

Abstract
The ability to localize agents to specific anatomic sites remains an important aspect in designing more efficient therapeutics. Light-activated therapies, in particular, allow for the focal ablation of target tissues and cells. In order to increase the specificity of these agents, stimuli-activated systems have been developed, which are non-phototoxic in the absence of activation. To this end, we propose a novel paradigm for excited state quenching and activation based upon the direct conjugation of quenching moieties to the porphyrinic macrocycle. Model compounds, based upon meso-(p-aminophenyl)porphyrins were synthesized bearing 1 to 4 sulfonamide-linked 2,4-dinitrobenzene. The singlet oxygen and fluorescence quantum yields of these compounds were obtained and compared, as well as the kinetics of activation with relevant activating agents. In addition, methods were developed to further modify the porphyrin in order to modulate the polarity and effect conjugation to biomolecules or nanoparticulate scaffolds. These systems may prove useful in the treatment of a number of disease states, such as cancer and bacterial infection.
INTRODUCTION
A plethora of methodologies have been developed in order to increase the efficacy of therapeutic regimes, while decreasing extraneous toxicity. One technique that has found clinical utility in the treatment of cancers1-3 and a number of other non-malignant ailments, such as age related macular degeneration,1-3 and cardiovascular disease,4,5 is based upon the photosensitized destruction of target tissues. While light-activated therapies can be highly efficacious, due to their focal nature, they suffer from drawbacks, such as photosensitivity and the limited penetration depth of light through heterogeneous tissues. A number of approaches have been investigated to overcome these issues, including the targeting of photosensitizers (PS) to tissues of interest with peptides, antibodies, and nanoparticulate delivery vehicles.6-13
Recently, a novel strategy utilizing activatable photosensitizers has been posited.8,13-17 In these systems the excited states of the porphyrinic chromophore are quenched via electron- or energy-transfer processes, and can be regained in the presence of certain enzymes or by preferential localization. We have previously reported the synthesis of two different activatable systems based upon aggregation induced quenching. In the first example, a polymeric nanoparticle preparation in which the potent photosensitizer, meso-tetraphenylporpholactol, was rendered non-phototoxic upon encapsulation.13 When incubated with lipid or cells, the photosensitizer was released and regained its phototoxicity. In vivo, this preparation was able to eradicate cancer in an entire cohort of mice. In the second system, multiple chlorin e6 chromophores were conjugated to a polylysine graft co-polymer backbone which is readily cleaved by Cathepsin B, a tumor-associated protease.18 Upon conjugation, the dyes exhibited an 86% decrease in fluorescence, and an 80% decrease in singlet oxygen generation, which could be regained by incubation with the protease.
One other strategy that has been investigated is the conjugation of quencher molecules to the porphyrinic macrocycle via linker moieties that do not participate in the π-conjugation of the chromophore.14-17 A number of quenchers have been investigated, including metalloporphyrins, dye molecules, or antioxidants. In some of these systems, the quencher molecule is separated from the chromophore by an enzyme cleavable linker. This allows for the spatial control of photosensitizer activation, as the construct will remain essentially non-fluorescent and non-phototoxic until it reaches the site of interest and is activated. These systems are highly useful in the treatment of diseases in which specific enzymes are upregulated, as compared to their expression in healthy tissues.
Analogous to the development of activatable photosensitizers, much research has been accomplished with regard to the development of activatable fluorescent probes, from which novel techniques can be learned and applied to porphyrinic systems. A number of groups have described the synthesis of fluorescent probes that are activated by thiols.19-21 The potential utility of these agents is that thiols represent a class of molecules responsible for the maintenance of cellular redox homeostasis, which may be altered in cases of disease or infection. In particular, the relative levels of thiols such as glutathione, cysteine, and homocysteine and their oxidized disulfide forms have been implicated in the development of specific diseases, including Alzheimer's, cancer, and cardiovascular disease.22-25 In their construct, the authors utilized a classic donor-π-acceptor architecture based upon the formation of a thiol-cleavable arenesulfonamide. The resulting probe exhibited superb stability in the presence of oxygen or nitrogen nucleophiles, yet was readily cleaved by thiols, such as cysteine, glutathione, and dithiothreitol. This probe demonstrated a 120-fold increase in fluorescence upon activation, and serves as a potential model for activatable photosensitizers.
Herein, we report a novel paradigm for excited state quenching based upon the conjugation of 2,4-dinitrobenzene (DNB) to the periphery of meso-tetraphenylporphyrins via thiol-labile sulfonamide linkages. These model systems, derived from mono-, di-, tri-, and tetra(p-aminophenyl)porphyrin have been synthesized and their photophysical properties have been investigated. In addition, methods to further functionalize the amines with moieties that allow for the modulation of the polarity of the porphyrin or conjugation to biomolecules or nanoparticles have been developed. These photosensitizer systems, while imparting specificity for thiol-containing analytes, serve as a template on which to base future substrate-specific agents.
RESULTS AND DISCUSSION
Synthesis and characterization of sulfonamidophenylporphyrins (SPP)
meso-p-Aminophenylporphyrins bearing 1 through 4 anilines have previously been reported for use in a number of applications.26 These macrocycles can be synthesized using a variety of methodologies. For mono-, di-, and tri(p-aminophenyl)porphyrins (1, 2c, 2t, and 3, respectively, Scheme 1), the most facile synthesis involved the nitration of meso-tetraphenylporphyrin (TPP) with NaNO2, followed by reduction with SnCl2·2H2O.26 The four derivatives including the cis- and trans-isomers of 2 (2c and 2t) were readily synthesized by adding varying ratios of NaNO2 to a TFA solution of TPP. While it would have been optimal to utilize the same strategy to synthesize the tetra(p-aminophenyl)porphyrin derivative (4), it is exceedingly difficult to add more than three nitro-substituents to the meso-phenyl groups.26 Instead, the meso-tetra(p-aminophenyl)porphyrin was synthesized by the condensation of p-nitrobenzaldehyde with pyrrole in refluxing propionic acid, followed by SnCl2·2H2O reduction of the nitro groups to the corresponding amines.26
Scheme 1.

