

Abstract
Sprouty proteins are potent receptor tyrosine kinase inhibitors that antagonize growth factor signaling and are involved in lung development. However, little is known about the regulation or targets of Sprouty-4 (Spry4) in lung cancer. Our study aimed to determine the role of Spry4 in NSCLC. We found that Spry4 mRNA expression was decreased in NSCLC cell lines and in dysplastic lung cell lines compared to a non-transformed cell line, suggesting that Spry4 has tumor suppressing activity. When Spry4 was stably transfected into H157 and H2122 NSCLC cell lines, decreased migration and invasion were observed. MMP-9 activity was decreased and expression of MMP inhibitors TIMP1 and CD82 were increased. Stable expression of Spry4 led to reduced cell growth and reduced anchorage independent growth in NSCLC cell lines, along with upregulation of tumor suppressors p53 and p21. Changes in epithelial and mesenchymal markers indicated that Spry4 expression induces a reversal of the epithelial to mesenchymal transition characteristic of tumor cells. Treatment of a non-transformed lung epithelial cell line with shRNA to Spry4 led to decreased expression of epithelial markers and increased cell growth, supporting the concept of Spry4 acting as a tumor suppressor. We demonstrated that activity of the Spry4 promoter is increased by Wnt7A/Fzd9 signaling through peroxisome proliferator activated receptor γ. These data present previously undescribed targets of Spry4 and suggest that Spry4 is a downstream target of Wnt7A/Fzd 9 signaling. Spry4 may have efficacy in the treatment of NSCLC.
Introduction
Sprouty (Spry) proteins were first identified as potent receptor tyrosine kinase (RTK) inhibitors that modulate tracheal branching in Drosophila (1, 2). Subsequent studies identified four vertebrate Spry proteins with highly conserved C-terminal and Spry domains and variable N-terminal domains (3). Interaction of Spry proteins with RTK pathways depends on the specific Spry and cellular context. Sprys have been shown to inhibit EGFR, FGFR, VEGFR, and PDGFR (4–6). Sprys can bind to Grb2 or SOS to disrupt the receptor complex and interfere with ERK/MAPK activation. Spry2 has been shown to interact with c-CBL to inhibit EGF-mediated ERK/MAPK signaling (3). Downregulation of Spry1 has been observed in breast and prostate cancer, of Spry2 in breast, prostate, and liver cancer, and of Spry4 in prostate cancer (7, 8). Spry4 appears to act as a marker of treatment response in gastrointestinal tumors and Spry1 presence is associated with a good prognosis in renal cell carcinoma patients (8). A protective role for Spry2 in the lung has been described in vitro, as well as in vivo in a urethane model of non-small cell lung cancer (NSCLC) and in a germline KRAS mutation model of lung cancer (9–11).
The Wnt family of proteins control diverse developmental pathways and act in cooperation with the Frizzled (Fzd) family of seven-membrane spanning G-coupled protein receptors. Increased activity of the canonical Wnt/β-catenin signaling pathway has been associated with oncogenic stimulation in several types of cancer (12–16). In contrast, our previous work has shown Wnt7A is lost in NSCLC and activation of Wnt7A signaling leads to reversal of the transformed phenotype in NSCLC (17). Wnt7A binds to the Fzd9 receptor and signals through ERK-5 to activate the tumor suppressor peroxisome proliferator-activated receptor γ (PPARγ), but the downstream targets of PPARγ are largely unknown (18, 19). PAPRγ and its synthetic agonists, such as ciglitazone and rosiglitazone, inhibit transformed growth and metastasis and promote epithelial differentiation and have demonstrated tumor prevention efficacy (20–25).
We previously observed that Spry4 expression is upregulated with activation of Wnt7A and Fzd9 by quantitative PCR (QPCR) and immunoblot in NSCLC, suggesting a role for Spry4 in a pathway outside of RTK signaling (17). However, the specific involvement of Spry4 in this non-canonical Wnt signaling pathway is unknown. Spry4 has been shown to inhibit FGF pathways in cell lines and mice, but unlike other members of the Sprouty family, Spry4 has not been shown to inhibit EGF signaling (5, 26, 27). Evaluation of mouse organogenesis has identified Spry4 expression in the lung epithelium of the developing embryo, but characterization of Spry4 expression in adult lung tissue still needs to be completed (28). There is clearly a need for further studies exploring the role of Spry4 in the context of the lung epithelium. In the study presented here, we examined Spry4 activity in NSCLC and found that it is lost in cancer cell lines and dysplastic cell lines. We identified Wnt7A/Fzd9 and PPARγ as regulators of Spry4 and described new targets of Spry4 known to be involved in suppressing tumor growth and metastasis. We demonstrated that re-expression of Spry4 results in decreased transformed cell growth, decreased migration and invasion, and increased differentiation of NSCLC cells.
Materials and Methods
Cell Culture and Retrovirus-mediated Gene Transfer
NSCLC and Beas2B (a human non-transformed lung epithelial cell line) cell lines were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum at 37°C in a humidified 5% CO2 incubator. The HBEC (human bronchial epithelial cells) cell line was cultured in Bronchial Epithelial Basal Media at 37°C in a humidified 5% CO2 incubator. Cell lines were obtained from the University of Colorado Cancer Center Cell Line Core in 1995, except the HBEC cell line, which was obtained in 2009 from Dr. Robert Doebele at the University of Colorado Denver. Morphology of all cells lines was monitored twice weekly and stocks of cell lines were passaged no more than ten times for use in experiments. The Spry4/PCDNA3 vector was kindly provided by Diane Harris (UCLA, Los Angeles, CA, USA). Spry4 was ligated into the retroviral vector LPCX (Clontech; Mountainview, CA, USA) and 3ug of the expression vector with Spry4 was packaged into a replication-defective retrovirus using 293t cells as described previously (17). H157 and H2122 cell lines were transduced, selected, and maintained in growth medium containing puromycin (1µg/ml). The H157 Wnt7A/Fzd9 cell line was previously described (17). The B2B Spry4 knockdown cell lines were created using Spry4 shRNA in a lentiviral vector from Open Biosystems (Huntsville, AL). 293t cells were transfected with packaging vectors and 1ug of the Spry4 lentiviral shRNA or negative lentiviral control plasmid and then B2B cells were transduced with the resulting viral media. Cell lines were selected and maintained with growth medium containing puromycin (1ug/ml).
Cell Growth, MTS assay, Soft Agar Assay, and Caspase Assay
For the cell growth assay, 50,000 cells were seeded in triplicate per well of a 24-well culture plate in complete growth medium. Cells were counted for six days using a hemocytomoeter. For the soft agar assay, 2,500 cells were seeded in triplicate in a 6-well plate and the assay was conducted as previously described (18). The data are presented as cloning efficiency: the mean number of colonies per well divided by the number of cells plated. Activity of Caspase 3/7 was analyzed using a Caspase-Glo 3/7 Assay (Promega; Madison, WI, USA). For the MTS assay (Promega), 500 cells per well were seeded in triplicate for each cell line. At 24, 48, and 72 hours, 20ul of MTS reagent was added to each well, incubated for 1 hour at 37C, and results analyzed in a 96-well plate reader at 490nm. The sample data was normalized to background readings of media only.
