

Abstract
The insulin/insulin growth factor (IGF) pathway is a critical mediator of longevity and aging. Efforts to extend longevity by altering the insulin/IGF pathway may have varying effects on other physiological processes. Reduced insulin/IGF levels may decrease the incidence of certain cancers as well as the risk of developing metastatic disease. However, it may also increase the risk of developing cardiovascular disease as well as cardiovascular related mortality. Pursuing the right insulin/IGF pathway targets will require striking a balance between inhibiting cancer cell development and progression and avoiding damage to tissues under normal insulin/IGF mediated control. This review will discuss the roles of the insulin/IGF pathway in aging and longevity and the development of cancer cell metastasis and considerations in taking insulin/IGF directed targets to the oncology clinic.
Keywords: Insulin Growth Factor, Cancer, Aging
Introduction
There is increasing evidence demonstrating that modulation of the insulin/Insulin-like growth factor (IGF) axis can alter aging phenotypes and longevity in animal models. Attempts to alter activation and regulation of this pathway for therapeutic benefit in patients with cancers are currently ongoing. While this is an extremely exciting area of research, the fact that the insulin/IGF pathway is responsible for regulating normal growth and development and does not act only at the local level raises concerns that altering insulin/IGF signaling may have unintended consequences on various diseases of aging. This underscores the importance of local modulation of events where a process may be beneficial in one organ or local microenvironment and detrimental in another.
The two most common diseases of aging are cardiovascular disease (CVD) and cancer. CVD is the leading cause of morbidity and mortality in the United States [1]. Cancer is the second leading cause of death in the United States with the majority of people dying from metastatic disease [1]. Metastatic dissemination requires tumor cells to gain access to the lymphatic and blood vascular systems and subsequent migration into and growth in secondary sites. The insulin/IGF pathway is known to play an important role in the development and prognosis of CVD. It is also an important mediator of cancer development and metastasis with roughly 25% of human malignancies having abnormal activation of this pathway [2].
Given this, a more complete understanding of the consequences of altering insulin/IGF signal transduction pathways is important to understanding not only the biology of aging as it relates to longevity but also to the development and progression of diseases like cancer and CVD. This review will focus on the role of the insulin/IGF axis in aging and how the changes in insulin/IGF concentrations and signaling that occur during ageing may be permissive for development and spread of cancer, and the development and progression of CVD.
Insulin and Type I Insulin Growth Factor Receptor Pathway
The insulin receptor (IR) and the type I IGF-I receptor (IGF-IR) are highly conserved structures that evolved from a single ancestral receptor (Figure 1). Structurally, they consist of two half-receptors which each has an extracellular component and a transmembrane subunit that possesses tyrosine kinase activity [3, 4]. The divergence of these receptors from one another is hypothesized to have resulted from more complex animals needing to independently regulate glucose uptake from cell survival and proliferation [4].
FIGURE 1. IGF-I, IGF-II, and Insulin receptors and their role.

The insulin-like growth factors (IGF-I and IGF-II) are ligands that bind to the IGF-I Receptor (IGF-R) and the IGF-II Receptor (IGF-2R), and also to the Insulin Receptor (IR). IGF-1R is a transmembrane tyrosine kinase consisting of two α-β subunits and has high affinity for both IGF-I and IGF-II. The α and β chains are covalently linked into dimers, originating a functional α2β2 heterotetrameric complex. IGF-2R is a single protein of ~300kDa, apparently with no intrinsic catalytic activity. It has a multifunctional protein binding for both IGF-II and mannose 6-phosphate ligands.
The mitogenic factors IGFs bind with high affinity both their receptors and to a family of secreted proteins termed IGF Binding Proteins (six of them are known, IGFBP-1 through -6), which are present in the circulation and in the extracellular space. IGFBPs regulate the biological effects of IGF ligands by transporting IGFs to their site of action, protecting them from degradation, or by sequestering IGFs from their receptors. These processes are further modulated by IGFBP proteases which, through high affinity cleavage of IGFBPs into fragments with lower affinity for IGFs, are able to increase free IGF availability.
Activation of IGF-1R results in phosphorylation of akt and adaptor proteins of the irs family or shc, which through their downstream signaling pathways can regulate apoptosis and lead to enhanced growth, survival, migration and metastasis in cancer cells. IGF2R accumulates vesicles that are internalized and regulates proliferation, tumor suppression, and placental development.
The Insulin Receptor (IR) mediates the effects of insulin, and its main physiological role appears to be metabolic regulation. There are two insulin receptor isoforms arising from alternative splicing of exon 11, which therefore either contain (IR-B isoform) or lack (IR-A) the exon 11. The isoforms are co-expressed in different tissues and many cancers, and have different affinities for insulin, IGF-II and IGF-I, with IR-A (overexpressed in several cancer cell types) binding both insulin and IGF-II with high affinities. In addition, there are also hybrid receptors, assembled with one α-β chain of IGF-1R and one α-β chain of IR-A or IR-B. IGF-II ligand can bind to IGF-2R, IGF-1R, IR-A and to both hybrid receptors.
In complex mammals, for example, insulin is primarily involved in regulating metabolic processes. It is secreted by the pancreas predominantly after food intake, binds to the IR and at physiological concentrations has low affinity for IGF-IR [5]. IGF-I influences survival, proliferation, apoptosis, and cell migration through autocrine, paracrine, and endocrine mechanisms (Figure 2). It is predominantly made by the liver but can also be produced by tumors [4]. IGF-I and IGF-II are ligands for the IGF-IR and have a relatively low affinity for the insulin receptor [6].
FIGURE 2. IGF-1 MEDIATED EFFECTS ON CANCER MIGRATION AND INVASION.
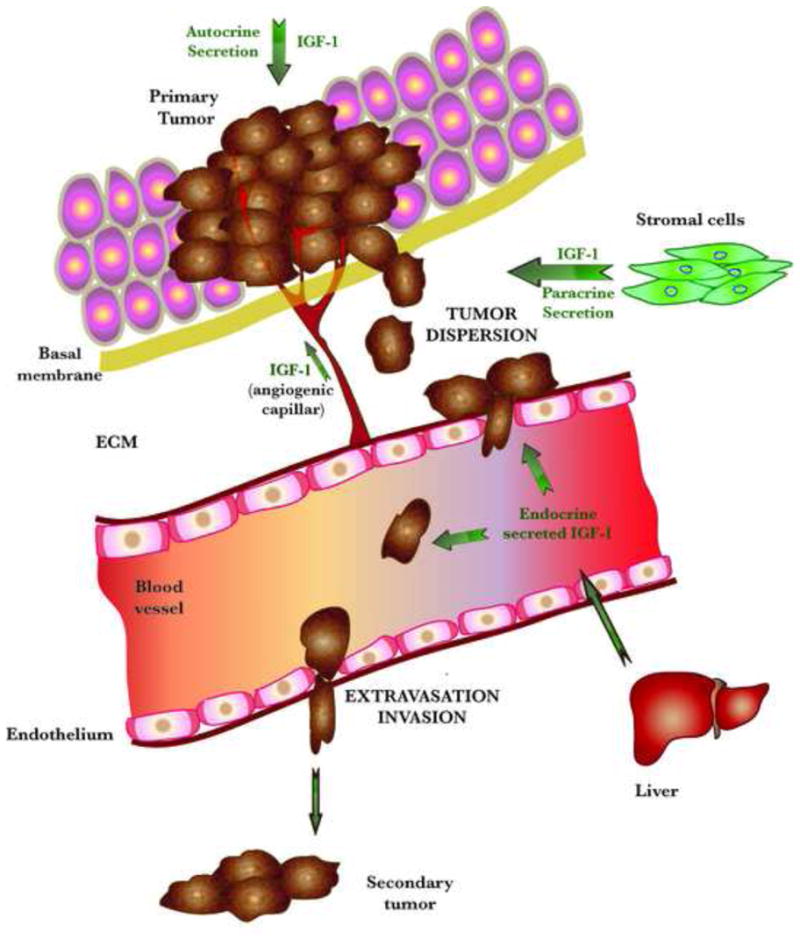
IGF-1R signaling is implicated in the major steps of the metastatic process, i.e., cell adhesion, migration, invasion, angiogenesis and metastatic growth in secondary sites.