Synthesis of aromatic SPP.
The mono- and di-sulfonamidophenylporphyrins (SPP, 1SAM, 2cSAM, and 2tSAM, respectively, Scheme 1) were synthesized via microwave-assisted syntheses. The advantage of this method is that it allows for relatively short reaction times with moderate to good yields.14,27 Dissolution of 1, 2c, or 2t and excess 2,4-dinitrobenzenesulfonyl chloride (DNsCl) in a mixture of CHCl3/pyridine followed by microwave irradiation gave the mono-SPP 1SAM in 70% yield, the cis-di-SPP 2cSAM in 57% yield, and the trans-di-SPP 2tSAM in 81% yield.
Our initial attempts to synthesize the tri-SPP derivative (3SAM) via the microwave-mediated procedure described above failed due to solubility issues associated with the use of a large excess DNsCl, as 8 equivalents per amine were utilized and the reaction is limited to a small reaction volume (8 mL). Hence, a milder, room temperature method was utilized. A solution of porphyrin 3 in anhydrous pyridine was treated with DNsCl (10 equiv) and allowed to stir 22 h, at which time the resulting crude mixture was purified by column chromatography affording the desired tri-SPP (3SAM) in 78% yield (Scheme 1). The tetra-SPP derivative, 4SAM, was synthesized by the same procedure, and resulted in a 50% isolated yield.
The presence of the sulfonamide substituents in the products was confirmed by 1H NMR spectroscopy. In all cases, the spectra showed a characteristic singlet corresponding to the sulfonamide protons at approximately 11.5 ppm, with additional signals in the aromatic region for the protons of the dinitrobenzene. In addition, a peak at ~ -2 ppm was observed, which is typically associated with the two pyrrolic amine protons in the core of the free base porphyrin. While the cis- and trans- isomers of the di-SPP were synthesized from the respective purified aminophenylporpyrin derivatives,26 the products can be readily distinguished by looking at the symmetry of their respective spectra (see Figure S1). Both products, 2cSAM and 2tSAM, possess C2 symmetry, yet have different axes of symmetry. For the cis isomer, 2cSAM, the symmetry axis goes through the pyrrolic β protons, which results in the appearance of a singlet for four of the eight β protons. In the trans isomer, on the other hand, the symmetry axis is through the meso-phenyl position, resulting in doublets for all of the β protons. The tri- and tetrasulfonamide derivatives, 3SAM and 4SAM, were rather unremarkable aside from their C2 and C4 symmetries, respectively.
The UV-vis absorption spectra of the aminophenylporphyrins (Figure 1A) demonstrated decreasing extinction coefficients with increasing number of amine substituents in DMF (Table 1). This is likely due to the formation of aggregates, which are not observed when the porphyrins are dissolved in THF (Table S1). Concomitant with this decrease in extinction coefficient is a bathochromic shift of the absorption spectrum. This translates into red-shifted fluorescence emission spectra, with shifts up to 17 nm as compared to TPP (Figure 1B). Interestingly, the fluorescence quantum yields of the aminophenylporphyrins increase with increasing amination. Previous reports have described enhanced emission properties resulting from the aggregation of the porphyrinic dyes,28 and we believe that this may be the origin of these observations.
Figure 1.

Absorption spectra of (A) aminophenyl and (C) sulfonamidophenyl porphyrins; Normalized emission spectra of (B) aminophenyl and (D) sulfonamidophenyl porphyrins excited at the respective Soret band maxima (See Table 1 below for values. All spectra acquired in DMF).
Table 1.
Photophysical properties of synthesized porphyrins.a
| Compound | Soret (nm) | log ε (L mol-1cm-1) | Fluorescence emission (nm) | ΦFl | Φ Δ |
|---|---|---|---|---|---|
| TPP | 417 | 5.7 | 651 | 0.15 | 0.64 |
| 1 | 418 | 5.5 | 664 | 0.05 | 0.22 |
| 2c | 424 | 5.3 | 674 | 0.13 | 0.37 |
| 2t | 424 | 5.5 | 674 | 0.14 | 0.42 |
| 3 | 432 | 5 | 682 | 0.24 | 0.47 |
| 4 | 434 | 4.9 | 693 | 0.24 | 0.53 |
| 1SAM | 418 | 5.2 | 653 | 0.02 | 0.11 |
| 2cSAM | 420 | 5.2 | 652 | 0.01 | 0.01 |
| 2tSAM | 421 | 4.6 | 652 | 0 | 0.01 |
| 3SAM | 422 | 5.4 | 651 | 0.01 | 0.05 |
| 4SAM | 440 | 5.3 | 653 | 0.00 | 0.01 |
| 7 | 424 | 4.9 | 660 | 0.17 | 0.57 |
| 8 | 422 | 5.1 | 656 | 0.08 | 0.23 |
| 1SAM-TE | 417 | 5.5 | 651 | 0.05 | 0.16 |
| 1SAM-AA | 418 | 5.5 | 651 | 0.03 | 0.23 |
| 1SAM-PS | 418 | 5.2 | 650 | 0.05 | 0.23 |
| 1SAM-mPEG | 417 | 4.5 | 650 | 0.05 | 0.19 |
| 4SAM-mPEG | 420 | 5.1 | 651 | 0.02 | 0.07 |
| 4-mPEG | 437 | 5.3 | 686 | 0.3 | 0.61 |
All data were collected in DMF at room temperature.
Upon sulfonamide formation, the Soret bands of the porphyrins return to approximately the same wavelength as TPP (Figure 1C) with equivalent extinction coefficients (Table 1). The contribution of the DNB to the absorption spectrum is seen in the shape of the Q bands, as well as a broadened Soret band. The only product that does not follow this pattern is the tetrasulfonamide 4SAM, which is bathochromically shifted with a severely broadened Soret band due to decreased solubility in DMF and aggregation. In all cases, the porphyrins exhibited excited state quenching, with negligible fluorescence emission or singlet oxygen generation.