Quantitative Real-Time PCR
RNA was extracted from cells with RNeasy (Qiagen Inc.; Valencia, CA, USA) and 5µg of RNA was converted to cDNA. RNA from primary dysplastic cell cultures was a gift from Dr. Dan Merrick (VAMC, Denver, CO, USA). Dysplastic cell cultures are arbitrarily numbered 1–13. Primer sequences for Spry4 were F 5’-CCAGGATGTCACCCAC CATTG-3’and R 5’-TGTGCTGCTGCTGCTC-3’ and for GAPDH were F 5’-GCCAAATATG ATGACATCAAGAAGG-3’ and R-5’GGTGTCGCTGTTGAAGTCAGAG-3’ (Integrated DNA Technologies; Coralville, IA, USA). Primer sets for p53, p21, CD82, TIMP1, KRT8, KRT18, and Vimentin were obtained from SABiosciences and are available on the manufacturer’s website (SABioscience; Frederick, MD, USA). PCR conditions were 95°C for 10 minutes, followed by 40 cycles of 95°C for 15 seconds and 60°C for 1 minute. Spry4 and GAPDH reactions included 2µl of cDNA, SYBRGreen Jumpstart Taq Readymix (Sigma Aldrich; St. Louis, MO, USA), 200nM primers, and 2mM MgCl2 in a 25µl volume. Superarray PCR required the RT2 Real-Time SYBRGreen PCR Master Mix (SABioscience) and 1µl of Superarray qRT-PCR Primer Assay in a 25µl volume. GAPDH was used to normalize all samples. The QPCR data is presented as fold-changes in normalized mRNA levels in control vs experimental samples and are the average of at least triplicate experiments with standard error presented as error bars.
Transfections and Luciferase Assays
The reporter plasmids PPAR Response Element (PPRE) (3µg) and Spry4 promoter-luciferase (3µg) (kindly provided by Dr. Warburton, USC, Los Angeles, CA, USA), expression plasmids wild-type PPARγ (3µg) and Spry4 (3µg), and β-galactosidase control plasmids (3µg) were transfected into H157 and H2122 cells using Lipofectamine Reagent (Invitrogen; Carlsbad, CA, USA). Truncated Spry4 luciferases were constructed from the full-length promoter luciferase (−4446) at −1182, −418, and −31 base pairs from the transcription start site. Truncated Spry4 luciferases −4446, −1182 and −418 were gifts from Dr. Warburton (39) and the −31 luciferase was constructed by the UCD DERC Molecular Biology Core. PPARγ inhibitor T0070907 (Cayman Chemical; Ann Arbor, MI, USA) was applied once at 25µM 24 hours after transfection. Cells were collected, washed with PBS, and resuspended in Luciferase Reporter Lysis Buffer (Promega). After centrifugation, luciferase activity was measured in the supernatant using a Luciferase Assay (Promega) and β-galactosidase activity was measured by absorbance. β-galactosidase was used for normalization. The data are presented as fold-change in relative light units/milliunits of β-galactosidase and represent the average of three independent experiments.
Immunoblot Analysis
The following antibodies were used for immunoblotting: Spry4 and Survivin (SantaCruzBiotech; Santa Cruz, CA, USA), Wnt7A (R&D Systsems; Minneapolis, MN, USA), E-cadherin (Cell Signaling; Danvers, MA, USA), GAPDH, CD82 and β-actin (Abcam; Cambridge, MA, USA). Cell extracts for all immunoblots were prepared in MAP kinase lysis buffer. Aliquots of lysates were resolved by 10% SDS-PAGE and transferred to nitrocellulose. The membranes were blocked in Tris-buffered saline with 3% bovine serum albumin and then incubated with primary antibody for 12–16 hours at 4°C. Membranes were washed in Tris-buffered saline, incubated with alkaline phosphatase-coupled secondary antibodies for 1 hour at 4°C, and visualized with LumiPhos reagent (Pierce; Rockford, IL, USA). β-actin or GAPDH were used as loading controls.
Three-dimensional Cell Culture, Collagen Invasion Assay, and Scratch Assay
Three-dimensional basement membrane cultures were established as previously described (57). Briefly, 5,000 cells/well were grown in 2% matrigel (BD Bioscience; San Jose, CA, USA) with EGF on a 50% matrigel base layer. The collagen invasion assay was performed as previously described (33). Briefly, 5,000 cells/well were seeded in 2% matrigel on a layer of 1:1 collagen (BD Bioscience) to matrigel mix. Culture growth recorded on day 5. For the scratch assay, cells were grown in complete growth medium until 90–100% confluent. A 3mm space was introduced across the diameter of each plate. At time zero, cells were treated with 1ug/ml mitomycin to inhibit cell proliferation. Cell migration was recorded at 24 and 48 hours. Images were captured using a 40× lens on a light microscope and a digital camera.
MMP-9 Activity Assay
The MMP-9 Human Biotrak assay (GE Healthcare; Piscataway, NJ, USA) was used to measure levels of active MMP-9 in NSCLC cell lines. Briefly, the assay used 100ul of cell culture supernatant a two-site ELISA sandwich format to measure endogenous levels of active MMP-9 relative to a set of standards. Absorbance was measured at 405 and is presented as fold-change in active MMP-9 levels interpolated from the standard curve.
Results
Spry4 expression is lost in NSCLC cell lines
Sixteen NSCLC cell lines were analyzed by QPCR and decreased Spry4 expression was detected in 21 cell lines relative to the non-transformed human lung epithelial cell line HBEC (The HBEC cell line was cultured in a different media than the NSCLC cell lines.) (Fig. 1A). Eleven out of 11 dysplastic lung cell cultures analyzed for Spry4 expression also had reduced levels of mRNA compared to B2B and HBEC cells (Fig. 1B). In Figure 1C, immunoblot data for NSCLC cell lines H157, H2122, A549, H661, and H1703, and the HBEC cell line demonstrate reduced expression of Spry4 in NSCLC cell lines compared to the non-transformed cell line.
Figure 1.


Spry4 expression is decreased in NSCLC cell lines and dysplastic lung cell culture. (A,B) QPCR for Spry4 mRNA in NSCLC cell lines and dysplastic lung epithelial cell culture. Dysplastic cell cultures are arbitrarily numbered 1–13. Cell line mRNA levels are presented as fold-reductions compared to the non-transformed lung epithelial cell line HBEC. Results are the average of triplicate experiments normalized to GAPDH. (C) Immunoblot analysis of Spry4 protein expression in NSCLC cell lines compared to a non-transformed lung epithelial cell line (HBEC). β-actin is included as a loading control.
Wnt7A/Fzd9 signaling increases Spry4 promoter activity through PPARγ in NSCLC
PPARγ activation leads to suppression of growth and restoration of epithelial differentiation in NSCLC and has been identified as a mediator of Wnt7A/Fzd9 tumor suppression signaling (18, 20). Previous data indicates that Spry4 expression is increased in response to Wnt7A/Fzd9 pathway activation, indicating that both PPARγ and Spry4 are downstream targets of Wnt7A/Fzd9 (17). We have previously described H157 and H2122 cell lines as lacking Wnt7a/Fzd9 and Fzd9 respectively and have generated cell lines with stable Wnt7a and/or Fzd9 expression (17). We transfected Spry4-luciferase into H157 and H2122 cells with stable Wnt7A and/or Fzd9 expression. In both cell lines, establishment of the Wnt7A/Fzd9 signaling pathway led to increased Spry4-luciferase activity (Fig. 2A). To establish the relationship between PPARγ and Spry4, H157 and H2122 cells were transiently transfected with empty vector, Spry4, or wild type PPARγ, along with a vector encoding a PPAR response element (PPRE) luciferase or a Spry4 promoter luciferase. In H157 and H2122 cells, expression of Spry4 did not lead to increased PPRE luciferase activity (Fig. 2B). Transfection of PPARγ into H157 and H2122 cells with or without T0070907 (T007), a PPARγ inhibitor, showed increased Spry4 luciferase activity with wild type PPARγ and decreased activity with T007 treatment (Fig. 2B). PPARλ did not activate the Spry4 luciferase (data not shown). To further connect PPARγ expression with Spry4 promoter activity, H157 and H2122 cells with stable expression of PAPRγ were transfected with full length (−4446) or one of three truncated Spry4 promoter luciferases (−1182, −418, and −31) (Fig. 2C). Activity of the Spry4 promoter decreased with sequences smaller than the 1182 truncation, suggesting that a sequence between 1182 and 31 base pairs from the transcription start site in the promoter is needed for the influence of PPARγ on Spry4 promoter activity.
