] or MPTES-modified silicon surfaces [17, 18]. Both surfaces are expected to be reactive towards the 5′-thiol functional group of the modified heterobifunctional DNA. We prepared two Au samples, one exposed to unmodified and another exposed to modified DNA, followed by reaction with MHA to block the unreacted Au surface and remove non-specifically bound DNA molecules. Both samples contained 100 nM of ssDNA in high salt PBS buffer (high ionic strength was necessary to ensure that the surface reaction can take place and charged DNA can approach the surface). In addition, the buffers contained 4 M Gu·HCl to denature the double-stranded DNA into single-stranded form for the attachment reaction. The XPS spectra of the Au surfaces reacted with 150-mer modified DNA showed high intensity nitrogen and phosphorous peaks, whereas the Au surface exposed to 100-mer unmodified DNA did not show any peaks due to nitrogen and phosphorous (Fig. 8). The N 1s peak indicated the presence of three chemically distinct forms (peaks at 398.9, 399.8, and 400.6 eV) as reported in the previous studies of surface immobilized DNA [19]. The phosphorous/nitrogen molar ratio for surface-attached DNA ranged from 0.23 to 0.34 (Table 1), near the expected value of 0.285 for our model sequence. Therefore, only thiol-modified DNA was capable of robust attachment to Au surfaces, consistent with the previous reports on this system. For sulfur, peaks corresponding to free thiol, Aubound thiol (thiolate), and oxidized sulfur were observed as is usually the case in similar DNA/short chain thiol systems [20]. The peak due to components in high oxidation state (S–O bonds) at 168–169 eV was not present on the surface reacted with 150-mer-modified DNA. It is plausible that in the MHA blocking step, which follows DNA attachment, the highly charged DNA, covalently linked to the gold surface, prevents either adsorption of oxidized negatively charged species (sulfonates or sulfates) or prevents formation of bilayers of MHA that would be susceptible to oxidation.
Fig. 8.
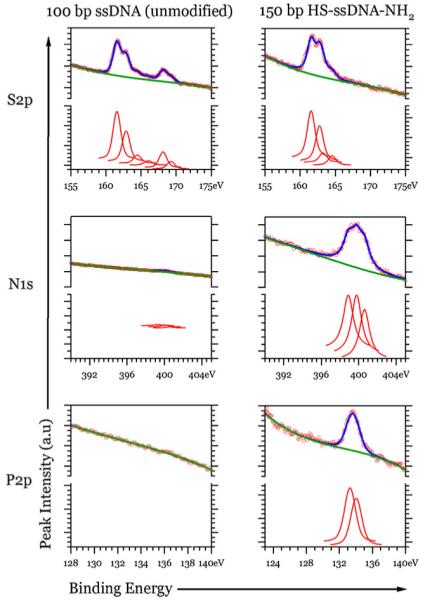
S 2p, N 1s, and P 2p XPS spectra of the gold surface reacted with unmodified and modified DNA followed by MHA (circles data, curves fits). Intensity scale in the region of each element is the same for both samples. The nitrogen and phosphorous are observed on the surface with modified DNA and not detectable (traces) for samples exposed to unmodified DNA. Three different species of nitrogen were present on the Au surface with covalently linked DNA. Both samples contained bound (161–163 eV) and unbound (164–165 eV) sulfur, but only the surface treated with 100-mer DNA showed oxidized sulfur (168–169 eV)
Table 1.