Synthesis and characterization of sulfonamidophenylporphyrin variants
While the above preparations involve the conjugation of the sulfonamide directly to the meso-phenyl groups, we also wanted to examine whether excited state quenching would still be observed if the DNB sulfonamides were separated from the porphyrin core by short aliphatic linkers. In order to achieve this, a solution of triaminophenylporphyrin porphyrin 3 in CHCl3/pyridine was treated with N,N'-dicyclohexylcarbodiimide (DCC) and N-Fmoc-amido-dPEG2-acid 5, followed by microwave irradiation. After workup, the Fmoc protecting group was removed by stirring intermediate 6 in DMF/piperidine at room temperature for 3 h. The resulting crude reaction mixture was purified by HPLC to afford the PEG-amine porphyrin derivative 7 in 73% yield (Scheme 2). The corresponding sulfonamide derivative was synthesized by dissolution of 7 in DMF/TEA followed by the treatment with excess DNsCl. The reaction mixture was stirred at room temperature for 2 days under an argon atmosphere, and then purified by preparative HPLC afforded the SPP derivative 8. The triaminophenylporphyrin 3 was chosen for this synthesis because of its high yield from TPP, as well as the almost complete quenching of the excited states when converted to 3SAM. As compared to the PEG-amine derivative 7, the fluorescence and singlet oxygen quantum yields of SPP 8 decreased by 50% (Table 1). This decrease was far less than what was demonstrated in 3SAM (ΦFl = 0.01, ΦΔ = 0.05), thereby illustrating that the removal of the DNB moieties from the porphyrin core has a detrimental effect on their quenching potential.
Scheme 2.

Synthesis of alkyl sulfonamidophenyl porphyrin 8.
Since the quenching efficiency of 8 was less than ideal, we decided to develop chemistries that would allow for the modulation of the polarity of the SPP or allow them to have reactive handles for conjugation to biomolecules or nanoparticles for in vitro or in vivo delivery. In order to do this, 1SAM was utilized as a model compound, and was modified by alkylation of the sulfonamide to generate a tertiary amine.
Alkylation can be effected by reacting an alkyl bromide with the SPP in dry acetone with freshly powdered anhydrous K2CO3. In order to incorporate a carboxylic acid functionality, 1SAM was reacted with tert-butyl bromoacetate. Upon complete conversion to the desired product the reaction mixture was purified by column chromatography affording a tertiary SPP (1SAM-TE) in 75% yield (Scheme 3). The ester group in the sulfonamidophenyl porphyrin 1SAM-TE was hydrolyzed by the treatment with CH2Cl2/TFA (4:1) at room temperature affording 1SAM-AA in quantitative yield. The protected acid was utilized in this synthesis due to the fact that the reaction between bromoacetic acid and the porphyrin did not result in product formation. This may be attributed to the fact that bromoacetic acid has low solubility in the reaction mixture.
Scheme 3.

Synthesis of tertiary sulfonamidophenyl porphyrins based upon 1SAM.
In order to modulate the polarity of the SPP, the addition of discrete PEG chains and alkyl sulfonates were investigated as model modifications. The PEG chain was appended to the SPP via reaction of 1SAM with 2-(2-(2-methoxyethoxy)ethoxy)ethyl 2-bromoacetate (PEG3-bromoacetate)29 for 7 h at room temperature in DMF in the presence of K2CO3. The desired product 1SAM-mPEG was recovered in 83% yield after purification. In order to introduce the sulfonate, a solution 1SAM in DMF was treated with K2CO3 and 3-bromopropanesulfonic acid, and was subjected to microwave irradiation, affording the desired tertiary SPP 1SAM-PS in 69% yield. The identity and purity of the products were confirmed by 1H NMR spectroscopy, with 1SAM-mPEG demonstrating the inclusion of the PEG chain via the appearance of multiplet for the twelve protons present in the methylene bridges from 3.25-4.25 ppm. The peak corresponding to the sulfonamide proton in 1SAM was also no longer present. In addition, a peak at 3.30 ppm was observed, corresponding to the terminal methoxy group. Similarly, for 1SAM-TE, there was no signal in the region where the sulfonamide NH-protons would be present, and the alkyl chain protons appear at 4.77 ppm.
As one last example, the synthesis of a maximally quenched amine-modified SPP based upon 4SAM was studied via introduction of the discrete PEG chains. A solution of porphyrin 4SAM in DMF was treated with K2CO3 and PEG3-bromoacetate at room temperature for 7 h to afford 4SAM-mPEG in 60% yield (Scheme 4). This derivative demonstrated a high degree of excited state quenching (ΦFl = 0.02, ΦΔ = 0.07), as compared to its non-quenched analog 4-mPEG (ΦFl = 0.3, ΦΔ = 0.61).
Scheme 4.

Synthesis of tertiary sulfonamidophenyl porphyrins based upon 4SAM.
Profiling of excited state activation
The activation of the synthesized SPP derivatives were investigated, as the ultimate utility of the quenched photosensitizers relies upon the recovery of excited state processes. Incubation of the SPP with thiols, specifically thiogycolic acid, resulted in cleavage of the sulfonamide bond, liberating the respective aminophenylporphyrin (Figure 2). In these reactions, a solution of the SPP (0.5 μM) in DMF was treated with a large excess of thiol (2.5 mM), and the kinetics were examined by monitoring the fluorescence intensity increase over time at the excitation and emission maxima of each resulting aminophenylporphyrin. While 2.5 mM thiol may seem excessive, relevant in vivo concentrations of thiols, such as glutathione, can be observed at millimolar concentrations.30 As expected, the time required to effect complete excited state recovery increases with increasing number of arylsulfonamides. The shape of the resulting curves is also informative, with 1SAM displaying a hyperbolic shape characteristic of a pseudo-first order reaction. Since 2SAM, 3SAM, and 4SAM must each proceed through intermediates, which also display differing degrees of quenching, and altered absorption and emission spectra, the line shape deviates from what is observed for 1SAM.
Figure 2.

Relative rate of thiogycolic acid mediated cleavage of aromatic sulfonamidophenyl porphyrins in DMF.