Figure 2.
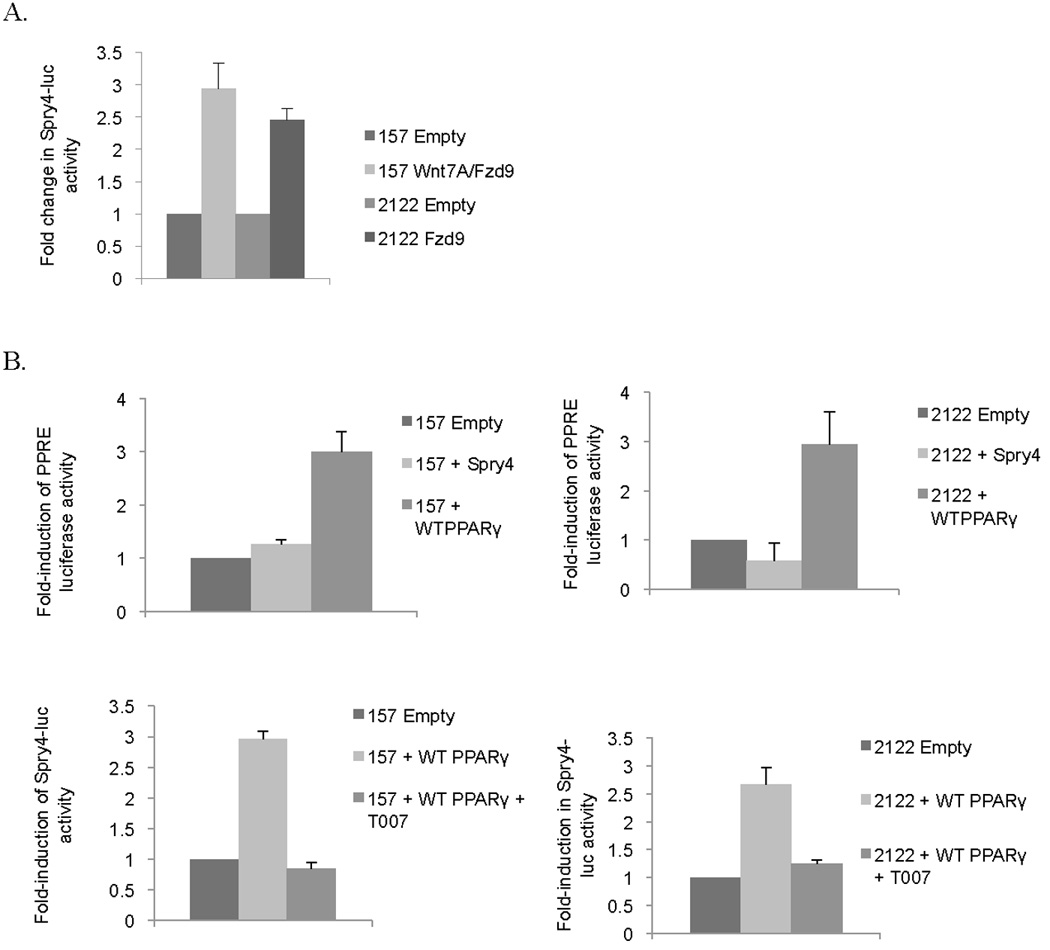

The Spry4 promoter is activated by PPARγ expression. (A) H157 and H2122 cells with stable Wnt7A and/or Fzd9 expression were transfected with Spry4-luciferase. Results are the average of triplicate experiments. (B) H157 and H2122 cells were transiently transfected with PPAR response element (PPRE) –luc, Spry4 and wild type PPARγ. H157 and H2122 cells were transfected with wild type PPARγ and Spry4 promoter luciferase and treated with T007, a PPARγ inhibitor. Results are the average of triplicate experiments. (C) H157 and H2122 cells with stable PPARγ expression were transfected with full length (−4446) and truncated versions of Spry4-luciferase (−1182, −418, −31). Results are the average of triplicate experiments. Change in activity is presented as fold-induction of the luciferase reporter.
Spry4 re-expression in NSCLC reduces cell growth and colony formation in soft agar and increases expression of tumor suppressors
Wnt7A/Fzd9 signaling through PPARγ has been shown to inhibit NSCLC cell growth and reduce colony formation in soft agar (17). We were interested whether Spry4 contributes to the growth suppression effects of Wnt7A/Fzd9. The role of Spry4 on NSCLC growth was assessed using H157 and H2122 cells stably transfected with Spry4 or an empty vector. Stable expression of Spry4 was confirmed by immunoblot (Fig. 3A). To investigate the influence of Spry4 on transformed cell growth, equal numbers of H157 and H2122 cells with Spry4 re-expression or empty vector control were plated in complete medium and cell numbers were measured for six days. Expression of Spry4 significantly reduced the growth rate of H157 and H2122 cells compared to empty vector control cells (Fig. 3B). Soft agar assays were used to evaluate the effect of Spry4 expression on colony formation, in which transformed cells are able to grow in an anchorage independent manner, but non-transformed cells are not. After 21 days of observation, stable expression of Spry4 inhibited colony formation compared to empty vector control cells (Fig. 3C). Apoptotic activity was evaluated in the context of Spry4 expression and there was no difference in Caspase 3/7 activity or Survivin protein level when compared to empty vector control cells (data not shown). To identify possible downstream targets of Spry4, QPCR was used to analyze mRNA expression of p53 and p21 in H2122 cells with Spry4 re-expression. p53 and p21 were increased with Spry4 expression compared to an empty vector control (Fig. 3D). p53 and its target p21 are well-known members of tumor suppression pathways in the lung (29). Increased expression of these genes may be related to the inhibition of proliferation observed with Spry4 re-expression. These results demonstrate that Spry4 expression reduces cell growth and anchorage-independent growth, two key characteristics of transformed cells.
Figure 3.


Stable expression of Spry4 reduces cell growth and anchorage independent growth in NSCLC cells. (A) Stable Spry4 expression in transfected H157 and H2122 cells was verified by immunoblot with a GAPDH loading control and by QPCR. (B) 50,000 cells from H157 and H2122 stably expressing Spry4 or an empty vector control were plated in triplicate in 6 wells and one well was counted each day. Results are the average of triplicate experiments. (C) 25,000 cells from H157 and H2122 stably expressing Spry4 or an empty vector control were seeded in triplicate in media with 0.3% agar on a base of 0.5% agar. Colonies were stained with nitroblue tetrazolium chloride at 21 days. Representative soft agar pictures are shown. Cloning efficiency is the ratio of counted colonies to seeded cells and is the average of triplicate experiments. (D) QPCR for p21 in H2122 cell lines stably expressing Spry4 compared to an empty vector control. Results are the average of triplicate experiments normalized to GAPDH.
Spry4 re-expression promotes a mesenchymal to epithelial transition
Since Spry4 expression was increased by PPARγ, we evaluated the effect of Spry4 on induction of epithelial differentiation. H157 and H2122 cells with stable Spry4 expression were grown in a 3-dimensional culture and observed for five days. Cell lines expressing Spry4 exhibited a change in morphologic architecture compared to cell lines expressing the empty vector, demonstrated by the restoration of an organized, spherical structure (Fig. 4A). E-cadherin is known to regulate the maintenance of an epithelial phenotype (30). Previous data have shown that E-cadherin is upregulated by Wnt7A/Fzd9 signaling and is involved in maintenance of an epithelial phenotype (18). Immuoblot data showed that expression of Spry4 leads to increased E-cadherin protein expression in H157 and H2122 cell lines compared to empty vector controls (Fig. 4B). In the context of Spry4 expression, markers of an epithelial state, KRT8 and KRT18, were increased by QPCR, while Vimentin, a marker of mesenchymal state, was decreased (Fig. 4C). This data suggests that Spry4 is involved in promoting the epithelial differentiation stimulated by Wnt7A/Fzd9 signaling.