Circulating IGF-1 is produced mainly in the liver (whereas insulin is produced in the pancreas). While insulin reaches the tumor cells endocrinously, IGF-1 is released in autocrine and paracrine manners, by the tumor cells and the mesenchymal cells in the close vicinity or within the tumor, respectively. IGF-1 present in the tumor microenvironment can be affected by several factors. The numerous blood vessels in the tumor provide an abundant source of IGF-1, and its cooperative signaling with integrins αvβ3 and αvβ5 is important in the regulation of neovascularization. Angiogenesis is also stimulated by IGF-induced expression of uPA (urokinase Plasminogen Activator). Excess IGF-1 can sometimes be synthesized by the tumor itself and can act directly on tumor cells that possess IGF receptors to stimulate their growth. Cancer cell replication is promoted also by the synergy of IGF-1 with hypoxia inducible factor 1 (H1F1). Cancer migration and metastasis can be stimulated as well. IGF-1 up-regulates the activity of matrix metalloproteinases (MMPs), required for the breakdown of the extracellular matrix (ECM), and alters metastasis by inducing ECM to form a site for attachment of the metastatic cells.
The increased IGF-IR levels in tumor cells might also promote extravasation, thus inducing invasion in distal organs. Metastatic cells will proliferate in sites in which growth factors can be supplied by the local microenvironment.
The signaling pathways downstream of the IR and the IGF-IR share many similarities. This has made it difficult to segregate the different bioactivities of insulin and IGFs in their different contexts. It is clear, however, that IGF-IR ligand binding leads to activation of cytosolic tyrosine kinases. This in turn leads to recruitment of adaptor proteins such as insulin receptor substrate-1 (IRS-1) and IRS-2 with subsequent activation of signaling via the phosphatidylinositol-3-kinase (PI3K)-AKT-FOXO and RAS/RAF/mitogen-activated protein kinase (MAPK) pathways [7].
Insulin/IGF-I Pathway and Aging in Worms and Flies
Our current understanding about the relationship between aging and the insulin/IGF-I pathway comes from studies on simple invertebrate models like the worm Caenorhabditis elegans and the fruit-fly Drosophila melanogaster. These models are useful scientific models because they are easy to manipulate genetically and have a single conserved insulin/IGF-I receptor. The insulin/IGF-I pathway was initially linked to life span in C. elegans when mutations in an insulin/IGF-I receptor ortholog, daf-2, doubled the its life span [8, 9]. Subsequent work showed that daf-2 mediated extensions in life span required the activity of a FOXO family transcription factor, daf-16 [10, 11]. In addition to increasing longevity, these insulin/IGF-I mutants also appeared to enjoy a longer “health span” including increased organism motility, for example, compared with controls [8].
In addition to C. elegans, mutations in the insulin/IGF-I receptor have also been shown to increase life span in Drosophila. One study, for instance, showed an 80% increase in life span in insulin/IGF-I receptor mutants compared with wild-type flies [12]. A 40% increase in lifespan was also seen in Drosophila carrying mutations in a downstream IRS-like signaling protein, chico [13]. Finally, similar to worms, FOXO appears critical to mediating life span extension in flies with studies showing FOXO overexpression can extend fly life span [14, 15]. The results of these invertebrate model studies were critical in proving that aging and longevity are controlled processes.
Insulin/IGF-I Pathway and Aging in Mice
The results presented above led multiple investigators to determine if insulin/IGF-I mediated regulation of aging and longevity also occurs in mammals. However, mammals have separate insulin and IGF-I receptors and this added genetic complexity makes the investigation of the specific role of the IGF-IR versus the IR harder to examine. Various groups have performed studies on mice specifically with altered IGF-IR signaling, opposed to either the IR or IR and IGF-IR changes. Holzenberger et al, for example, showed that heterozygous IGF-IR knockout mice live on average 26% longer than their wild-type littermates, with this life span extension being limited to female mice [16]. In addition, these knockouts were also more resistant to oxidative stress compared with wild-types. While these results are encouraging they have also been criticized because of the shortened survival in the control group [17]. Another example of alterations in IGF-I signaling leading to longevity changes comes from a group that established a cohort of mice that harbor a hypomorphic IGF-I mutation (Midi mice). These mice had an 18% increase in life span relative to controls [18]. Finally, a more recent example by Kappeler et al demonstrated that mice with heterozygous brain-specific IGF-I receptor knockout mutations had significantly longer life spans compared with controls (914 versus 836 days, p<0.05) but unchanged maximum life spans [19].
Other groups have investigated the affects of alterations in insulin signaling via disruption of the insulin receptor in adipose tissue [20]. These Fat Insulin Receptor Knockout (FIRKO) mice have been shown to outlive normal wild-type animals by approximately 18%.
Other evidence for insulin/IGF-I involvement in aging comes from studies in animals with mutations upstream of insulin and IGF-I. These include growth hormone (GH) deficient, GH-resistant, and hypopituitary mouse models [21–25]. GH stimulates IGF-I production in the liver and as would be expected, GH receptor mutants have decreased IGF-I levels. Studies using these models have demonstrated life span extensions of approximately 50%. This dichotomous finding raises the probability that life span extension is not solely related to IGF-I signaling but is also dependent on other mechanisms.
Groups also have studied the impact of aging and longevity by altering IGF-I signaling downstream of its receptor. Mice that overexpress Klotho, a hormone that inhibits intracellular insulin and IGF-I signaling, have significantly prolonged lives compared with wild-type mice [26]. In another study, Selman et al found that female mice with IRS-1 mutations live longer than wild-type mice and exhibit features of delayed aging like decreased age-related motor coordination and immune function changes [27]. They also reported that IRS-2 deleted mice were short-lived whereas IRS-1 +/− and IRS-2 +/− mice had normal life spans.
These findings were in contrast to those reported by Taguchi et al who showed IRS-2 +/− mice had about a 20% increase in life span, independent of sex, compared with wild-type mice [28]. The discordant results between these two studies may be related to varying statistical analysis of longevity data and different backcrosses to the C57Bl/6 strain. Also, the different fat content of the diets (5% versus 9%) may suggest that heterozygous IRS-2 deletions are simply more protective against the potentially life-shortening effects of a high fat diet.
Alterations in other components of the IGF-I signaling pathway, including IGF-I binding proteins can also influence aging and longevity. Pregnancy-associated plasma protein-A (PAPP-A) is a metalloproteinase that degrades inhibitory IGF-I binding proteins leading to increased IGF-I bioavailability [29]. Experiments with PAPP-A knock-out mice demonstrate a 30% increase in longevity in female and male mice. Importantly, they also found a reduced incidence of spontaneous tumors compared with wild-type mice [30]. In fact, 12/17 animals had extensive tumors (primarily liver, lung, kidney and colon) and an enlarged spleen or lymph nodes compared with small solitary liver lesions in 3/20 knockout mice. The authors hypothesize that their finding of increased life span may be related to decreased local tissue availability of IGF-I. PAPP-A mediated control of local IGF-I levels has been shown in skeletal muscle model but whether this mechanism is occurring in this tumor model has not yet been investigated [31].
Thus, inhibition of downstream targets of IGF-I signaling alters aging and longevity in multiple models. It also appears that selectively inhibiting IRS-2 in specific tissues can mediate similar results. Taguchi et al reported that IRS-2 homozygous or heterozygous deletions selective for the brain increased life span by 14% and 18%, respectively, compared with controls. These mice also had features of delayed aging [28]. This is in contrast to the findings for global knockouts described above.
In summary, these studies show that modifying insulin/IGF-I signaling either upstream or downstream of its receptor can alter aging and longevity in invertebrates and mammals. They also show that selectively inhibiting IGF-I signaling in specific tissues can lead to similar results as inhibiting IGF-I indiscriminately throughout the body. What remains to be determined is whether the mechanisms elucidated in these preclinical models are recapitulated in humans and to dissect the mechanisms underlying the phenotypic response to the locally active genetic event in the brain compared to the whole organism knockdowns.