In order to examine the specificity of the sulfonamide cleavage reaction, the activation of the tertiary SPP, 4SAM-mPEG, was investigated with a number of potential substrates, including thiols (thioglycolic acid [TGA], L-cysteine [Cys], dithiothreitol [DTT], L-glutathione [GSH], cysteamine, penicillamine), non-thiol amino acids (Lysine) and oxidants (H2O2, tert-butyl hydroperoxide, potassium superoxide and NaOCl, peroxynitrite). The porphyrin was also reacted with pig liver esterase, a hydrolytic enzyme. The solutions of sulfonamidophenyl porphyrin (1 μM) in DMF and analytes (1 mM, 1000 equiv) in HEPES buffer (pH = 7.4) were incubated at 37 °C for 15 min in DMF/HEPES (1:1, 2 mL, pH = 7.4). Each solution of porphyrin 4SAM-mPEG was then excited at 437 nm while monitoring the fluorescence intensity 686 nm. As has been illustrated previously,19,21 incubation of the SPP with amino acids that do not bear a thiol group and oxidants do not result in cleavage of the sulfonamide bond (Figure 3). Conversely, L-cysteine, DTT, TGA, and GSH all result in activation, although to varying degrees. At physiological pH, L-cysteine and DTT both bear overall neutral charges, whereas GSH and TGA are essentially anionic, which allows us to intimate that the charge of the molecule may alter its ability to cleave the sulfonamide bond. This is further supported by the fact that cysteamine, which should by positively charged in the utilized buffer system, does not react at all. In addition to charge effects, the sulfonamide cleavage must also contend with steric constraints. As compared to TGA, GSH is much larger and bulkier, which diminishes its ability to interact with the sulfonamide, resulting in a lesser degree of activation. Similarly, penicillamine, which is structurally similar to cysteine, except for the inclusion of two methyl groups on the amino acid side chain, does not react at all. This is likely due to the steric bulk adjacent to the thiol. This apparent selectivity for small, anionic or neutral charged thiol-containing analytes may serve to further increase specificity for in vivo applications, as these molecules are not expected to interact with proteins or oxidized thiols.
Figure 3.
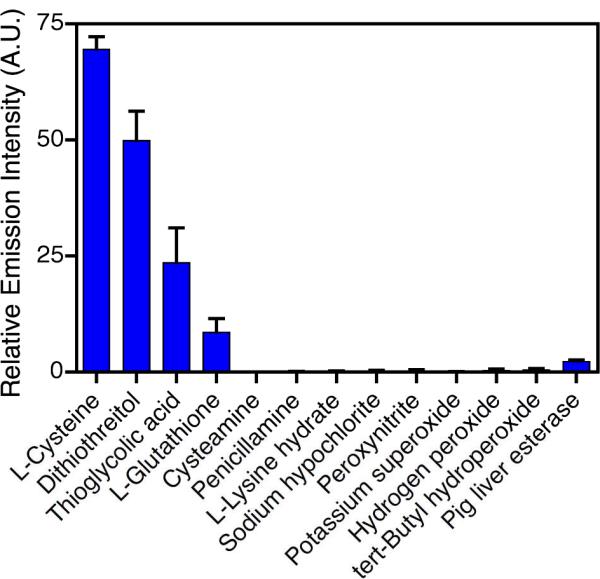
Response of tertiary sulfonamide 4SAM-mPEG to select analytes. Relative emission intensity of 0.5 μM in DMF/HEPES (1:1, 2 mL, 10 mM, pH = 7.4) at 686 nm (λex = 437 nm) after incubation at 37 °C for 15 min in the presence of 0.5 mM (final concentration) analyte. Pig liver esterase was utilized at a concentration of 2 units/mL.
CONCLUSIONS
We have thus demonstrated that the conjugation of dinitrobenzene to the porphyrin macrocycle via a thiol-cleavable sulfonamide bond resulted in chromophores exhibiting excited state quenching. The polarity of these dyes can be modulated by the conversion of the sulfonamide to the tertiary amine via reaction with hydrophilic alkyl bromides. In addition, a conjugatable linker can be introduced by reaction with tert-butylbromoacetate, followed by hydrolytic cleavage of the t-butyl ester. The SPP are readily cleaved by a number of biologically relevant thiols, including glutathione and cysteine, and as such may demonstrate utility in vivo. Although the compounds synthesized herein demonstrated a novel paradigm for excited state quenching of porphyrinic photosensitizers, other derivatives can be envisioned bearing moieties activated by oxidants or other stimuli, and as such, are the continuing focus of our research.
EXPERIMENTAL SECTION
General procedure for the microwave-assisted synthesis of 1SAM, 2cSAM and 2tSAM
To a solution of porphyrin (~30 mg) in CHCl3/pyridine (2.5 mL, 9:1) was added 2,4-dinitrobenzenesulfonyl chloride (8 equiv per amine). The reaction mixture was subjected to microwave irradiation with the following settings: T = 120 °C, t = 25 min, power = 300 W, Pmax = off. Upon completion, methanol (5 mL) was added to the reaction mixture, followed by the addition of CH2Cl2 (20 mL) and water (10 mL). The organic layer was separated, dried over anhydr MgSO4, filtered and evaporated to dryness. The solid residue was purified as detailed below.
5-[4-N-(2,4-dinitrobenzene)sulfonamidophenyl]-10,15,20-triphenylporphyrin (1SAM)
The solid residue was purified by flash chromatography (silica, CH2Cl2) and all fractions containing the product were combined and evaporated to dryness to afford a dark purple solid (30 mg, 70%). UV/Vis (DMF) λmax (log ε) 418 (5.25), 516 (4.06), 558 (3.91), 650 (3.60) nm; λem (DMF) 652 nm; 1H NMR (500 MHz, [D7]DMF) δ -2.77 (br s, 2H), 7.78 (d, J = 8.0 Hz, 2H), 7.89 (m, 9H), 8.30 (br m, 8H), 8.71 (d, J = 9.0 Hz, 1H), 8.90 (br m, 9H), 9.13 (s, 1H), 11.46 (s, 1H) ppm; 13C NMR (125 MHz, [D7]DMF) δ 119.5, 120.0, 120.5, 120.6, 120.65, 120.67, 123.4, 127.2, 127.8, 129.4, 132.8, 135.6, 137.2, 141.9, 148.7, 150.8 ppm; ESI-MS obsd 860; HRMS (ESI+ of [MH+], CH3CN): m/z calcd for C50H33N7O6S: 860.2286, found : 860.2300; HPLC tR = 18.6 min (using a gradient of 60% to 0% of buffer A over 25 min).