Figure 4.

Spry4 re-expression induces a more epithelial phenotype in NSCLC cells. (A) 5,000 cells from H157 and H2122 stably expressing Spry4 or an empty vector control were seeded in media with 4% matrigel on top of a 1:1 matrigel and media base layer. Cell morphology was recorded on day 5. Data is representative of triplicate experiments. (B) Western blot analysis of E-cadherin in cell lysates from H157 and H2122 cell lines with stable Spry4 expression compared to an empty vector control. Loading control is β-actin and data are representative of triplicate experiments. (C) QPCR for KRT8, KRT18, and Vimentin in H157 and H2122 cell lines stably expressing Spry4 compared to an empty vector control. Results are the average of triplicate experiments and are normalized to GAPDH.
Spry4 reduces migration and invasion in NSCLC and reduces MMP-9 activity
Spry4 has been observed to inhibit migration in prostate, pancreatic cells, and endothelial cells, but effects of Spry4 on NSCLC migration and invasion have not been investigated (7, 31, 32). To assess the effects of Spry4 on cell motility, we performed a scratch assay. A 3mm space was created on confluent plates of H157 and H2122 cells expressing Spry4 or the empty vector. Cells were treated at time zero with 1ug/ml mitomycin to inhibit proliferation and the experiment was conducted in triplicate. The cells were photographed at 24 and 48 hours to record movement of cells into the space created by the lesion. H157 and H2122 cells expressing Spry4 exhibited reduced motility compared to empty vector transfected cell lines (Fig. 5A). Invasiveness of cells with stable Spry4 expression was examined using a 3-dimensional collagen/matrigel matrix, as depicted in figure 5B. In a chambered glass slide, a layer of 1:1 collagen to matrigel was applied, followed by a mix of cells, media, and matrigel (33). Photographs of cells were taken from above the wells, where cell extensions into the lower collagen layer can be observed. After five days of growth, Spry4 expressing cell lines demonstrated reduced invasion into the collagen/matrigel matrix compared to empty vector transfected cell lines (Fig. 5B). The reduction of cell mobility and invasion demonstrated with Spry4 expression suggests that Spry4 may act on downstream targets to inhibit metastasis.
Figure 5.


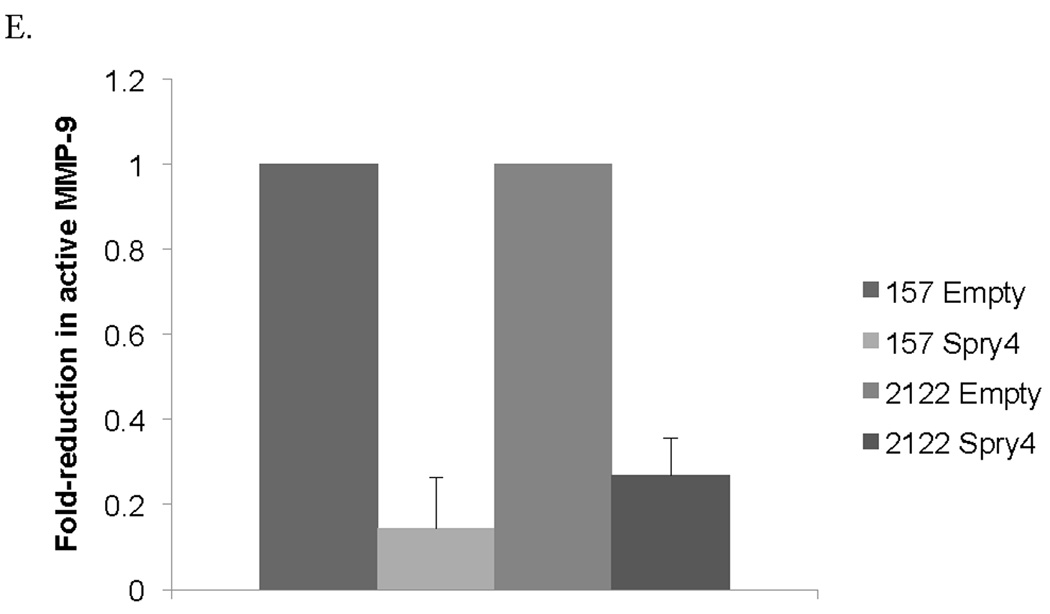
Re-expression of Spry4 inhibits migration and invasion in NSCLC cells. (A) A 3mm space was created across the diameter of plates of H157 and H2122 cells stably expressing Spry4 or an empty vector control and migration was recorded at 24 and 48 hours. Results are representative of triplicate experiments. (B) The 3D invasion assay uses a layer of collagen beneath a layer of matrigel and media containing the cells of interest. 5,000 cells from H157 and H2122 stably expressing Spry4 or an empty vector control were seeded in triplicate in media with 4% matrigel on top of a 1:1 matrigel and collagen base layer. Cell invasion was recorded on day 5. (C) Expression levels of CD82 and TIMP1 were assessed by QPCR in H157 and H2122 cell lines stably expressing Spry4 or an empty vector control. Results are the average of triplicate experiments normalized to GAPDH. (D) Increased expression of CD82 was confirmed by western blot analysis of cell lysates from H157 and H2122 cell lines stably expressing Spry4 compared to an empty vector control. Loading control is β-actin. (e) H157 cells stably expressing Spry4, H2122 cells stably expressing Spry4, and corresponding empty vector controls were seeded in a 96-well plate and analyzed with the MMP-9 Human Biotrak Assay. Absorbance was measured and a standard curve was used to calculate the amount of active MMP-9 in the samples. Results are the average of triplicate experiments with standard error.
We were also interested in previously unidentified targets of Spry4 that might be related to migration or invasion. CD82 is a tetraspanin cell surface glycoprotein identified based on its function as a migration suppressor independent of tumor growth and has been identified as a marker of good prognosis in NSCLC (34, 35). We were interested in a possible role for CD82 in the inhibition of migration and invasion observed with Spry4 re-expression. Spry4 expression resulted in increased CD82 mRNA levels and increased CD82 protein expression by immunoblot (Fig. 5C,D). CD82 mRNA expression was decreased by QPCR in 11 NSCLC cell lines compared to compared to non-transformed B2B cell line (data not shown). Nine of those cells lines also had decreased expression of Spry4 compared to B2B, suggesting a correlation between reduced levels of CD82 and Spry4. CD82 has been associated with reduced MMP-9 protein and enzymatic activity in NSCLC (36). Levels of active MMP-9 were assayed using an ELISA kit and were found to be decreased in Spry4 stably expressing H157 and H2122 cells compared to empty vector controls (Fig. 5E). Reduced activity of MMP-9 was also observed in the H157 cell line with re-expressed Wnt7A and Fzd9 (data not shown). TIMP1, a negative regulator of MMP-9, was upregulated by QPCR and has been suggested as the link between CD82 and decreased MMP-9 activity (Fig. 5C) (36). These targets of Spry4 may be responsible for the reduction of migration and invasion that was observed with Spry4 re-expression.