IGF-I and Aging in Humans
Rigorous studies to evaluate the relevance of insulin/IGF in human aging and longevity have been limited by challenges in accruing appropriate patient populations and controls. However, some success has been achieved by Suh et al who conducted a case controlled study comparing the levels of IGF-I in the offspring of centenarians to age and sex matched controls without a family history of longevity [32, 33]. They found that female offspring of centenarians had 35% higher serum IGF-I levels compared with female controls, while there were no differences between male cohorts. To investigate whether elevated IGF-I levels were caused by increased production or as a result of IGF-I insensitivity, the authors looked to see if there were height differences between the groups. Female offspring were significantly shorter compared to controls whereas the heights of males were similar. Based on this, it appears that elevated IGF-I levels in females is a compensatory response to reduced IGF-IR signaling. Sequence analysis of IGF-I and IGF-IR genes showed significant overrepresentation of heterozygous mutations in the IGF-IR gene amongst centenarians compared with controls. These mutations were associated with high serum IGF-I levels and reduced IGF-IR activity as measured in transformed lymphocytes. While a small sample size, this study shows that alterations in IGF-I pathway signaling can be correlated with human longevity in a similar manner to that seen in preclinical models.
Other insights regarding the relationship between insulin/IGF and human longevity comes from studying patients with Laron syndrome. This is a condition that is caused by deletions or mutations in the GH receptor or post-receptor pathways and results in low IGF-I levels [33]. While none of the studies on these patients are large enough to draw definitive conclusions they do provide important observations regarding the consequences of altered IGF-I levels on longevity and diseases of aging.
Longevity data, for instance, have been reported from a cohort of 65 Israeli patients as well as a cohort of about 135 Ecuadorian patients with Laron syndrome. It appears that these patients do reach old age with the oldest reported patient being 78 years old [34]. This is despite the fact that these patients tend to be markedly obese, hyperlipidemic, and develop Type 2 diabetes [34]. It is also interesting to note that these patients tend not to develop cancer at the same frequency as people with normal levels of IGF-I [35]. So, while there are not extensive amounts of data there is a growing body of literature suggesting that the insulin/IGF pathway is involved in longevity in humans.
Given this, groups have started to investigate ways to modify insulin/IGF-I signaling in the hopes of modifying human longevity. One of the more interesting approaches to modify IGF-I levels in humans has been calorie restriction. Calorie restriction is the most consistent preclinical method known to increase longevity. While the biochemical mechanisms responsible for this phenomenon are unknown, it is thought to be, at least partially, related to reduced IGF-I signaling. Preclinical models have demonstrated calorie restriction leads to an approximately 40% decrease in serum IGF-I [36].
Fontana et al have conducted a number of calorie restriction studies on patients and have found that, unlike in rodents, 1 year or 6 years of calorie restriction in humans does not reduce total or free IGF-I levels if protein intake is high [37]. However, patients on a protein restricted diet do have significantly lower IGF-I serum levels compared with patients on higher protein diets. They also experience a cardioprotective benefit but no data have been reported yet on life span or the incidence of disease including cancer. Based on their clinical studies, it appears that changing protein intake may be more powerful than calorie intake in altering serum IGF-I levels.
This study raises multiple issues regarding the challenges that we will face in attempting to translate our findings from mice to humans. The first is the length of calorie restriction necessary to see the affects of increased longevity. Mice have a life span of about 3 years and 1 to 2 years of calorie restriction constitutes the majority of their life span. A similar study in human would have to be started during early middle age and continued indefinitely, not a study that can successfully be executed in the current day and age.
Challenges to translating findings from mice to human, other than the length of calorie restriction, will include the fact that mammalian mutants used in longevity studies have genetic alterations that can affect development at very early points in utero, continuing through to and beyond puberty. If the major role of increasing longevity is early in life then it may not actually be feasible to translate these findings into a human intervention. Given this, efforts to alter insulin/IGF action in post-puberty experimental animals should be actively pursued.
Other issues concern the fact that targets other than IGF-I may be critical mediators of the increased longevity seen with dietary restriction. Multiple groups are investigating nutrient sensors of dietary restriction that may mediate the life-extending effects of calorie restriction. Examples of targets that have been directly implicated with aging include: sirtuins, the mammalian target of rapamycin (mTOR), and the hexosamine biosynthesis pathway [38]. Further, it is interesting that caloric restriction, reduced glucose availability, will upregulate pathways involved in angiogenesis, invasion, and alterations of the tissue that create a locally permissive microenvironment for tumor progression and dissemination. Lastly, aside from calorie restriction, another promising method of modulating IGF levels includes alternate day fasting. This approach has been shown to increase longevity and reduce the development of cancer in multiple animal models [39]. It remains to be seen, however, if this approach can or will be effective in humans.
IGF and Cancer
Until now, we have only considered the effects of altering insulin/IGF signaling on aging and longevity. However, insulin/IGF are important in carcinogenesis and cancer dissemination [40]. Understanding how alterations in IGF signaling impact metastasis is relevant, especially given that the majority of the morbidity and mortality of cancer is associated with metastatic disease. This is particularly important if future human longevity studies are designed to alter insulin/IGF signaling in patients during late adulthood, a time in life where the incidence of cancer increases. While some of the risk of altering the natural history of carcinogenesis and cancer cell metastasis by modulating insulin/IGF signaling is theoretical, there is also some evidence to suggest that it is not. One study of patients with prostate cancer suggests that IGF-I concentrations may be related less to the process of carcinogenesis and more to the probability of whether early lesions progress or not [41]. Therefore, understanding the relationship between IGF modulation and cancer metastasis is important to consider.
We will focus on the involvement of the IGF pathway in the first step that leads to successful metastasis--invasion [42]. A significant portion of a cell s ability to acquire an invasive phenotype is related to its ability to migrate through the basement membrane and subsequently into the bloodstream. The process of migration starts when a cell is stimulated by a motility- or scatter-promoting factor that then orients and polarizes the cell along an axis of movement. After establishing directionality, extracellular matrix changes and cell reconfiguration enables forward movement through cellular protrusions, pseudopods or lamellopodia, and formation of adhesive contacts at the cell s leading edge. Simultaneously, detachment at the back of the cell occurs via actin disassembly [43]. This is an energy requiring process and the insulin/IGF system interfaces with it at multiple points. While this general process is similar for all cells, the specific details vary greatly between different cell types and environmental conditions.
Overall, this coordinated sequence of events requires extensive temporal and spatial cooperation and is regulated by many pathways including IGF-I (Figure 2). IGF-I is involved in the downstream activation of multiple pathways, including phosphatidylinositol 3 -kinase (PI3K), which is important in mediating, amongst other things, cell-cell adhesions, crosstalk with integrins, and cytoskeleton reorganization.
IGF-I and Mechanisms of Cancer Cell Migration
IGF-I and Intercellular Adhesion Molecules
Normally cells are tightly held together by adhesion molecules and in order to migrate they have to disrupt these cell-cell connections. Cells are attach through a superfamily of transmembrane glycoproteins collectively known as cell adhesion molecules. Cadherins are a subfamily of this group that exhibit calcium-dependent binding. They are single transmembrane-spanning proteins the cytoplasmic domain of which has a binding site for cytosolic proteins including beta-catenin, which in turn binds to alpha-catenin, which interacts with the cytoskeleton. E-cadherin is a prototypic member of this subfamily and is expressed in most epithelial tissues [44]. Reduced levels of E-cadherin have been demonstrated in many cancers including head and neck, esophagus, skin, thyroid, lung, breast, stomach, liver, kidney, pancreas, colon, prostate, and the female genital tract, and have been associated with tumor progression through increased cellular spreading and migration [45, 46]. Control of these E-cadherin-adhesion complexes is regulated by multiple signaling pathways, including IGF [47].