5,10-Bis-[4-N-(2,4-dinitrobenzene)sulfonamidophenyl]-15,20-diphenylporphyrin (2cSAM)
The solid residue was chromatographed [silica, CH2Cl2 → CH2Cl2/methanol (49:1)] and all fractions containing the product were combined and evaporated to dryness to afford a purple solid 2cSAM
Alternate synthesis of 2cSAM
A sample of porphyrin 2c (26 mg, 0.04 mmol)26 was dissolved in anhydrous pyridine (2 mL) followed by the treatment with 2,4-dinitrobenzenesulfonyl chloride (0.11 g, 0.41 mmol). The reaction mixture was subjected to stirring 27 h at room temperature. Upon completion, 1M HCl (5 mL) was added to the reaction mixture, followed by the addition of CH2Cl2 (25 mL). The organic layer was separated, dried over anhydr MgSO4, filtered and evaporated to dryness. The solid residue was chromatographed [silica, CH2Cl2 → CH2Cl2/methanol (49:1)] and all fractions containing the product were combined and evaporated to dryness to afford a purple solid 2cSAM (25 mg, 57%): UV/Vis (DMF) λmax (log ε) 420 (5.18), 520 (4.18), 568 (4.20), 660 (3.88) nm; λem (DMF) 652 nm; 1H NMR (500 MHz, [D7]DMF) δ -2.81 (brs, 2H), 7.77 (d, J = 8.0 Hz, 4H), 7.89 (d, J = 7.0 Hz, 6H), 8.29 (m, 8H), 8.71 (d, J = 9.0 Hz, 2H), 8.88 (m, 8H), 8.90 (d, J = 2.0 Hz, 2H), 9.12 (d, J = 2.0 Hz, 2H), 11.47 (s, 2H) ppm; 13C NMR (125 MHz, [D7]DMF) δ 119.6, 120.5, 120.6, 120.7, 127.2, 132.8, 135.6, 137.2, 141.9, 148.7, 150.8 ppm; ESI-MS obsd 1105; HRMS (ESI+ of [MH+], CH3CN): m/z calcd for C56H36N10O12S2: 1105.2028, found : 1105.2019; HPLC tR = 19.04 min (using a gradient of 60% to 0% of buffer A over 25 min).
5,15-Bis-[4-N-(2,4-dinitrobenzene)sulfonamidophenyl]-10,20-diphenylporphyrin (2tSAM)
The solid residue was chromatographed [silica, CH2Cl2 → CH2Cl2/methanol (49:1)] and all fractions containing the product were combined and evaporated to dryness to afford a purple solid 2tSAM
Alternate synthesis of 2tSAM
A sample of porphyrin 2t (13 mg, 0.02 mmol)26 was dissolved in anhydrous pyridine (2 mL) followed by addition of 2,4-dinitrobenzenesulfonyl chloride (54 mg, 0.20 mmol). The reaction mixture was subjected to stirring 18 h at room temperature. Upon completion, 1M HCl (3 mL) was added to the reaction mixture, followed by the addition of CH2Cl2 (15 mL). The organic layer was separated, dried over anhydr MgSO4, filtered and evaporated to dryness. The solid residue was chromatographed [silica, CH2Cl2 → CH2Cl2/methanol (49:1)] and all fractions containing the product were combined and evaporated to dryness to afford a purple solid 2tSAM (18 mg, 81%): UV/Vis (DMF) λmax (log ε) 421 (4.57), 520 (3.50), 567 (3.45), 657 (3.21) nm; 1H NMR (500 MHz, [D7]DMF) δ -2.79 (br s, 2H), 7.78 (d, J = 8.0 Hz, 4H), 7.88 (d, J = 6.5 Hz, 6H), 8.29 (m, 8H), 8.72 (d, J = 9.0 Hz, 2H), 8.89 (br m, 10H), 9.13 (d, J = 2.0 Hz, 2H), 11.49 (s, 2H) ppm; 13C NMR (125 MHz, [D7]DMF) δ 115.9, 120.5, 121.6, 128.1, 128.4, 133.8, 136.5, 138.2, 142.8, 149.7, 151.8 ppm; ESI-MS obsd 1105; HRMS (ESI+ of [MH+], CH3CN): m/z calcd for C56H36N10O12S2: 1105.2028, found: 1105.2016; HPLC tR = 18.15 min (using a gradient of 60% to 0% of buffer A over 25 min).
5,10,15-Tris-[4-N-(2,4-dinitrobenzene)sulfonamidophenyl]-20-phenylporphyrin (3SAM)
A solution of porphyrin 3 (23 mg, 35 μmol)26 in anhydrous pyridine (3.0 mL) was treated with 2,4-dinitrobenzenesulfonyl chloride (93 mg, 0.35 mmol). The reaction mixture was stirred at room temperature for 22 h. Upon completion, 1 M HCl (4 mL) was added to the reaction mixture, followed by the addition of CH2Cl2 (20 mL) and THF (10 mL). The organic layer was separated, dried over anhydr MgSO4, filtered and evaporated to dryness. The solid residue was chromatographed [silica, CH2Cl2 → CH2Cl2/methanol (49:1)] and all fractions containing the product were combined and evaporated to dryness followed by precipitation from CH2Cl2 and hexanes to afford a dark purple solid (37 mg, 78%): UV/Vis (DMF) λmax (log ε) 422 (5.42), 570 (5.00) nm; λem (DMF) 653 nm; 1H NMR (500 MHz, [D7]DMF) δ -2.83 (br s, 2H), 7.77 (d, J = 8.5 Hz, 6H), 7.89 (d, J = 7.5 Hz, 4H), 8.28 (m, 9H), 8.72 (m, 3H), 8.88 (br m, 10H), 9.13 (br s, 2H), 11.48 (s, 3H) ppm; 13C NMR (125 MHz, [D7]DMF) δ 119.7, 119.8, 120.8, 120.9, 127.4, 133.0, 134.8, 135.7, 136.8, 137.4, 148.9, 151.0 ppm; ESI-MS obsd 1350; HRMS (ESI+ of [MH+], CH3CN): m/z calcd for C62H39N13O18S3: 1350.1771, found: 1350.1752; HPLC tR = 17.36 min (using a gradient of 60% to 0% of buffer A over 25 min).