Knockdown of Spry4 expression by shRNA leads to reversed expression of EMT markers and increased cell growth
To assess the effects of loss of Spry4 expression on a non-transformed lung epithelial cell line, B2B cells were stably transfected with shRNA or an shRNA negative control. Reduction of Spry4 expression was confirmed by immunoblot and QPCR (Fig. 6 A,B). Expression of Spry4 shRNA led to decreased expression of epithelial markers KRT8 and KRT18, decreased expression of migration suppressor CD82, and decreased expression of growth suppressors p53 and p21 (Fig. 6B). Increased expression of mesenchymal marker Vimentin was also observed with Spry4 knockdown (Fig. 6B). In a cell growth assay, loss of Spry4 expression by shRNA led to increased cell growth of B2B cells compared to a negative control. Increased cell growth was observed by an MTS cell proliferation assay when Spry4 expression was knocked down by shRNA in H157 and H2122 cell lines stably expressing Wnt7a/Fzd9 (Fig. 6C). In the context of Wnt7a/Fzd9 expression in lung epithelial cells, Spry4 expression is important for the inhibition of cell growth. These data suggest that loss of Spry4 expression in lung epithelial cells could be one step leading to transformation or increased migration and invasion.
Figure 6.


Treatment of a non-transformed lung epithelial cell line, Beas2B (B2B), with shRNA to Spry4 leads to changes in EMT markers and increased cell growth. (A) Immunoblot for expression of Spry4 and E-cadherin in B2B cells treated with shRNA to Spry4. Loading control is βactin. (B) QPCR was used to measure Spry4, KRT8, KRT18, CD82, p53, p21, and Vimentin mRNA levels in B2B cells treated with Spry4 shRNA. Results are the average of triplicate experiments normalized to GAPDH. (C) 20,000 cells from B2B cells treated with shRNA to Spry4 or an shRNA control were plated in 6 wells and one well was counted each day. Data shown is the average of triplicate experiments. (D) 500 cells from each cell line were seeded per well in triplicate. At 24, 48, and 72 hours, 20ul of MTS reagent (Promega) was added to each well, incubated for 1 hour at 37C, and analyzed at 490nm. Results are presented as the average of triplicate experiments.
Discussion
In this study, we were interested in identifying regulators and targets of Spry4 in NSCLC, about which little was previously known. In a non-canonical Wnt signaling pathway, Wnt7A and Fzd9 have previously been shown to act in a tumor suppressive manner to reduce cell growth and promote epithelial cell differentiation in NSCLC (17). Wnt7A/Fzd9 activates PPARγ through an ERK5-dependent pathway and signals to downstream targets such as E-cadherin (18). Based on our study, Spry4 appears to act as a tumor suppressor in NSCLC. Spry4 promoter activity is increased by PPARγ, a downstream target of Wnt7A/Fzd9 signaling, and affects cell growth, differentiation, motility, and invasion. Figure 7 depicts our proposed signaling model for the Wnt7a and Spry4 pathway. Identification of changes in the expression of genes known to be important in tumor suppression, differentiation, metastasis, and invasion supports the biologic effects observed with Spry4 re-expression. Of potential clinical significance is our observation that Spry4 is frequently lost in NSCLC and dysplastic lung epithelial cells.
Figure 7.
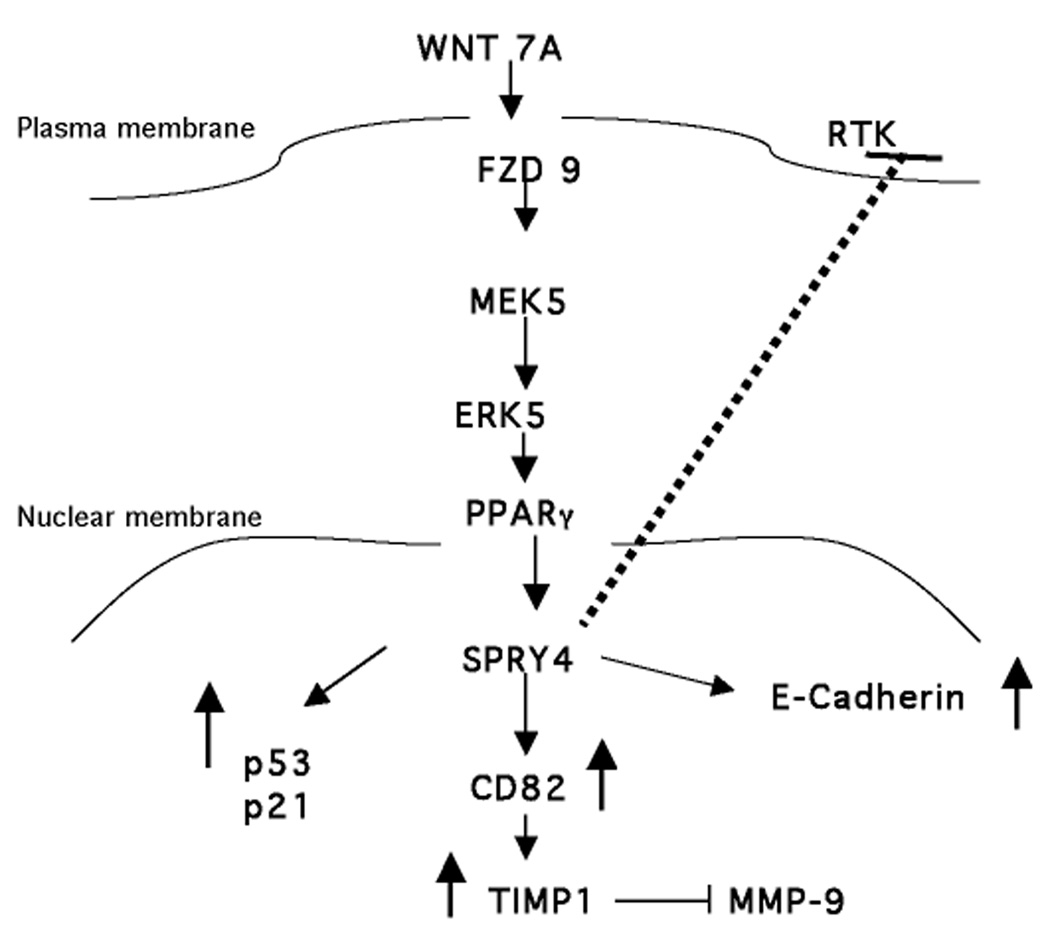
A model for Wnt7A/Fzd9 activation of Spry4 through PPARγ. Expression of Spry4 results in decreased MMP-9 activity, through CD82 and TIMP1, and increased E-cadherin, p21, and p53 expression.
The regulation of Spry4 expression is still largely undetermined. Tyrosine phosphorylation induced by growth factors has been identified as a positive regulator of Spry, though the phosphorylation is specific to cell type, growth factor, and individual Spry (37). Spry4 can be induced by FGF, EGF, and PDGF, but unlike Spry1 and Spry2, it does not appear to be phosphorylated in response to growth factor treatment (37, 38). However, phosphorylation of mouse Spry4 at Tyr53 is required for inhibition of FGF induced ERK activation (27). We have demonstrated that reintroduction of the Wnt7A/Fzd9 signaling pathway in NSCLC cell lines leads to increased activity of the Spry4 promoter. Loss of Spry4 expression by shRNA in cell lines with stable Wnt7a/Fzd9 expression led to increased cell growth, further strengthening the signaling link between Wnt7A and Spry4. Our data also demonstrate that Spry4 is regulated by PPARγ signaling, a novel mode of regulation of Spry4 expression outside of the RTK pathway. We have shown that truncation of the Spry4 promoter leads to reduced activation of Spry4-luc in PPARγ expressing cell lines, however, no consensus site for PPARγ exists within the promoter region. There is one half DR1 binding site near −1600bp but its functionality is unknown. Analysis of the Spry4 promoter region has identified many transcription factor binding sites, suggesting complicated regulation of the Spry4 promoter (39). In our study, the observation of a more gradual decrease in activity, as opposed to a drastic change with a single truncation, suggests that there may be multiple factors working in collaboration to regulate the Spry4 promoter. Further functional analysis is required to determine the regulatory connection between PPARγ and Spry4.