The stimulatory effects of IGF-I on modulating cell-cell adhesions have been observed in mouse fibroblasts, glioblastoma, colorectal, and breast cancer cells [48–51]. Some of the most extensive work has been done using breast cancer cell models. In vitro studies show that breast cancer cells that are E-cadherin negative or treated with an antiE-cadherin antibody have diminished levels of IGF-IR mediated cell-cell adhesion [48, 52]. However, when cells co-express IGF-IR and E-cadherin, IGF-IR dependent adhesion is restored [48, 52]. Another study by Pennisi et al showed that MCF-7 cells treated with antisense IGF-IR treatment resulted in reduced aggregation and increased motility. These effects were shown to at least be partially mediated through decreased E-cadherin levels [53]. Lastly, changes in E-cadherin appear to be at least one pathway involved in escaping IGF-IR inhibition. In a transgenic breast cancer mouse model, where the IGF-IR is under the control of a doxycycline-inducible promoter, tumors that grew independent of IGF-IR activation escaped through upregulation of transcriptional repressors of E-cadherin [54].
While IGF-IR is involved in regulating cell-cell adhesion proteins, the consequences of IGF-IR activation depend on the cellular context. For example, breast cancer cells grown in monolayer are stimulated by IGF-I to migrate but not when cultured as three-dimensional spheroids [48, 52]. Similar results have been demonstrated in colorectal cancer cells [50, 55].
IGF and Integrins
Beyond changes in E-cadherin and other cell-cell adhesion molecules, in order to move forward a cell must also be able to “grab on” to the extracellular matrix (ECM). This process is mediated by integrins which help mediate cell adhesion to the ECM. Integrins are made up of heterodimeric transmembrane proteins that bind to a wide range of ECM molecules including fibronectin, fibrinogen, von Willebrand factor, vitronectin, and proteolysed forms of collagen and laminin [56]. Following stimulation they activate focal adhesion kinase (FAK) or c-Src, both cytosolic tyrosine kinases, ultimately leading to upregulation of pathways involved in cytoskeletal reorganization and migration [56].
Multiple studies have shown a role for IGF-IR in regulating integrin activation. The most well studied example is crosstalk between IGF-IR and α5β3 integrin, the vitronectin receptor [57, 58]. Ligand occupancy of this receptor can stimulate motility of a variety of cells and its overexpression has been implicated in the metastatic phenotype of tumor cells [59].
Our understanding of the mechanisms behind the interactions between IGF-IR and integrins are predominantly based on work done in smooth muscle cells. When these cells are inhibited with a monoclonal antibody to α5β3 integrin, the ability of IGF-I to stimulate smooth muscle cell migration is markedly reduced [60]. Conversely, stimulation of the integrin pathway using vitronectin or osteopontin enhances the ability of IGF-I to promote smooth muscle cell migration [61]. Mechanistically, it appears that in response to IGF-I, the src homology 2 domain containing protein tyrosine phosphatase substrate-1 (SHPS-1) is phosphorylated which in turn leads to recruitment of SHP-2, a tyrosine phosphatase. Recruitment of SHP-2 to the cell membrane appears necessary for full activation of IGF-I downstream signaling events including the PI3K pathway [62].
IGF binding proteins (IGFBPs) can also modulate integrin signaling and cellular migration in a variety of cells. Their effects, similar to cell-cell adhesion, vary based upon the cellular context. For example, IGFBP-1 reduces motility in a breast cancer cell model while in human trophoblast cells IGFBP-1 can promote migration independently of IGF-I [58, 63]. The interaction of the IGF-I pathway and IGF-I independent IGFBP-1 signaling is an important new area of investigation.
To move forward after disruption of cell-cell adhesions and “grabbing on” to the ECM, cells must generate forward protrusion of a leading edge(s) through spatially localized actin polymerization and crosslinking [64]. Ultimately, a dominant pseudopod or lamellopod is selected and defines the direction of the ultimate cellular motility. This process of cytoskeleton reorganization is also regulated through IGF-IR signaling and IGF-IR cross talk with integrins and other motility-associated receptors.
IGF and Cytoskeletal Changes
Multiple reports have demonstrated IGF-IR activation alters actin cytoskeleton organization. For example, KB epidermoid carcinoma cells exposed to IGF-I undergo rapid actin cytoskeletal changes [65]. In another study, Guvakova et al showed that overexpressing IGF-IR in MCF-7 cells could disrupt the polarized cell monolayer via rapid disassembly of actin fibers and accumulation of short actin projections [66]. The mechanisms involved in IGF mediated actin cytoskeletal reorganization require activation of downstream signaling pathways including PI3K. Proper activation of these pathways involves focal adhesion-associated signaling proteins. IGF-I induces phosphorylation of these proteins, including focal adhesion kinase (FAK), paxillin, and p130Cas. IGF-I promotes localization of focal adhesion proteins to distinct sites of the plasma membrane that are in close contact with the ECM. Furthermore, rapamycin inhibition of mammalian target of rapamycin (mTOR) in human rhabdomyosarcoma, Ewing sarcoma, glioblastoma, and prostate cancer, prevented IGF-I stimulated F-actin reorganization. This outcome was also correlated with decreased phosphorylation of FAK, paxillin, and p130Cas [67]. However, similar to that seen with cadherins and integrins, the actions of IGF-I on tyrosine phosphorylation also are context dependent [66, 68, 69].
Clinical Evidence for a Relationship between Insulin/IGF and Carcinogenesis and Metastasis
IGF mediated mechanisms involved in cell migration include interactions with cadherins, integrins, and cytoskeletal changes. Whether it is necessary to inhibit a select target or a combination of targets related to these pathways to delay or halt metastatic disease is not known at this time. However, increased IGF-I concentrations promote both carcinogenesis and the development of invasive and metastatic disease. Multiple prospective studies have demonstrated a relationship between increased concentrations of circulating IGF-I with a higher risk of developing prostate, breast, and colorectal cancers [41, 70, 71]. Other studies have illustrated associations between increased expression of IGF-IR, IGF-I, and IGF-II and metastasis [72]. Examples include overexpression of IGF-II as a reliable predictor of colorectal carcinoma metastasis to the liver [73], a significant correlation between higher IGF-IR levels and the incidence of liver metastasis in patients with uveal melanoma [74], and significantly higher chances of developing metastatic disease in synovial sarcoma patients with elevated levels of IGF-IR expression [75].
Multiple studies have also demonstrated an increased risk of cancer in patients with higher C-peptide/insulin levels. This includes an increased risk of colorectal cancer [76], pancreatic cancer [76, 77], post-menopausal breast cancer [78], and endometrial cancer [79]. The “metabolic” phenotype of type II diabetes with attendent insulin resistance and high insulin concentrations, obesity, and obesity-related pathologies are a risk factor for endometrioid endometrial cancer. Lastly, while studies on patients with diabetes are confounded by many variables these patients are known to have higher risks of developing cancer (including pancreas, liver, breast, colorectal, urinary tract and female reproductive organs) [80]. There is an increasing body of literature supporting a role for insulin/IGF in the development and progression of cancer. While we may not know the ideal targets to pursue within the IGF pathway, there is a growing body of clinical studies and novel agents in cancer patients designed to target the insulin/IGF pathway.
Strategies to Interfere with Insulin/IGF Signaling in Cancer
Some of the earliest efforts to target the insulin/IGF pathway include investigations with octreotide. Preclinical studies using this approach were encouraging but their findings did not translate into a clinical benefit [4, 81]. It is thought that prolonged exposure to octreotide leads to patients developing tolerance to its GH and IGF-I suppressing effects. IGF-IR targeting has been more successful, and its effectiveness in cancer treatment is supported by both preclinical evidence as well as early clinical trial results.
One strategy that has recently entered clinical trials has been using neutralizing antibodies against the IGF-I receptor. The broadest experience to date has been with CP-751,871 (figitumomab). Evidence suggests that this drug in combination with paclitaxel and carboplatin in squamous cell nonsmall cell lung cancer (NSCLC) may improve treatment response; a 54% objective response rate was observed in a Phase II study [82]. These data have lead to the initiation and accrual of patients to a series of phase III studies collectively called the ADVancing IGF-IR in Oncology (ADVIGO) studies. These include ADVIGO 1016 and ADVIGO 1018. ADVIGO 1016 is testing figitumomab in combination with carboplatin and paxlitaxel in NSCLC patients, while ADVIGO 1018 is investigating figitumomab in combination with eroltinib in patients with advanced Stage IIIB or metastatic NSCLC.