5,10,15,20-Tetrakis-[4-N-(2,4-dinitrobenzene)sulfonamidophenyl]porphyrin (4SAM)
A solution of porphyrin 4 (0.13 g, 0.20 μmol)31 in pyridine (40 mL) was treated with 2,4-dinitrobenzenesulfonyl chloride (0.43 g, 1.6 mmol). The reaction mixture was stirred at room temperature for 24 h. Upon completion, 1M HCl (30 mL) was added to the reaction mixture, followed by the addition of CH2Cl2 (100 mL) and MeOH (25 mL). The organic layer was separated and concentrated. The solid residue was purified by passing through a pad of silica gradually eluting with CH2Cl2, acetone, and acetone/MeOH/TEA (95:4.5:0.5) affording a dark red solid. The solid residue was dissolved in acetone followed by precipitation with diethyl ether yielding a red solid precipitate (0.16 g, 50%): UV/Vis (DMF) λmax (log ε) 440 (5.3) nm, 577 (4.52), 663 (4.18); λem (DMF) 653 nm; 1H NMR (500 MHz, [D6]DMSO) δ -3.04 (br s, 2H), 7.54 (d, J = 8.5 Hz, 8H), 8.13 (d, J = 8.0 Hz, 8H), 8.52 (d, J = 8.5 Hz, 4H), 8.75, (s, 8H), 8.76 (d, J = 2.5 Hz, 2H), 8.78 (d, J = 2.0 Hz, 2H), 9.03 (d, J = 2.5 Hz, 4H), 11.51 (s, 4H) ppm; 13C NMR (125 MHz, [D6]DMSO) δ 119.1, 119.8, 120.4, 127.4, 131.9, 135.2, 136.0, 136.4, 138.0, 147.9, 150.2 ppm; ESI-MS obsd 1596.6; HRMS (ESI+ of [MH+], CH3CN): m/z calcd for C68H42N16O24S4: 1595.1513, found: 1595.1490; HPLC tR = 16.68 min (using a gradient of 60% to 0% of buffer A over 25 min).
5,10,15-Tris-[4-N-(PEG)2amidophenyl]-20-phenylporphyrin (7)
A solution of porphyrin 3 (53 mg, 0.08 mmol)26 in CHCl3/pyridine (5 mL, 4:1) was treated with DCC (0.12 g, 0.60 mmol) and N-Fmoc-amido-dPEG2-acid (5, 0.21 g, 0.52 mmol). The reaction mixture was subjected to microwave irradiation with the following settings: T = 100 °C, t = 40 min, power = 300 W, Pmax = off. Upon completion, the reaction mixture was filtered to remove DCU and concentrated. The resulting solid residue was chromatographed [silica, CH2Cl2 → CH2Cl2/methanol (49:1)] and all fractions containing the product were combined and evaporated to dryness to afford porphyrin 6 as a purple solid (0.14 g, 97%): ESI-MS obsd 1805, calcd 1802.8 (C110H102N10O15). The title compound was used in the next step without further characterization. A sample of porphyrin 6 (0.14 g, 0.08 mmol) was treated with DMF/piperidine (20 mL, 3:1). The reaction mixture was stirred at room temperature for 3 h. Upon complete removal of the Fmoc protecting groups the reaction mixture was concentrated. The resulting residue was purified by preparative HPLC (using a gradient of 80% to 20% of buffer A) and all fractions containing the product were combined and evaporated to dryness to afford porphyrin 7 as a dark green solid (66 mg, 73% overall yield in two steps): UV/Vis (DMF) λmax (log ε) 424 (4.92), 518 (3.36), 554 (3.35), 596 (2.96), 650 (2.90) nm; λem (DMF) 660 nm; 1H NMR (500 MHz, [D4]MeOH) δ 2.89 (t, J = 6.0 Hz, 6H), 3.17 (t, J = 5.0 Hz, 6H), 3.79 (br m, 18H), 4.01 (t, J = 6.0 Hz, 6H), 8.04 (br m, 3H), 8.31 (d, J = 8.0 Hz, 6H), 8.55 (m, 8H), 8.81 (br m, 8H) ppm; 13C NMR (125 MHz, [D4]MeOH) δ 38.7, 40.9, 68.1, 68.3, 71.5, 71.6, 120.7, 123.6, 129.6, 130.8, 131.2, 137.3, 138.9, 139.8, 141.5, 142.2, 173.0 ppm; ESI-MS obsd 1136; HRMS (ESI+ of [MH+], CH3CN): m/z calcd for C65H72N10O9: 1137.5557, found: 1137.5582; HPLC tR = 7.91 min (using a gradient of 80% to 20% of buffer A over 20 min).