Inhibition of FGF-induced activation of the ERK pathway by Spry4 can occur through binding with Sos1 and is enhanced by heterodimerization with Spry1 (40). In the VEGF pathway, Spry4 can bind to Raf1 and inhibit Ras-independent inhibition of ERK activation (5). Spry4 has been shown to interact with testicular protein kinase 1 (TESK1) and lead to inhibition of integrin-mediated cell spreading (41). Beyond these examples, the targets of Spry4 are largely unknown. We have demonstrated that Spry4 re-expression leads to increased expression of tumor suppressors p53 and p21. p53 is a well-known negative growth regulator and is frequently lost in NSCLC (42). p21 negatively regulates cell cycle progression and loss of p21 results in the failure of cells to undergo cell cycle arrest in response to p53 activation (43). Upregulation of these tumor suppressors may be related to the observed decrease in cell proliferation and anchorage independent growth with Spry4 expression.
We observed increased expression of CD82 and TIMP1 and decreased active MMP-9 levels in cells expressing Spry4. We have also observed that active MMP-9 is decreased in H157 cells with stable Wnt7A/Fzd9 expression, though not as significantly; this provides further evidence of a signaling link between Spry4 and Wnt7A. CD82 is ubiquitously expressed in normal tissues and is downregulated in a variety of cancers; in lung cancer, CD82 expression has been associated with good prognosis and decreased metastasis and invasive potential (34, 35). In an orthotopic lung tumor model, CD82 dramatically reduced lymph node metastases (44). CD82 has been shown to upregulate TIMP1 in NSCLC, leading to suppression of invasion and metastases by inactivating MMP-9 (36). These previous studies support a role for CD82 in inhibition of invasion and metastasis in our study. Additionally, the CD82 promoter contains a binding motif for p53, which may act synergistically with junB to induce CD82 promoter activity (45). Increased levels of p53 in our study may be leading to the increased levels of CD82 and subsequently the decreased migration and invasion.
A mesenchymal to epithelial transition mediated by Wnt7A/Fzd9 may be acting through Spry4 (17). In normal cells, cadherin-dependent contacts between cells promote proper development and maintenance of normal epithelia (30). Epithelial tumor cells gradually lose their normal phenotype and acquire more mesenchymal characteristics, with increased motility and invasion (46). Our findings support a role for Spry4 as a downstream target of Wnt7A/Fzd9 in the mesenchymal to epithelial transition; Spry4 expression induced a more differentiated epithelial phenotype in a 3-dimensional culture, induced expression of E-cadherin, KRT8, and KRT18, and suppressed migration and invasion. It is important to note that, although Spry4 is a key mediator of the effects of Wnt7A/Fzd9 pathway, the pathway is known to signal to other targets which play a role in its biologic effects (47–49).
Spry proteins have been found to participate in multiple pathways related to RTK signaling, dependent upon cellular context and the specific RTK. Sprys have been shown to antagonize the Ras-ERK-MAPK pathway by inhibiting activation of Ras and Raf1 or to agonize EGF-mediated ERK MAPK signaling through an interaction with c-CBL (3). Spry4 however, does not bind c-CBL and thus, has not been linked to agonist activity (37). EGFR inhibition by Spry4 was observed in one study using NIH3T3 cells, but not in several other studies (6, 26, 27, 40). In mouse studies, overexpression of Spry4 led to severe pulmonary hypoplasia and loss of Spry4 led to sustained ERK signaling in response to FGF (26, 50). The canonical Wnt/β-catenin signaling pathway has been shown to interact with FGF signaling at GSK3β (51). However, Wnt7A/Fzd9 signaling does not activate β-catenin signaling, so any interaction with growth factor pathways would occur through an alternate pathway (17, 52). It may be possible that the Wnt7A/Fzd9 pathway is interacting with growth factor signaling pathways through Spry4 in a manner opposite of the canonical Wnt/β-catenin pathway, leading to tumor suppression instead of promotion. Spry4 could also be activated independent of RTK inhibition depending on the context. Studies are necessary to clarify the range of RTK inhibition by Spry4, effects of possible dimerization between Sprys on RTK inhibition, and potential interaction between Wnt and growth factor signaling, all in the context of NSCLC.
Loss of Spry4 expression by shRNA in a non-transformed lung epithelial cell line led to increased cell growth and decreased expression of epithelial markers in our study. These data suggest that Spry4 may influence the transformation of normal lung epithelial cells and may also suppress EMT in the lung epithelium. Spry4 is lost in NSCLC cells and dysplastic lung epithelial cells lines, similar to Wnt7A, however, the timing of loss is unknown (53, 54). Methylation of Spry4 has been detected in prostate cancer tissue (7). Wnt5a, another non-canonical, tumor-suppressing Wnt, is methylated in colorectal cancer and leukemia, suggesting the possibility that Wnt7A may also be methylated in NSCLC (55, 56). Further research needs to be done to establish whether loss of Wnt7A and Spry4 are early or late events in lung tumorigenesis and what the mechanism of loss is. We have presented data suggesting that Spry4 is downstream of PPARγ in the Wnt7A/Fzd9 tumor suppression pathway and that it acts to reduce cell growth, inhibit anchorage independent growth, suppress migration and invasion, and restore a non-transformed epithelial phenotype. Pharmacological activation of Spry4 may be a future target for treatment of NSCLC.
Acknowledgements
This work was supported by a Merit Award from the United States Department of Veterans Affairs, NIH grant #K22CA113700, NIH SPORE in Lung Cancer program Career Development Award #46345-AEFCCTR, and an NIH T32 Training Grant through the Cardiovascular Pulmonary Program of the University of Colorado Denver. The Spry4 luciferase −31 was constructed by the UCD DERC Molecular Biology Core: NIH P30DK57516.