Composition of the surface layer formed in reactions of the end-modified DNA with Au surfaces determined from the XPS spectra
| Surface reaction | Atomic percent |
Intensity of Au peak (relative) |
Ratio P/N | Nearest neighbor distance (nm) |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
| N | O | P | Sa |
C | ||||||
| S–Au | S–H | S–O | ||||||||
| 100 nM ssDNA in PBS+4 M Gu·HCl (ssDNA on the surface) |
3.4 | 13.6 | 1.2 | 2.8 | 0.7 | 0.0 | 78.4 | 1.00 | 0.35 | 7.7 |
| 50 nM dsDNA in PBS (dsDNA on the surface) | 5.3 | 15.9 | 1.3 | 2.4 | 0.5 | 0.1 | 74.5 | 0.82 | 0.25 | 8.2 |
| 50 nM dsDNA in PBS, denatured by treatment with 4 M Gu·HCl (ssDNA on the surface) |
3.2 | 20.6 | 0.74 | 3.2 | 0.5 | 1.0 | 70.7 | 1.62 | 0.23 | 8.6 |
Atomic percent of S takes into account attenuation by the MHA monolayer
We evaluated different denaturing buffers in the ssDNA surface attachment reaction by quantifying surface density of the DNA obtained for reactions from buffers containing 4 M Gu·HCl, 8 M urea, and 0.5 M NaOH. Surface density was estimated using the nitrogen/sulfur molar ratio and assuming formation of a complete thiol monolayer (MHA and HS-DNA) resulting in 0.214 nm2 surface area per sulfur atom [21]. All buffer compositions were effective in depositing ssDNA on the Au surface, providing practically the same (±7%) ssDNA density. Comparison between immobilization of single- and double-stranded thiol-modified 150-mers indicated very similar surface density for both types of reaction conditions. In the monolayer formed from the denaturing buffer, an average intermolecular distance of 7.7 nm between surface immobilized DNA molecules is very close to 8–10 nm diameter of the 150-mer ssDNA in solution estimated from the radius of gyration (Rg~4–5 nm for 150-mer). Such a dense surface coverage would imply formation of a monolayer of ssDNA during 1-h deposition from denaturing buffer using 100 nM DNA concentration. In the monolayer formed from the buffer preserving native double-stranded state, average intermolecular distance of 8.2 nm is also consistent with formation of the monolayer of the DNA molecules. The 150-mer dsDNA can be described as a rigid rod with dimensions of 2.2 nm×50 nm (diameter × length). Assuming formation of the monolayer of dsDNA molecules lying flat on the Au surface, the final average nearest neighbor distance is estimated at (2.2 nm×50 nm)1/2=10.6 nm. dsDNA on the surface could be converted to ssDNA by washing the sample with a denaturing buffer (containing 4 M Gu·HCl). XPS spectra showed a reduced amount of nitrogen and phosphorus, an increase in the amount of carbon and oxygen fractions, and a higher intensity from the Au substrate (due to decreased attenuation of photoelectrons in a thinner DNA overlayer). As expected, the calculated spacing between DNA strands was not affected.
To demonstrate reactivity of the amine group at the free end of the surface-attached DNA, we carried out a reaction of the amino group with a fluorescent dye molecule and characterized the resulting fluorescence. Since fluorescence cannot be detected near metal surfaces, we used thiolated DNA attached to maleimide-modified silicon surfaces rather than DNA immobilized onto gold surfaces. Silane chemistry was used to create an MPTES films on silicon wafer, resulting in thicknesses of 18.2–20.6 Å (with 0.5–2.0 Å error for multiple measurements on the same sample) and 72±2.0° for advancing (34±2.0° for receding) contact angle of water. Both the film thickness measurements (expected thickness for MPTES monolayer was 8–9 Å) and the large hysteresis in contact angles pointed to a disordered (and likely multilayer) surface created under our experiential conditions. Since only a small fraction of the maleimide terminal groups are needed for the reaction with the thiol of the heterobifunctionally modified DNA, these surfaces were adequate for the covalent attachment of HS-DNA-NH2 and provided a reasonable density of surface immobilized DNA to carry out a subsequent reaction between the fluorescent dye and terminal amino group of the DNA.
When a synthetic 27-mer DNA oligomer having a FAM (5,6-carboxyfluorescein) fluorescent label, but without a thiol group, was deposited on the MPTES sample, the change in thickness was barely detectable at 1.1±2.8 Å. The sample did not show significant fluorescence above the background (Fig. 9). Reacting MPTES film first with MHA, which should block all available terminal maleimide groups in the film, and then with DNA—labeled with TAMRA (5-carboxytetramethylrhodamine) and having a thiol group—resulted in a thickness increase of 2.4±1.5 Å and still displayed fluorescence near the background level.
Fig. 9.