5,10,15-Tris-[4-N-(2,4-dinitrobenzene)sulfonamido(PEG)2amidophenyl]-20-phenylporphyrin (8)
A solution of porphyrin 7 (23 mg, 0.02 μmol) in DMF/TEA (5 mL, 9:1) was treated with 2,4-dinitrobenzenesulfonyl chloride (0.11 g, 0.40 mmol). The reaction mixture was stirred at room temperature for 2 days under argon atmosphere. Upon completion, methanol was added to the reaction mixture, followed by the addition of ethyl acetate and water. The organic layer was separated, dried over anhydr MgSO4, filtered and evaporated to dryness. The solid residue was chromatographed [silica, CH2Cl2/methanol (99:1 → 19:1)] and all fractions containing the product were combined and evaporated to dryness to afford a orange-red solid as a second fraction which was further purified by preparative HPLC (using a gradient of 60 to 0 of buffer A with flow rate 21 mL/min; buffer A = 100:0.1 water/TFA, buffer B = 90:10:0.1 CH3CN/water/TFA) and all fractions containing the product were combined and evaporated to dryness to afford a light green solid (5 mg, 14%): UV/Vis (DMF) λmax (log ε) 422 (5.15), 518 (3.75), 554 (3.63), 594 (3.26), 650 (3.30) nm; λem (DMF) 656 nm; 1H NMR (500 MHz, [D7]DMF) δ -2.75 (br s, 2H), 2.84 (t, J = 6.0 Hz, 6H), 3.39 (m, 6H), 3.61 (br m, 18H), 3.91 (t, J = 6.0 Hz, 6H), 7.90 (m, 3H), 8.23 (br m, 11H), 8.33 (m, 2H), 8.50 (br m, 6H), 8.75 (m, 3H), 8.95 (br m, 9H), 10.49 (s, 3H) ppm; 13C NMR (125 MHz, [D7]DMF) δ 38.0, 43.7, 67.3, 69.7, 70.2, 70.3, 117.8, 120.5, 127.6, 132.3, 135.3, 139.3, 140.1, 148.4, 150.3, 170.2 ppm; ESI-MS obsd 1826; HRMS (ESI+ of [MH+], CH3CN): m/z calcd for C83H78N16O27S3: 1827.4457, found: 1828.4423; HPLC tR = 13.76 min (using a gradient of 60% to 0% of buffer A over 25 min).
5-[4-(N-(2,4-dinitrobenzene)sulfonamidophenyl-N-tertiarybutoxycarbonylmethyl)]-10,15,20-triphenylporphyrin (1SAM-TE)
A solution of 1SAM (26 mg, 0.03 mmol) in anhydrous acetone (6 mL) was treated with freshly powdered anhydrous K2CO3 (21 mg, 0.15 mmol, 5 equiv). The resulting mixture was treated with tert-butyl bromoacetate (35 μL, 0.24 mmol, 8 equiv) and subjected to stirring at room temperature. The progress of the reaction was monitored by LCMS analysis. Upon completion after 6 h, the reaction mixture was diluted with water (10 mL) and CH2Cl2 (25 mL). The organic layer was separated, dried over anhydr MgSO4, filtered and evaporated to dryness. Since LCMS analysis showed formation of a single product, the solid residue was dissolved in CH2Cl2 and precipitated with hexanes affording a dark purple solid (22 mg, 75%): UV/Vis (DMF) λmax (log ε) 417 (5.50), 513 (4.16), 549 (3.85), 590(3.61), 645 (3.51) nm; λem (DMF) 651 nm; 1H NMR (500 MHz, CDCl3) δ -2.80 (br s, 2H), 1.58 (s, 9H), 4.77 (s, 2H), 7.77 (br m, 11H), 8.22 (d, J = 6.0 Hz, 8H), 8.29 (d, J = 9.0 Hz, 1H), 8.54 (m, 2H), 8.76 (d, J = 4.0 Hz, 2H), 8.86 (s, 4H), 8.89 (d, J = 5.0 Hz, 2H) ppm; 13C NMR (125 MHz, CDCl3) δ 28.1, 55.0, 83.2, 117.9, 119.6, 120.4, 120.7, 125.7, 126.7, 127.6, 127.9, 133.7, 134.6, 135.7, 138.0, 138.2, 142.0, 143.6, 148.3, 150.0, 167.8 ppm; ESI-MS obsd 974.7; HRMS (ESI+ of [MH+], CH3CN): m/z calcd for C56H43N7O8S: 974.2967, found: 974.2980; HPLC tR = 24.95 min (using a gradient of 60% to 0% of buffer A over 25 min).
5-[4-(N-(2,4-dinitrobenzene)sulfonamidophenyl-N-carboxymethyl)]-10,15,20-triphenylporphyrin (1SAM-AA)
A sample of porphyrin 1SAM-TE (10 mg, 0.01 mmol) in CH2Cl2 (2 mL) was treated with TFA (0.5 mL). The reaction mixture was stirred for 2 h. To the resulting mixture water (5 mL) and ethyl acetate (10 mL) were added. The organic layer was separated, washed with a saturated aq NaHCO3 solution. The organic layer was dried over anhydr MgSO4, filtered and evaporated to dryness. The resulting residue was re-dissolved in minimum volume of ethyl acetate followed by the addition of hexanes. The precipitate thus formed was filtered affording a red solid (9 mg, quant): UV/Vis (DMF) λmax (log ε) 418 (5.50), 514 (5.09), 549 (4.79), 591 (4.56), 647 (4.51) nm; λem (DMF) 651 nm; 1H NMR (500 MHz, [D6]DMSO) δ -2.93 (s, 2H), 4.51 (s, 2H), 7.84 (br m, 12H), 8.21 (br, 9H), 8.82 (br m, 9H) ppm; 13C NMR (125 MHz, [D6]DMSO) δ 28.6, 119.7, 120.0, 121.4, 122.7, 123.4, 126.9, 128.6, 131.5, 134.2, 135.0, 141.1 ppm; ESI-MS obsd 918.5; HRMS (ESI+ of [MH+], CH3CN): m/z calcd for C52H35N7O8S: 918.2341, found: 918.2313; HPLC tR = 20.19 min (using a gradient of 60% to 0% of buffer A over 25 min).