Abbreviations
- B2B
Beas2B
- Fzd9
Frizzled 9
- NSCLC
Non-small Cell Lung Cancer
- PPARγ
Peroxisome Proliferator Activated Receptor γ
- PPRE
PPAR Response Element
- QPCR
Quantitative PCR
- Spry4
Sprouty-4
References
- 1.Hacohen N, Kramer S, Sutherland D, Hiromi Y, Krasnow MA. sprouty encodes a novel antagonist of FGF signaling that patterns apical branching of the Drosophila airways. Cell. 1998;92(2):253–263. doi: 10.1016/s0092-8674(00)80919-8. [DOI] [PubMed] [Google Scholar]
- 2.Casci T, Vinos J, Freeman M. Sprouty, an intracellular inhibitor of Ras signaling. Cell. 1999;96(5):655–665. doi: 10.1016/s0092-8674(00)80576-0. [DOI] [PubMed] [Google Scholar]
- 3.Mason JM, Morrison DJ, Basson MA, Licht JD. Sprouty proteins: multifaceted negative-feedback regulators of receptor tyrosine kinase signaling. Trends Cell Biol. 2006;16(1):45–54. doi: 10.1016/j.tcb.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 4.Gross I, Bassit B, Benezra M, Licht JD. Mammalian sprouty proteins inhibit cell growth and differentiation by preventing ras activation. J Biol Chem. 2001;276(49):46460–46468. doi: 10.1074/jbc.M108234200. [DOI] [PubMed] [Google Scholar]
- 5.Sasaki A, Taketomi T, Kato R, et al. Mammalian Sprouty4 suppresses Ras-independent ERK activation by binding to Raf1. Nat Cell Biol. 2003;5(5):427–432. doi: 10.1038/ncb978. [DOI] [PubMed] [Google Scholar]
- 6.Leeksma OC, Van Achterberg TA, Tsumura Y, et al. Human sprouty 4, a new ras antagonist on 5q31, interacts with the dual specificity kinase TESK1. Eur J Biochem. 2002;269(10):2546–2556. doi: 10.1046/j.1432-1033.2002.02921.x. [DOI] [PubMed] [Google Scholar]
- 7.Wang J, Thompson B, Ren C, Ittmann M, Kwabi-Addo B. Sprouty4, a suppressor of tumor cell motility, is down regulated by DNA methylation in human prostate cancer. Prostate. 2006;66(6):613–624. doi: 10.1002/pros.20353. [DOI] [PubMed] [Google Scholar]
- 8.Lo TL, Fong CW, Yusoff P, et al. Sprouty and cancer: the first terms report. Cancer Lett. 2006;242(2):141–150. doi: 10.1016/j.canlet.2005.12.032. [DOI] [PubMed] [Google Scholar]
- 9.Minowada G, Miller YE. Overexpression of Sprouty 2 in mouse lung epithelium inhibits urethane-induced tumorigenesis. Am J Respir Cell Mol Biol. 2009;40(1):31–37. doi: 10.1165/rcmb.2008-0147OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sutterluty H, Mayer CE, Setinek U, et al. Down-regulation of Sprouty2 in non-small cell lung cancer contributes to tumor malignancy via extracellular signal-regulated kinase pathway-dependent and -independent mechanisms. Mol Cancer Res. 2007;5(5):509–520. doi: 10.1158/1541-7786.MCR-06-0273. [DOI] [PubMed] [Google Scholar]
- 11.Shaw AT, Meissner A, Dowdle JA, et al. Sprouty-2 regulates oncogenic K-ras in lung development and tumorigenesis. Genes Dev. 2007;21(6):694–707. doi: 10.1101/gad.1526207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giles RH, van Es JH, Clevers H. Caught up in a Wnt storm: Wnt signaling in cancer. Biochim Biophys Acta. 2003;1653(1):1–24. doi: 10.1016/s0304-419x(03)00005-2. [DOI] [PubMed] [Google Scholar]
- 13.Howe LR, Brown AM. Wnt signaling and breast cancer. Cancer Biol Ther. 2004;3(1):36–41. doi: 10.4161/cbt.3.1.561. [DOI] [PubMed] [Google Scholar]
- 14.Austinat M, Dunsch R, Wittekind C, Tannapfel A, Gebhardt R, Gaunitz F. Correlation between beta-catenin mutations and expression of Wnt-signaling target genes in hepatocellular carcinoma. Mol Cancer. 2008;7(1):21. doi: 10.1186/1476-4598-7-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dahl C, Guldberg P. The genome and epigenome of malignant melanoma. APMIS. 2007;115(10):1161–1176. doi: 10.1111/j.1600-0463.2007.apm_855.xml.x. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki H, Toyota M, Caraway H, et al. Frequent epigenetic inactivation of Wnt antagonist genes in breast cancer. Br J Cancer. 2008 doi: 10.1038/sj.bjc.6604259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Winn RA, Marek L, Han SY, et al. Restoration of Wnt-7a expression reverses non-small cell lung cancer cellular transformation through frizzled-9-mediated growth inhibition and promotion of cell differentiation. J Biol Chem. 2005;280(20):19625–19634. doi: 10.1074/jbc.M409392200. [DOI] [PubMed] [Google Scholar]
- 18.Winn RA, Van Scoyk M, Hammond M, et al. Antitumorigenic effect of Wnt 7a and Fzd 9 in non-small cell lung cancer cells is mediated through ERK-5-dependent activation of peroxisome proliferator-activated receptor gamma. J Biol Chem. 2006;281(37):26943–26950. doi: 10.1074/jbc.M604145200. [DOI] [PubMed] [Google Scholar]
- 19.Michalik L, Desvergne B, Wahli W. Peroxisome-proliferator-activated receptors and cancers: complex stories. Nat Rev Cancer. 2004;4(1):61–70. doi: 10.1038/nrc1254. [DOI] [PubMed] [Google Scholar]
- 20.Bren-Mattison Y, Van Putten V, Chan D, Winn R, Geraci MW, Nemenoff RA. Peroxisome proliferator-activated receptor-gamma (PPAR(gamma)) inhibits tumorigenesis by reversing the undifferentiated phenotype of metastatic non-small-cell lung cancer cells (NSCLC) Oncogene. 2005;24(8):1412–1422. doi: 10.1038/sj.onc.1208333. [DOI] [PubMed] [Google Scholar]
- 21.Tsubouchi Y, Sano H, Kawahito Y, et al. Inhibition of human lung cancer cell growth by the peroxisome proliferator-activated receptor-gamma agonists through induction of apoptosis. Biochem Biophys Res Commun. 2000;270(2):400–405. doi: 10.1006/bbrc.2000.2436. [DOI] [PubMed] [Google Scholar]
- 22.Chang TH, Szabo E. Induction of differentiation and apoptosis by ligands of peroxisome proliferator-activated receptor gamma in non-small cell lung cancer. Cancer Res. 2000;60(4):1129–1138. [PubMed] [Google Scholar]
- 23.Leung WK, Bai AH, Chan VY, et al. Effect of peroxisome proliferator activated receptor gamma ligands on growth and gene expression profiles of gastric cancer cells. Gut. 2004;53(3):331–338. doi: 10.1136/gut.2003.021105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xin B, Yokoyama Y, Shigeto T, Futagami M, Mizunuma H. Inhibitory effect of meloxicam, a selective cyclooxygenase-2 inhibitor, and ciglitazone, a peroxisome proliferator-activated receptor gamma ligand, on the growth of human ovarian cancers. Cancer. 2007;110(4):791–800. doi: 10.1002/cncr.22854. [DOI] [PubMed] [Google Scholar]
- 25.Osawa E, Nakajima A, Wada K, et al. Peroxisome proliferator-activated receptor gamma ligands suppress colon carcinogenesis induced by azoxymethane in mice. Gastroenterology. 2003;124(2):361–367. doi: 10.1053/gast.2003.50067. [DOI] [PubMed] [Google Scholar]
- 26.Taniguchi K, Ayada T, Ichiyama K, et al. Sprouty2 and Sprouty4 are essential for embryonic morphogenesis and regulation of FGF signaling. Biochem Biophys Res Commun. 2007;352(4):896–902. doi: 10.1016/j.bbrc.2006.11.107. [DOI] [PubMed] [Google Scholar]
- 27.Sasaki A, Taketomi T, Wakioka T, Kato R, Yoshimura A. Identification of a dominant negative mutant of Sprouty that potentiates fibroblast growth factor- but not epidermal growth factor-induced ERK activation. J Biol Chem. 2001;276(39):36804–36808. doi: 10.1074/jbc.C100386200. [DOI] [PubMed] [Google Scholar]