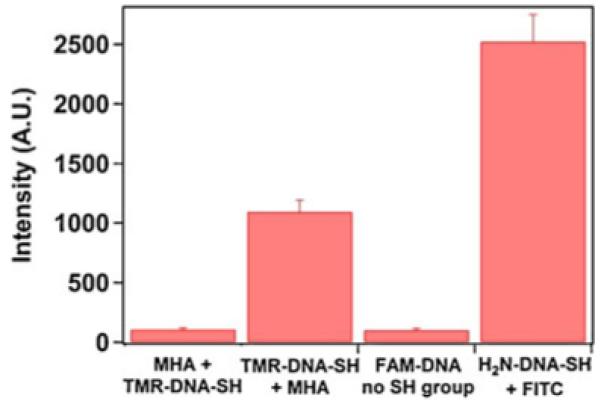
Fluorescence of the maleimide functionalized Si wafers after reactions with modified DNA. The two controls were (1) MPTES surface blocked with MHA and then reacted with 100 nM of 5′-TAMRA and 3′-thiol-modified DNA (MHA+TMR-DNA-SH) and (2) MPTES surface reacted with 100 nM 5′-FAM oligonucleotide without a 3′-thiol group (FAM-DNA no SH group). One hundred nanomolar 5′-TAMRA and 3′-thiol-modified DNA deposited on a maleimide silane surface followed by the reaction with MHA (TMR-DNA-SH+MHA) showed an approximately 10-fold increase in intensity compared to the two controls. One hundred nanomolar 5′-thiol and 3′-amine-modified 150-mer model DNA deposited on MPTES and reacted with FITC displayed a 20-fold increase in fluorescence compared to the two controls. Treatment of MPTES surfaces by FITC solution without the DNA attachment step resulted only in background fluorescence
On the other hand, using synthetic heterobifunctional 40-mer DNA oligomer having a fluorescent label (TAMRA) and a thiol group resulted in 2.7±2.3 Å change in film thickness and strong fluorescence. Therefore, availability of maleimide groups on the surface and thiol groups in the DNA ensured robust attachment of the DNA oligomers. Similarly, reaction of 150-mer-modified DNA followed by a blocking step with MHA and a reaction with FITC produced a change in thickness of 4.0±0.7 Å. The sample displayed high fluorescence (Fig. 9), indicating that amino groups in the surface-attached bifunctional DNA, obtained by our single-step modification procedure, are available for further reactions. These groups can be used for covalent attachment of molecular or nanoscopic markers for sensing applications or microscopic probes for direct manipulation of the DNA molecules, e.g., in optical or magnetic tweezers experiments.
Conclusions
With the development of next-generation sequencing strategies, single molecule sequencing is a valuable approach to minimize reagent consumption and increase the throughput by monitoring sequencing using high density of surface immobilized DNA strands. These methods benefit from robust DNA modification techniques that can install desired chemical functionality with high yield and at a low cost. The sequencing chip should be particularly robust when covalent attachment chemistry is used. In this work, we demonstrated efficient heterobifunctional modification of dsDNA fragments having an arbitrary sequence by a simple two-step procedure. These steps comprise A-tailing of the blunt-ended DNA followed by ligation with synthetic T-tailed adapters carrying the desired functional groups (amine and thiol). The critical ligation step was extremely sensitive to enzyme concentration, and high enzyme loading was critical to achieving ligation at both ends of the dsDNA fragment. The modification procedure was also effective in modifying sheared and end-repaired genomic DNA.
Another two-step follow-up procedure can be applied to select for the desired bifunctionalized product. This additional procedure consists of a PCR step with a single (same sequence for forward and reverse) modified primer (which installs a 5′-end modification) followed by the addition of an extra modified base by terminal deoxynucleotidyl transferase (which installs a 3′-end modification). Both functional groups in the modified DNA are chemically active as demonstrated by the DNA surface attachment via a 5′-thiol group and subsequent labeling of the surface immobilized DNA with a fluorophore via a 3′-amino group. Additional research on optimal enzymes and conditions could further improve the efficiency and yield of the method.
Acknowledgments
This work was supported by National Institutes of Health grant R21 HG004141. We have used XPS facilities supported by the Center for Advanced Materials and Nanotechnology of Lehigh University. We thank Dr. Alfred Miller for performing XPS experiments and Ms. Vanessa Kern for help with analysis of the XPS spectra. We are grateful to Dr. Carl Fuller of Life Technologies for insightful discussions.
Contributor Information
Hana I. Lim, Chemistry Department, Lehigh University, 6 E. Packer Ave., Bethlehem, PA 18015, USA
Piercen M. Oliver, Chemistry Department, Lehigh University, 6 E. Packer Ave., Bethlehem, PA 18015, USA
Jutta Marzillier, Department of Biological Sciences, Lehigh University, 111 Research Drive, Bethlehem, PA 18015, USA.