Other agents targeting the IGF-R extracellular domain have been investigated in Phase I studies. These include AVE-1642, AMG-479 (one complete response in a Ewing s sarcoma patient), IMC-A12 and MK-0646 (both resulting in two patients with stable disease for more than 9 and 12 months, respectively) [83–86]. A common adverse effect noted in studies of figitumomab and these newer agents is hyperglycemia, often accompanied by insulin resistance.
Another strategy to block the IGF-IR is through the inhibition of its tyrosine kinase activity. The high sequence homology of the kinase domains contained in all the IGF system receptors might lead to the risk of indiscriminate inhibition. Yet, there are several agents under clinical and preclinical investigation with higher affinity and thus selectivity for the IGF-IR kinase, such as NVP-AEW541, NVP-ADW743, and picropodophyllin (PPP) [87, 88]. The only non-monoclonal antibody therapy under clinical development is based on INSM18 [89], a small molecule inhibitor of IGF-IR that also targets the HER2 receptor tyrosine kinase.
Another possible strategy is to target IGF-I ligand bioavailability by either using ligand-neutralizing monoclonal antibodies or recombinant insulin growth factor binding proteins (IGFBPs), such as rhIGFBP3. Agents of this type are entering clinical investigations. There are multiple strategies being tested in the clinic to modulate insulin/IGF signaling. Which targets and agents will succeed and move forward for cancer therapy remains to be seen.
It also appears that while reducing IGF levels may be beneficial both in terms of longevity as well as reducing the incidence of cancer and the development of metastatic disease that prolonged exposure to low IGF levels may have unintended consequences. This includes the possibility of negatively affecting the incidence and natural history of CVD.
IGF and Cardiovascular Disease
CVD is the leading cause of morbidty and mortality in the United States and is one of the most prevalent diseases of older individuals, the same subgroup known to have increased. Multiple observational studies have demonstrated a correlation between circulating IGF-I concentrations and the development of CVD [90, 91]. One of the largest of these was a nested case-control study performed within a larger prospective study on cardiovascular epidemiology [90]. Serum IGF-I and IGFBP-3 concentrations were measured for all patients. Patients in the low quartile of IGF-I at baseline were found over a 15 year follow-up period to have had a significantly higher risk of ischemic heart disease (relative risk 1.94, 95% CI: 1.03 to 3.66) compared with the high IGF-I quartile group. Patients in the high IGFBP-3 quartile had an adjusted relative risk of 2.16 (95% CI, 1.18 to 3.95) of having ischemic heart disease while patients with low IGF-I and high IBGBP-3 levels had an even higher risk of developing ischemic heart disease. The increased risk in ischemic heart disease is on par with that seen with other well known cardiovascular risk factors, such as cholesterol and blood pressure. While this study does not establish causality, it provides the basis for a strong association between the IGF pathway constituents and the development of ischemic heart disease.
Stronger evidence for a causal role of IGF-I in the development of CVD comes from patients with and without a defined polymorphism in the promoter region of the IGF-I gene [92]. Patients who did not have this wild-type 192 base pair allele had 18% lower serum IGF-I concentrations. Noncarriers also had a 1.7 increased relative risk of type 2 diabetes and myocardial infarction compared with carriers. Studies like this lend evidence to the hypothesis that low concentrations of IGF-I are adversely involved in the pathogenesis of diabetes and CVD.
Beyond low IGF-I concentrations being implicated in the development of CVD they have also been associated with poorer outcomes. One study prospectively examined the association of serum IGF-I concentrations with cardiovascular disease mortality in over 1,000 men and women between 51 to 98 years of age (mean, 74 years old) [91]. Serum IGF-I concentrations were measured at study entry and patients were followed for 9 to 13 years. They found that the relative risk of ischemic heart disease mortality was 38% higher for every 40 ng/ml (1 standard deviation) decrease in IGF-I concentration using a proportional hazards model and adjusting for CVD risk factors. The conclusions of the authors were that, independent of prevalent ischemic heart disease and cardiovascular risk factors, low baseline concentrations of IGF-I increase the risk of fatal ischemic heart disease among elderly men and women. Based on the current evidence it appears that low IGF-I levels are an unfavorable factor in regards to the development and prognosis of CVD.
Conclusions
Will Targeting the Insulin Growth Factor Help us or Hurt us?
It appears that while lower levels of IGF may be beneficial in terms of extending life span, reducing carcinogenesis, and the development of cancer metastasis it may also be detrimental in terms of both developing CVD and increasing cardiovascular related mortality. A review of data from 16 different prospective case-control studies that evaluated the relationship between baseline concentrations of IGF-I and the risk of subsequently developing disease found that patients in the upper quartile compared with the lowest quartile had an increased risk of developing cancer (prostate, breast, lung and colorectal); whereas, patients with IGF-I concentations in the lowest quartile had an increased risk of developing ischemic heart disease and diabetes [93]. Based on this information it seems reasonable to hypothesize that each individual is likely to have an ideal insulin/IGF axis setpoint which balances the competing risks of diseases associated with aging versus longevity [94]. This has led some to believe that because there is a higher relative incidence of cancer in early adult life and an increasing prevalence of CVD with age that reduced levels of IGF-I during early adulthood followed by higher than normal levels during later adulthood may be ideal [95].
From an evolutionary standpoint it is actually counterintuitive to think that the normal physiological decline in serum IGF-I concentration with age is actually not optimal. One possible explanation for this is antagonistic pleiotropy. This concept states that while certain traits may be beneficial during the reproductive years they may be detrimental later on in life. Interestingly, this possibility is in contrast to recent work in C. elegans suggesting that tumor suppression does not always have to come at the expense of longevity. Genes regulated by FOXO/daf-16 can promote longevity and tumor resistance [96]. While not entirely clear at this time it is possible that the genes identified in this study may be essential to regulating tumor growth in cancers with abnormal insulin/IGF-I signaling such as breast and prostate cancers, and glioblastoma [97]. Further work in this area has to be done to determine the clinical relevance of these findings.
A coupling between longevity and tumor resistance genes may also be consistent with dauer. Dauer is a growth-arrested larval state that pre-pubescent C. elegans can enter under harsh environmental conditions. It is partially mediated through down regulation of insulin/IGF-I signaling that in turn leads to upregulation of stress-response genes. It is possible that while this response may have originally developed to allow an organism to protect itself from reproducing in a harsh environment, the same genes that are upregulated to protect against endogenous stress may also protect against aging and therefore have been selected to regulate longevity. In humans, one could hypothesize that low levels of insulin/IGF-I pathway activation could activate stress resistance genes and shift cells from a growth state, which may negatively affect their long-term survival, to a maintenance state, that may positively affect their long-term survival and delay aging related processes, such as cancer [98]. Whether this process or antagonistic pleiotropy better explain the role of insulin/IGF in aging and longevity is unclear at this time.
Ultimately, whether targeting the insulin/IGF factor to promote longevity is going to be beneficial is unclear at this time. The insulin/IGF pathway is an essential component of many normal and abnormal processes throughout the human body. While we have focused on aging, cancer, and CVD this pathway is also important in the central nervous system and the kidneys. As such, figuring out how to balance insulin/IGF levels to promote longevity while not negatively impacting other normal processes will be challenging.
If we try to learn from our clinical experience with radiation therapy, one of the oldest targeted agents used in the clinic, it took us some time to realize that the dose of radiation the patient receives per day can have a profound influence on the development of late term toxicities. This emphasizes the point that while the acute effects of many targeted agents are quickly being realized, the long term effects of them are largely unknown. As there are likely many more patients enrolled on cancer clinical trials aimed at altering insulin/IGF than those on endocrine, GH deficiencies, anti-aging or longevity studies it is imperative for oncologists to consider more than overall and progression free survival as study endpoints. Altering insulin/IGF signaling has the potential to have significant late term toxicities and it is critical that these patients be followed for extended periods of time. This is likely particularly important in children as well as patients kept on maintenance therapy.