- 28.Zhang S, Lin Y, Itaranta P, Yagi A, Vainio S. Expression of Sprouty genes 1, 2 and 4 during mouse organogenesis. Mech Dev. 2001;109(2):367–370. doi: 10.1016/s0925-4773(01)00526-3. [DOI] [PubMed] [Google Scholar]
- 29.Coopman PJ, Mueller SC. The Syk tyrosine kinase: a new negative regulator in tumor growth and progression. Cancer Lett. 2006;241(2):159–173. doi: 10.1016/j.canlet.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 30.Acloque H, Thiery JP, Nieto MA. The physiology and pathology of the EMT. Meeting on the epithelial-mesenchymal transition. EMBO Rep. 2008;9(4):322–326. doi: 10.1038/embor.2008.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lee SH, Schloss DJ, Jarvis L, Krasnow MA, Swain JL. Inhibition of angiogenesis by a mouse sprouty protein. J Biol Chem. 2001;276(6):4128–4133. doi: 10.1074/jbc.M006922200. [DOI] [PubMed] [Google Scholar]
- 32.Jaggi F, Cabrita MA, Perl AK, Christofori G. Modulation of endocrine pancreas development but not beta-cell carcinogenesis by Sprouty4. Mol Cancer Res. 2008;6(3):468–482. doi: 10.1158/1541-7786.MCR-07-0255. [DOI] [PubMed] [Google Scholar]
- 33.Xiang B, Muthuswamy SK. Using three-dimensional acinar structures for molecular and cell biological assays. Methods Enzymol. 2006;406:692–701. doi: 10.1016/S0076-6879(06)06054-X. [DOI] [PubMed] [Google Scholar]
- 34.Liu WM, Zhang XA. KAI1/CD82, a tumor metastasis suppressor. Cancer Lett. 2006;240(2):183–194. doi: 10.1016/j.canlet.2005.08.018. [DOI] [PubMed] [Google Scholar]
- 35.Tonoli H, Barrett JC. CD82 metastasis suppressor gene: a potential target for new therapeutics? Trends Mol Med. 2005;11(12):563–570. doi: 10.1016/j.molmed.2005.10.002. [DOI] [PubMed] [Google Scholar]
- 36.Jee BK, Park KM, Surendran S, et al. KAI1/CD82 suppresses tumor invasion by MMP9 inactivation via TIMP1 up-regulation in the H1299 human lung carcinoma cell line. Biochem Biophys Res Commun. 2006;342(2):655–661. doi: 10.1016/j.bbrc.2006.01.153. [DOI] [PubMed] [Google Scholar]
- 37.Mason JM, Morrison DJ, Bassit B, et al. Tyrosine phosphorylation of Sprouty proteins regulates their ability to inhibit growth factor signaling: a dual feedback loop. Mol Biol Cell. 2004;15(5):2176–2188. doi: 10.1091/mbc.E03-07-0503. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 38.Ozaki K, Kadomoto R, Asato K, Tanimura S, Itoh N, Kohno M. ERK pathway positively regulates the expression of Sprouty genes. Biochem Biophys Res Commun. 2001;285(5):1084–1088. doi: 10.1006/bbrc.2001.5295. [DOI] [PubMed] [Google Scholar]
- 39.Ding W, Bellusci S, Shi W, Warburton D. Genomic structure and promoter characterization of the human Sprouty4 gene, a novel regulator of lung morphogenesis. Am J Physiol Lung Cell Mol Physiol. 2004;287(1):L52–L59. doi: 10.1152/ajplung.00430.2003. [DOI] [PubMed] [Google Scholar]
- 40.Ozaki K, Miyazaki S, Tanimura S, Kohno M. Efficient suppression of FGF-2-induced ERK activation by the cooperative interaction among mammalian Sprouty isoforms. J Cell Sci. 2005;118(Pt 24):5861–5871. doi: 10.1242/jcs.02711. [DOI] [PubMed] [Google Scholar]
- 41.Tsumura Y, Toshima J, Leeksma OC, Ohashi K, Mizuno K. Sprouty-4 negatively regulates cell spreading by inhibiting the kinase activity of testicular protein kinase. Biochem J. 2005;387(Pt 3):627–637. doi: 10.1042/BJ20041181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robles AI, Linke SP, Harris CC. The p53 network in lung carcinogenesis. Oncogene. 2002;21(45):6898–6907. doi: 10.1038/sj.onc.1205563. [DOI] [PubMed] [Google Scholar]
- 43.Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005;65(10):3980–3985. doi: 10.1158/0008-5472.CAN-04-3995. [DOI] [PubMed] [Google Scholar]
- 44.Takeda T, Hattori N, Tokuhara T, Nishimura Y, Yokoyama M, Miyake M. Adenoviral transduction of MRP-1/CD9 and KAI1/CD82 inhibits lymph node metastasis in orthotopic lung cancer model. Cancer Res. 2007;67(4):1744–1749. doi: 10.1158/0008-5472.CAN-06-3090. [DOI] [PubMed] [Google Scholar]
- 45.Marreiros A, Dudgeon K, Dao V, et al. KAI1 promoter activity is dependent on p53, junB and AP2: evidence for a possible mechanism underlying loss of KAI1 expression in cancer cells. Oncogene. 2005;24(4):637–649. doi: 10.1038/sj.onc.1208216. [DOI] [PubMed] [Google Scholar]
- 46.Boyer B, Valles AM, Edme N. Induction and regulation of epithelial-mesenchymal transitions. Biochem Pharmacol. 2000;60(8):1091–1099. doi: 10.1016/s0006-2952(00)00427-5. [DOI] [PubMed] [Google Scholar]
- 47.Xu Y, Farmer SR, Smith BD. Peroxisome proliferator-activated receptor gamma interacts with CIITA × RFX5 complex to repress type I collagen gene expression. J Biol Chem. 2007;282(36):26046–26056. doi: 10.1074/jbc.M703652200. [DOI] [PubMed] [Google Scholar]
- 48.Bren-Mattison Y, Meyer AM, Van Putten V, et al. Antitumorigenic effects of peroxisome proliferator-activated receptor-gamma in non-small-cell lung cancer cells are mediated by suppression of cyclooxygenase-2 via inhibition of nuclear factor-kappaB. Mol Pharmacol. 2008;73(3):709–717. doi: 10.1124/mol.107.042002. [DOI] [PubMed] [Google Scholar]
- 49.Lee SY, Hur GY, Jung KH, et al. PPAR-gamma agonist increase gefitinib's antitumor activity through PTEN expression. Lung Cancer. 2006;51(3):297–301. doi: 10.1016/j.lungcan.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 50.Perl AK, Hokuto I, Impagnatiello MA, Christofori G, Whitsett JA. Temporal effects of Sprouty on lung morphogenesis. Dev Biol. 2003;258(1):154–168. doi: 10.1016/s0012-1606(03)00106-4. [DOI] [PubMed] [Google Scholar]
- 51.Katoh M. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer Biol Ther. 2006;5(9):1059–1064. doi: 10.4161/cbt.5.9.3151. [DOI] [PubMed] [Google Scholar]
- 52.Karasawa T, Yokokura H, Kitajewski J, Lombroso PJ. Frizzled-9 is activated by Wnt-2 and functions in Wnt/beta -catenin signaling. J Biol Chem. 2002;277(40):37479–37486. doi: 10.1074/jbc.M205658200. [DOI] [PubMed] [Google Scholar]
- 53.Calvo R, West J, Franklin W, et al. Altered HOX and WNT7A expression in human lung cancer. Proc Natl Acad Sci U S A. 2000;97(23):12776–12781. doi: 10.1073/pnas.97.23.12776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kirikoshi H, Katoh M. Expression of WNT7A in human normal tissues and cancer, and regulation of WNT7A and WNT7B in human cancer. Int J Oncol. 2002;21(4):895–900. [PubMed] [Google Scholar]
- 55.Ying J, Li H, Yu J, et al. WNT5A exhibits tumor-suppressive activity through antagonizing the Wnt/beta-catenin signaling, and is frequently methylated in colorectal cancer. Clin Cancer Res. 2008;14(1):55–61. doi: 10.1158/1078-0432.CCR-07-1644. [DOI] [PubMed] [Google Scholar]
- 56.Roman-Gomez J, Jimenez-Velasco A, Cordeu L, et al. WNT5A, a putative tumour suppressor of lymphoid malignancies, is inactivated by aberrant methylation in acute lymphoblastic leukaemia. Eur J Cancer. 2007;43(18):2736–2746. doi: 10.1016/j.ejca.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 57.Debnath J, Muthuswamy SK, Brugge JS. Morphogenesis and oncogenesis of MCF-10A mammary epithelial acini grown in three-dimensional basement membrane cultures. Methods. 2003;30(3):256–268. doi: 10.1016/s1046-2023(03)00032-x. [DOI] [PubMed] [Google Scholar]