Dmitri V. Vezenov, Chemistry Department, Lehigh University, 6 E. Packer Ave., Bethlehem, PA 18015, USA
References
- 1.Kahvejian A, Quackenbush J, Thompson JF. Nat. Biotechnol. 2008;26:1125–1133. doi: 10.1038/nbt1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schloss JA. Nat Biotechnol. 2008;26:1113–1115. doi: 10.1038/nbt1008-1113. [DOI] [PubMed] [Google Scholar]
- 3.Heller MJ. Annu Rev Biomed Eng. 2002;4:129–153. doi: 10.1146/annurev.bioeng.4.020702.153438. [DOI] [PubMed] [Google Scholar]
- 4.Metzker ML. Nat Rev Genet. 2010;11:31–46. doi: 10.1038/nrg2626. [DOI] [PubMed] [Google Scholar]
- 5.Goddard JM, Erickson D. Anal Bioanal Chem. 2009;394:469–479. doi: 10.1007/s00216-009-2731-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fuller CW, Middendorf LR, Benner SA, Church GM, Harris T, et al. Nat Biotechnol. 2009;27:1013–1023. doi: 10.1038/nbt.1585. [DOI] [PubMed] [Google Scholar]
- 7.Harris TD, Buzby PR, Babcock H, Beer E, Bowers J, et al. Science. 2008;320:106–109. doi: 10.1126/science.1150427. [DOI] [PubMed] [Google Scholar]
- 8.Neuman K, Lionnet T, Allemand J. Annu Rev Mater Res. 2007;37:33–67. [Google Scholar]
- 9.Nordhoff E, Schurenberg M, Thiele G, Lubbert C, Kloeppel KD, et al. Int J Mass Spectrom. 2003;226:163–180. [Google Scholar]
- 10.Shaltout RM, Loy DA, Wheeler DR. Mater. Res. Soc. Symp. Proc. Vol. 576. San Francisco, CA: 1999. Maleimide functionalized siloxane resins; pp. 15–20. [Google Scholar]
- 11.Krebs CJ, Khan SM, Mollard B, Robins DM. Anal Biochem. 2006;350:313–315. doi: 10.1016/j.ab.2005.10.049. [DOI] [PubMed] [Google Scholar]
- 12.Nordhoff E, Luebbert C, Thiele G, Heiser V, Lehrach H. Nucleic Acids Res. 2000;28:e86. doi: 10.1093/nar/28.20.e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang K, Taranenko NI, Allman SL, Chang LY, Chen CH. Rapid Commun Mass Spectrom. 1994;8:727–730. doi: 10.1002/rcm.1290080913. [DOI] [PubMed] [Google Scholar]
- 14.Taranenko NI, Hurt R, Zhou JZ, Isola NR, Huang H, et al. J Microbiol Methods. 2002;48:101–106. doi: 10.1016/s0167-7012(01)00314-1. [DOI] [PubMed] [Google Scholar]
- 15.Levicky R, Herne TM, Tarlov MJ, Satija SK. J Am Chem Soc. 1998;120:9787–9792. [Google Scholar]
- 16.Steel AB, Levicky RL, Herne TM, Tarlov MJ. Biophys J. 2000;79:975–981. doi: 10.1016/S0006-3495(00)76351-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang YY, Prokein T, Hinz M, Seliger H, Goedel WA. Anal Biochem. 2005;344:216–223. doi: 10.1016/j.ab.2005.05.041. [DOI] [PubMed] [Google Scholar]
- 18.Vaidya AA, Norton ML. Langmuir. 2004;20:11100–11107. doi: 10.1021/la048509l. [DOI] [PubMed] [Google Scholar]
- 19.Dinsmore MJ, Lee JS. J Inorg Biochem. 2008;102:1599–1606. doi: 10.1016/j.jinorgbio.2008.02.007. [DOI] [PubMed] [Google Scholar]
- 20.Lee CY, Canavan HE, Gamble LJ, Castner DG. Langmuir. 2005;21:5134–5141. doi: 10.1021/la0472302. [DOI] [PubMed] [Google Scholar]
- 21.Alves CA, Smith EL, Porter MD. J Am Chem Soc. 1992;114:1222–1227. [Google Scholar]