In the future, we will hopefully be able to have a balanced discussion with patients regarding the possible tradeoffs of initiating treatment to alter insulin/IGF pathway signaling but for now we simply do not know enough. Ultimately, the insulin/IGF axis is a common pathway between aging and cancer and improving our understanding of it promises to help us better balance the pros and cons of manipulating it on the entire human body versus just a specific disease process.
Acknowledgments
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Available from: http://www.cdc.gov/nchs/deaths.htm.
- 2.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 3.Adams TE, et al. Structure and function of the type 1 insulin-like growth factor receptor. Cell Mol Life Sci. 2000;57(7):1050–93. doi: 10.1007/PL00000744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pollak M. Insulin, insulin-like growth factors and neoplasia. Best Pract Res Clin Endocrinol Metab. 2008;22(4):625–38. doi: 10.1016/j.beem.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 5.Belfiore A. The role of insulin receptor isoforms and hybrid insulin/IGF-I receptors in human cancer. Curr Pharm Des. 2007;13(7):671–86. doi: 10.2174/138161207780249173. [DOI] [PubMed] [Google Scholar]
- 6.Gauguin L, et al. Structural basis for the lower affinity of the insulin-like growth factors for the insulin receptor. J Biol Chem. 2008;283(5):2604–13. doi: 10.1074/jbc.M709220200. [DOI] [PubMed] [Google Scholar]
- 7.Baserga R, Peruzzi F, Reiss K. The IGF-1 receptor in cancer biology. Int J Cancer. 2003;107(6):873–7. doi: 10.1002/ijc.11487. [DOI] [PubMed] [Google Scholar]
- 8.Kenyon C, et al. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366(6454):461–4. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- 9.Kimura KD, et al. daf-2, an insulin receptor-like gene that regulates longevity and diapause in Caenorhabditis elegans. Science. 1997;277(5328):942–6. doi: 10.1126/science.277.5328.942. [DOI] [PubMed] [Google Scholar]
- 10.Lin K, et al. daf-16: An HNF-3/forkhead family member that can function to double the life-span of Caenorhabditis elegans. Science. 1997;278(5341):1319–22. doi: 10.1126/science.278.5341.1319. [DOI] [PubMed] [Google Scholar]
- 11.Paradis S, Ruvkun G. Caenorhabditis elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3 kinase to the DAF-16 transcription factor. Genes Dev. 1998;12(16):2488–98. doi: 10.1101/gad.12.16.2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tatar M, et al. A mutant Drosophila insulin receptor homolog that extends lifespan and impairs neuroendocrine function. Science. 2001;292(5514):107–10. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- 13.Clancy DJ, et al. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292(5514):104–6. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- 14.Hwangbo DS, et al. Drosophila dFOXO controls lifespan and regulates insulin signalling in brain and fat body. Nature. 2004;429(6991):562–6. doi: 10.1038/nature02549. [DOI] [PubMed] [Google Scholar]
- 15.Junger MA, et al. The Drosophila forkhead transcription factor FOXO mediates the reduction in cell number associated with reduced insulin signaling. J Biol. 2003;2(3):20. doi: 10.1186/1475-4924-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holzenberger M, et al. IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature. 2003;421(6919):182–7. doi: 10.1038/nature01298. [DOI] [PubMed] [Google Scholar]
- 17.Liang H, et al. Genetic mouse models of extended lifespan. Exp Gerontol. 2003;38(11–12):1353–64. doi: 10.1016/j.exger.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 18.Sell CLS. Aging in the IGF1 hypomorphic mice. American Aging Association 36th Annual Meeting; San Antonio, TX. 2007. [Google Scholar]
- 19.Kappeler L, et al. Brain IGF-1 receptors control mammalian growth and lifespan through a neuroendocrine mechanism. PLoS Biol. 2008;6(10):e254. doi: 10.1371/journal.pbio.0060254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bluher M, Kahn BB, Kahn CR. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science. 2003;299(5606):572–4. doi: 10.1126/science.1078223. [DOI] [PubMed] [Google Scholar]
- 21.Brown-Borg HM, et al. Dwarf mice and the ageing process. Nature. 1996;384(6604):33. doi: 10.1038/384033a0. [DOI] [PubMed] [Google Scholar]
- 22.Coschigano KT, et al. Assessment of growth parameters and life span of GHR/BP gene-disrupted mice. Endocrinology. 2000;141(7):2608–13. doi: 10.1210/endo.141.7.7586. [DOI] [PubMed] [Google Scholar]
- 23.Flurkey K, et al. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci U S A. 2001;98(12):6736–41. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ikeno Y, et al. Delayed occurrence of fatal neoplastic diseases in ames dwarf mice: correlation to extended longevity. J Gerontol A Biol Sci Med Sci. 2003;58(4):291–6. doi: 10.1093/gerona/58.4.b291. [DOI] [PubMed] [Google Scholar]
- 25.Vergara M, et al. Hormone-treated snell dwarf mice regain fertility but remain long lived and disease resistant. J Gerontol A Biol Sci Med Sci. 2004;59(12):1244–50. doi: 10.1093/gerona/59.12.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kurosu H, et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309(5742):1829–33. doi: 10.1126/science.1112766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selman C, et al. Evidence for lifespan extension and delayed age-related biomarkers in insulin receptor substrate 1 null mice. Faseb J. 2008;22(3):807–18. doi: 10.1096/fj.07-9261com. [DOI] [PubMed] [Google Scholar]
- 28.Taguchi A, Wartschow LM, White MF. Brain IRS2 signaling coordinates life span and nutrient homeostasis. Science. 2007;317(5836):369–72. doi: 10.1126/science.1142179. [DOI] [PubMed] [Google Scholar]
- 29.Boldt HB, Conover CA. Pregnancy-associated plasma protein-A (PAPP-A): a local regulator of IGF bioavailability through cleavage of IGFBPs. Growth Horm IGF Res. 2007;17(1):10–8. doi: 10.1016/j.ghir.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 30.Conover CA, Bale LK. Loss of pregnancy-associated plasma protein A extends lifespan in mice. Aging Cell. 2007;6(5):727–9. doi: 10.1111/j.1474-9726.2007.00328.x. [DOI] [PubMed] [Google Scholar]
- 31.Rehage M, et al. Transgenic overexpression of pregnancy-associated plasma protein-A increases the somatic growth and skeletal muscle mass in mice. Endocrinology. 2007;148(12):6176–85. doi: 10.1210/en.2007-0274. [DOI] [PubMed] [Google Scholar]
- 32.Suh Y, et al. Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci U S A. 2008;105(9):3438–42. doi: 10.1073/pnas.0705467105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laron Z. Growth hormone insensitivity (Laron syndrome) Rev Endocr Metab Disord. 2002;3(4):347–55. doi: 10.1023/a:1020905725012. [DOI] [PubMed] [Google Scholar]
- 34.Laron Z. The GH-IGF1 axis and longevity. The paradigm of IGF1 deficiency. Hormones (Athens) 2008;7(1):24–7. doi: 10.14310/horm.2002.1111034. [DOI] [PubMed] [Google Scholar]
- 35.Shevah O, Laron Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: a preliminary report. Growth Horm IGF Res. 2007;17(1):54–7. doi: 10.1016/j.ghir.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 36.Fontana L, Klein S. Aging, adiposity, and calorie restriction. JAMA. 2007;297(9):986–94. doi: 10.1001/jama.297.9.986. [DOI] [PubMed] [Google Scholar]
- 37.Fontana L, et al. Long-term effects of calorie or protein restriction on serum IGF-1 and IGFBP-3 concentration in humans. Aging Cell. 2008;7(5):681–7. doi: 10.1111/j.1474-9726.2008.00417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barzilai N, Bartke A. Biological approaches to mechanistically understand the healthy life span extension achieved by calorie restriction and modulation of hormones. J Gerontol A Biol Sci Med Sci. 2009;64(2):187–91. doi: 10.1093/gerona/gln061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Varady KA, Hellerstein MK. Alternate-day fasting and chronic disease prevention: a review of human and animal trials. Am J Clin Nutr. 2007;86(1):7–13. doi: 10.1093/ajcn/86.1.7. [DOI] [PubMed] [Google Scholar]
- 40.Pollak M. Insulin and insulin-like growth factor signalling in neoplasia. Nat Rev Cancer. 2008;8(12):915–28. doi: 10.1038/nrc2536. [DOI] [PubMed] [Google Scholar]
- 41.Chan JM, et al. Insulin-like growth factor-I (IGF-I) and IGF binding protein-3 as predictors of advanced-stage prostate cancer. J Natl Cancer Inst. 2002;94(14):1099–106. doi: 10.1093/jnci/94.14.1099. [DOI] [PubMed] [Google Scholar]
- 42.Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12(8):895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- 43.Ridley AJ, et al. Cell migration: integrating signals from front to back. Science. 2003;302(5651):1704–9. doi: 10.1126/science.1092053. [DOI] [PubMed] [Google Scholar]
- 44.Patel SD, et al. Cadherin-mediated cell-cell adhesion: sticking together as a family. Curr Opin Struct Biol. 2003;13(6):690–8. doi: 10.1016/j.sbi.2003.10.007. [DOI] [PubMed] [Google Scholar]
- 45.Birchmeier W, Behrens J. Cadherin expression in carcinomas: role in the formation of cell junctions and the prevention of invasiveness. Biochim Biophys Acta. 1994;1198(1):11–26. doi: 10.1016/0304-419x(94)90003-5. [DOI] [PubMed] [Google Scholar]
- 46.Conacci-Sorrell M, Zhurinsky J, Ben-Ze’ev A. The cadherin-catenin adhesion system in signaling and cancer. J Clin Invest. 2002;109(8):987–91. doi: 10.1172/JCI15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rivard N. Phosphatidylinositol 3-kinase: a key regulator in adherens junction formation and function. Front Biosci. 2009;14:510–22. doi: 10.2741/3259. [DOI] [PubMed] [Google Scholar]
- 48.Guvakova MA, Surmacz E. Overexpressed IGF-I receptors reduce estrogen growth requirements, enhance survival, and promote E-cadherin-mediated cell-cell adhesion in human breast cancer cells. Exp Cell Res. 1997;231(1):149–62. doi: 10.1006/excr.1996.3457. [DOI] [PubMed] [Google Scholar]
- 49.Morford LA, et al. Insulin-like growth factors (IGF) enhance three-dimensional (3D) growth of human glioblastomas. Cancer Lett. 1997;115(1):81–90. doi: 10.1016/s0304-3835(97)04717-4. [DOI] [PubMed] [Google Scholar]
- 50.Playford MP, et al. Insulin-like growth factor 1 regulates the location, stability, and transcriptional activity of beta-catenin. Proc Natl Acad Sci U S A. 2000;97(22):12103–8. doi: 10.1073/pnas.210394297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Valentinis B, et al. Anti-apoptotic signaling of the IGF-I receptor in fibroblasts following loss of matrix adhesion. Oncogene. 1999;18(10):1827–36. doi: 10.1038/sj.onc.1202471. [DOI] [PubMed] [Google Scholar]
- 52.Mauro L, et al. Role of the IGF-I receptor in the regulation of cell-cell adhesion: implications in cancer development and progression. J Cell Physiol. 2003;194(2):108–16. doi: 10.1002/jcp.10207. [DOI] [PubMed] [Google Scholar]
- 53.Pennisi PA, et al. Reduced expression of insulin-like growth factor I receptors in MCF-7 breast cancer cells leads to a more metastatic phenotype. Cancer Res. 2002;62(22):6529–37. [PubMed] [Google Scholar]
- 54.Jones RA, CC, Wood GA, Petrik JJ, Moorehead RA. Reversibility and recurrence of IGF-IR-induced mammary tumors. Oncogene. 2009;28(21):2152–62. doi: 10.1038/onc.2009.79. [DOI] [PubMed] [Google Scholar]
- 55.Garrouste F, et al. Prevention of cytokine-induced apoptosis by insulin-like growth factor-I is independent of cell adhesion molecules in HT29-D4 colon carcinoma cells-evidence for a NF-kappaB-dependent survival mechanism. Cell Death Differ. 2002;9(7):768–79. doi: 10.1038/sj.cdd.4401022. [DOI] [PubMed] [Google Scholar]
- 56.Hood JD, Cheresh DA. Role of integrins in cell invasion and migration. Nat Rev Cancer. 2002;2(2):91–100. doi: 10.1038/nrc727. [DOI] [PubMed] [Google Scholar]
- 57.Tai YT, et al. Insulin-like growth factor-1 induces adhesion and migration in human multiple myeloma cells via activation of beta1-integrin and phosphatidylinositol 3′-kinase/AKT signaling. Cancer Res. 2003;63(18):5850–8. [PubMed] [Google Scholar]
- 58.Zhang X, Yee D. Insulin-like growth factor binding protein-1 (IGFBP-1) inhibits breast cancer cell motility. Cancer Res. 2002;62(15):4369–75. [PubMed] [Google Scholar]
- 59.Samanna V, et al. Alpha-V-dependent outside-in signaling is required for the regulation of CD44 surface expression, MMP-2 secretion, and cell migration by osteopontin in human melanoma cells. Exp Cell Res. 2006;312(12):2214–30. doi: 10.1016/j.yexcr.2006.03.022. [DOI] [PubMed] [Google Scholar]
- 60.Gockerman A, PT, Jones JI, Clemmons DR. Insulin-like growth factor (IGF)-binding proteins inhibit the smooth muscle cell migration responses to IGF-I and IGF-II. Endocrinology. 1995;36(10):4168–73. doi: 10.1210/endo.136.10.7545099. [DOI] [PubMed] [Google Scholar]
- 61.Zheng B, Clemmons DR. Blocking ligand occupancy of the alphaVbeta3 integrin inhibits insulin-like growth factor I signaling in vascular smooth muscle cells. Proc Natl Acad Sci U S A. 1998;95(19):11217–22. doi: 10.1073/pnas.95.19.11217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Clemmons DR, et al. Role of the integrin alphaVbeta3 in mediating increased smooth muscle cell responsiveness to IGF-I in response to hyperglycemic stress. Growth Horm IGF Res. 2007;17(4):265–70. doi: 10.1016/j.ghir.2007.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gleeson LM, et al. Insulin-like growth factor-binding protein 1 stimulates human trophoblast migration by signaling through alpha 5 beta 1 integrin via mitogen-activated protein Kinase pathway. J Clin Endocrinol Metab. 2001;86(6):2484–93. doi: 10.1210/jcem.86.6.7532. [DOI] [PubMed] [Google Scholar]
- 64.Lauffenburger DA, Horwitz AF. Cell migration: a physically integrated molecular process. Cell. 1996;84(3):359–69. doi: 10.1016/s0092-8674(00)81280-5. [DOI] [PubMed] [Google Scholar]
- 65.Kadowaki T, et al. Insulin-like growth factors, insulin, and epidermal growth factor cause rapid cytoskeletal reorganization in KB cells. Clarification of the roles of type I insulin-like growth factor receptors and insulin receptors. J Biol Chem. 1986;261(34):16141–7. [PubMed] [Google Scholar]
- 66.Guvakova MA, Surmacz E. The activated insulin-like growth factor I receptor induces depolarization in breast epithelial cells characterized by actin filament disassembly and tyrosine dephosphorylation of FAK, Cas, and paxillin. Exp Cell Res. 1999;251(1):244–55. doi: 10.1006/excr.1999.4566. [DOI] [PubMed] [Google Scholar]
- 67.Liu L, CL, Chung J, Huang S. Rapamycin inhibits F-actin reorganization and phosphorylation of focal adhesion proteins. Oncogene. 2008;27(37):4998–5010. doi: 10.1038/onc.2008.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Konstantopoulos N, Clark S. Reduced cell attachment and phosphorylation of focal adhesion kinase associated with expression of a mutant insulin receptor. J Biol Chem. 1996;271(46):28960–8. doi: 10.1074/jbc.271.46.28960. [DOI] [PubMed] [Google Scholar]
- 69.Leventhal PS, FE Tyrosine phosphorylation and enhanced expression of paxillin during neuronal differentiation in vitro. J Biol Chem. 1996;271(11):5957–60. doi: 10.1074/jbc.271.11.5957. [DOI] [PubMed] [Google Scholar]
- 70.Renehan AG, et al. Insulin-like growth factor (IGF)-I, IGF binding protein-3, and cancer risk: systematic review and meta-regression analysis. Lancet. 2004;363(9418):1346–53. doi: 10.1016/S0140-6736(04)16044-3. [DOI] [PubMed] [Google Scholar]
- 71.Ma J, et al. Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J Natl Cancer Inst. 1999;91(7):620–5. doi: 10.1093/jnci/91.7.620. [DOI] [PubMed] [Google Scholar]
- 72.Pollak MN. Insulin-like growth factors and neoplasia. Novartis Found Symp. 2004;262:84–98. discussion 98–107, 265–8. [PubMed] [Google Scholar]
- 73.Barozzi C, et al. Relevance of biologic markers in colorectal carcinoma: a comparative study of a broad panel. Cancer. 2002;94(3):647–57. doi: 10.1002/cncr.10278. [DOI] [PubMed] [Google Scholar]
- 74.All-Ericsson C, et al. Insulin-like growth factor-1 receptor in uveal melanoma: a predictor for metastatic disease and a potential therapeutic target. Invest Ophthalmol Vis Sci. 2002;43(1):1–8. [PubMed] [Google Scholar]
- 75.Xie Y, et al. Expression of insulin-like growth factor-1 receptor in synovial sarcoma: association with an aggressive phenotype. Cancer Res. 1999;59(15):3588–91. [PubMed] [Google Scholar]
- 76.Pisani P. Hyper-insulinaemia and cancer, meta-analyses of epidemiological studies. Arch Physiol Biochem. 2008;114(1):63–70. doi: 10.1080/13813450801954451. [DOI] [PubMed] [Google Scholar]
- 77.Michaud DS, et al. Prediagnostic plasma C-peptide and pancreatic cancer risk in men and women. Cancer Epidemiol Biomarkers Prev. 2007;16(10):2101–9. doi: 10.1158/1055-9965.EPI-07-0182. [DOI] [PubMed] [Google Scholar]
- 78.Gunter MJ, et al. Insulin, insulin-like growth factor-I, and risk of breast cancer in postmenopausal women. J Natl Cancer Inst. 2009;101(1):48–60. doi: 10.1093/jnci/djn415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gunter MJ, et al. A prospective evaluation of insulin and insulin-like growth factor-I as risk factors for endometrial cancer. Cancer Epidemiol Biomarkers Prev. 2008;17(4):921–9. doi: 10.1158/1055-9965.EPI-07-2686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vigneri P, et al. Diabetes and cancer. Endocr Relat Cancer. 2009 doi: 10.1677/ERC-09-0087. [DOI] [PubMed] [Google Scholar]
- 81.Weckbecker G, et al. Somatostatin analogue octreotide enhances the antineoplastic effects of tamoxifen and ovariectomy on 7,12-dimethylbenz(alpha)anthracene-induced rat mammary carcinomas. Cancer Res. 1994;54(24):6334–7. [PubMed] [Google Scholar]
- 82.Karp DD, et al. Phase II study of the anti-insulin-like growth factor type 1 receptor antibody CP-751,871 in combination with paclitaxel and carboplatin in previously untreated, locally advanced, or metastatic non-small-cell lung cancer. J Clin Oncol. 2009;27(15):2516–22. doi: 10.1200/JCO.2008.19.9331. [DOI] [PubMed] [Google Scholar]
- 83.Tolcher AW, et al. Phase I and pharmacokinetic study of YM155, a small-molecule inhibitor of survivin. J Clin Oncol. 2008;26(32):5198–203. doi: 10.1200/JCO.2008.17.2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Higano C, et al. A phase I, first in man study of weekly IMCA12, a fully human insulin like growth factor-I receptor IgG1 monoclonal antibody, in patients with advanced solid tumors. Proceedings ASCO, 2007; 2007. p. A269. [Google Scholar]
- 85.Tolcher AW, et al. Phase I pharmacokinetic and biologic correlative study of mapatumumab, a fully human monoclonal antibody with agonist activity to tumor necrosis factor-related apoptosis-inducing ligand receptor-1. J Clin Oncol. 2007;25(11):1390–5. doi: 10.1200/JCO.2006.08.8898. [DOI] [PubMed] [Google Scholar]
- 86.Atzori F, et al. A phase I, pharmacokinetic (PK) and pharmacodynamic (PD) study of weekly (qW) MK-0646, an insulin-like growth factor-1 receptor (IGFIR) monoclonal antibody (MAb) in patients (pts) with advanced solid tumors. J Clin Oncol. 2008;26:157s. doi: 10.1158/1078-0432.CCR-10-3336. [DOI] [PubMed] [Google Scholar]
- 87.Mukohara T, et al. Sensitivity of breast cancer cell lines to the novel insulin-like growth factor-1 receptor (IGF-1R) inhibitor NVP-AEW541 is dependent on the level of IRS-1 expression. Cancer Lett. 2009;282(1):14–24. doi: 10.1016/j.canlet.2009.02.056. [DOI] [PubMed] [Google Scholar]
- 88.Vasilcanu R, et al. Picropodophyllin induces downregulation of the insulin-like growth factor 1 receptor: potential mechanistic involvement of Mdm2 and beta-arrestin1. Oncogene. 2008;27(11):1629–38. doi: 10.1038/sj.onc.1210797. [DOI] [PubMed] [Google Scholar]
- 89.Harzstark AL, et al. A phase I trial of nordihydroguareacetic acid (NDGA) in patients with non-metastatic prostate cancer and rising PSA. J Clin Oncol. 2007;2518:15500s. [Google Scholar]
- 90.Juul A, et al. Low serum insulin-like growth factor I is associated with increased risk of ischemic heart disease: a population-based case-control study. Circulation. 2002;106(8):939–44. doi: 10.1161/01.cir.0000027563.44593.cc. [DOI] [PubMed] [Google Scholar]
- 91.Laughlin GA, et al. The prospective association of serum insulin-like growth factor I (IGF-I) and IGF-binding protein-1 levels with all cause and cardiovascular disease mortality in older adults: the Rancho Bernardo Study. J Clin Endocrinol Metab. 2004;89(1):114–20. doi: 10.1210/jc.2003-030967. [DOI] [PubMed] [Google Scholar]
- 92.Vaessen N, et al. A polymorphism in the gene for IGF-I: functional properties and risk for type 2 diabetes and myocardial infarction. Diabetes. 2001;50(3):637–42. doi: 10.2337/diabetes.50.3.637. [DOI] [PubMed] [Google Scholar]
- 93.Juul A. Serum levels of insulin-like growth factor I and its binding proteins in health and disease. Growth Horm IGF Res. 2003;13(4):113–70. doi: 10.1016/s1096-6374(03)00038-8. [DOI] [PubMed] [Google Scholar]
- 94.Janssen JA, Lamberts SW. Igf-I and longevity. Horm Res. 2004;62(Suppl 3):104–9. doi: 10.1159/000080508. [DOI] [PubMed] [Google Scholar]
- 95.Yang J, Anzo M, Cohen P. Control of aging and longevity by IGF-I signaling. Exp Gerontol. 2005;40(11):867–72. doi: 10.1016/j.exger.2005.08.001. [DOI] [PubMed] [Google Scholar]
- 96.Pinkston-Gosse J, Kenyon C. DAF-16/FOXO targets genes that regulate tumor growth in Caenorhabditis elegans. Nat Genet. 2007;39(11):1403–9. doi: 10.1038/ng.2007.1. [DOI] [PubMed] [Google Scholar]
- 97.Brunet A. Aging and cancer: killing two birds with one worm. Nat Genet. 2007;39(11):1306–7. doi: 10.1038/ng1107-1306. [DOI] [PubMed] [Google Scholar]
- 98.Kenyon C. The plasticity of aging: insights from long-lived mutants. Cell. 2005;120(4):449–60. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]