5-[4-N-(2,4-dinitrobenzene)sulfonamido-N-3-sulfopropyl]-10,15,20-triphenylporphyrin (1SAM-PS)
A solution of porphyrin 1SAM (22 mg, 0.025 mmol) in anhydrous DMF (4 mL) was treated with freshly powdered anhydrous K2CO3 (14 mg, 0.10 mmol, 4 equiv) and 3-bromopropanesulfonic acid sodium salt (34 mg, 0.15 mmol, 6 equiv). The reaction mixture was subjected to microwave irradiation with the following settings: T = 70 °C, t = 10 min, power = 300 W, Pmax = off. The reaction was monitored by HPLC analysis. Upon completion, the solvent was evaporated and the resulting residue was purified by preparative HPLC (using a gradient of 60 to 0 of buffer A with flow rate 21 mL/min; buffer A = 100:0.1 water/TFA, buffer B = 90:10:0.1 CH3CN/water/TFA). All fractions containing the product were combined and evaporated to dryness to afford porphyrin 1SAM-PS as a dark green solid (17 mg, 69%): UV/Vis (DMF) λmax (log ε) 418 (5.2), 514 (3.8), 547 (3.48), 590 (3.3), 645 (3.2) nm; λem (DMF) 650 nm; 1H NMR (500 MHz, CD2Cl2) δ -0.54 (s, 2H), 1.25 (s, 2H). 2.82 (s, 2H), 2.92 (s, 2H), 7.98 (br m, 11H), 8.32 (br m, 4H), 9.01 (br m, 15H) ppm; ESI-MS obsd 982.4; HRMS (ESI+ of [MH+], CH3CN): m/z calcd for C53H39N7O9S2: 982.2323, found: 982.230; HPLC tR = 15.00 min (using a gradient of 60% to 0% of buffer A over 25 min).
5-[4-N-(2,4-dinitrobenzene)sulfonamido-N-(PEG)2methyleneamidophenyl]-10,15,20-triphenylporphyrin (1SAM-mPEG)
A solution of porphyrin 1SAM (22 mg, 0.025 mmol) in anhydrous DMF (5 mL) was treated with freshly powdered anhydrous K2CO3 (17 mg, 0.12 mmol, 5 equiv). The resulting mixture was treated with PEG3-bromoacetate (57 mg, 0.20 mmol, 8 equiv)29 and subjected to stirring at room temperature. The progress of the reaction was monitored by LCMS analysis. Upon completion after 7 h, the reaction mixture was diluted with water (15 mL) and CH2Cl2 (25 mL). The aqueous layer was separated and extracted with ethyl acetate (15 mL). The combined organic layers were dried over anhydr MgSO4, filtered and evaporated to give a viscous residue. The residue was dissolved in CH2Cl2 and precipitated with hexanes affording a solid crude residue. The crude residue was purified by column chromatography [silica, CH2Cl2 → CH2Cl2/methanol (99:1)] following further purification by preparative HPLC (using a gradient of 60 to 0 of buffer A with flow rate 21 mL/min; buffer A = 100:0.1 water/TFA, buffer B = 90:10:0.1 CH3CN/water/TFA) and all fractions containing the product were combined and evaporated to dryness to afford a light green solid (22 mg, 83%): UV/Vis (DMF) λmax (log ε) 417 (4.46), 514 (3.16), 547 (2.87), 589 (2.71), 645 (2.62) nm; λem (DMF) 650 nm; 1H NMR (500 MHz, CDCl3) δ 3.30 (s, 3H), 3.49 (m, 2H), 3.58 (m, 2H), 3.63 (m, 2H), 3.67 (m, 2H), 3.77 (m, 2H), 4.42 (m, 2H), 4.95 (s, 2H), 8.02 (br m, 11H), 8.32 (d, J = 9.0 Hz, 1H), 8.58 (br m, 10H), 8.66 (br m, 8H) ppm; 13C NMR (125 MHz, CDCl3) δ 13.5, 17.9, 22.3, 22.9, 29.3, 31.5, 33.0, 53.8, 58.2, 64.8, 68.4, 70.0, 71.4, 119.4, 122.7, 126.0, 127.6, 128.0, 128.2, 129.0, 129.7, 133.2, 137.3, 138.1, 138.8, 139.4, 144.8, 145.3, 145.4, 145.6, 147.8, 149.7, 168.4 ppm; ESI-MS obsd 1064.1; HRMS (ESI+ of [MH+ + Na/2], CH3CN): m/z calcd for C59H49N7O11S: 1064.3284, found: 1064.3273; HPLC tR = 20.81 min (using a gradient of 60% to 0% of buffer A over 25 min).
5,10,15,20-Tetrakis-[4-N-(2,4-dinitrobenzene)sulfonamido-N-(PEG)2methyleneamidophenyl]porphyrin (4SAM-mPEG)
A solution of porphyrin 4SAM (20 mg, 0.012 mmol) in anhydrous DMF (3 mL) was treated with freshly powdered anhydrous K2CO3 (19 mg, 0.14 mmol, 12 equiv). The resulting mixture was treated with PEG3-bromoacetate (51 mg, 0.18 mmol, 15 equiv) and subjected to stirring at room temperature. The progress of the reaction was monitored by LCMS analysis. Upon completion after 7 h, the reaction mixture was diluted with water (15 mL) and CH2Cl2 (30 mL). The organic layer was separated, dried over anhydr MgSO4, filtered and evaporated to give a viscous residue. The residue was precipitated by adding diethyl ether affording a solid crude residue. The crude residue was purified by preparative HPLC (using a gradient of 60 to 0 of buffer A with flow rate 21 mL/min; buffer A = 100:0.1 water/TFA, buffer B = 90:10:0.1 CH3CN/water/TFA) and all fractions containing the product were combined and evaporated to dryness to afford a light green solid (17 mg, 60%): UV/Vis (DMF) λmax (log ε) 420 (5.09), 515 (3.59), 550 (3.29), 590 (3.15), 646 (3.04); λem (DMF) 651 nm; 1H NMR (500 MHz, CDCl3) δ 3.37 (s, 12H), 3.57 (m, 8H), 3.68 (m, 8H), 3.75 (br m, 16H), 3.85 (m, 8H), 4.47 (m, 8H), 4.97 (s, 8H), 8.05 (d, J = 6.5 Hz, 8H), 8.36 (d, J = 8.5 Hz, 4H), 8.53-8.68 (br m, 24H) ppm; 13C NMR (125 MHz, CDCl3) δ 14.1, 18.4, 22.7, 23.3, 29.3, 31.9, 33.4, 54.2, 59.0, 65.1, 68.9, 70.4, 71.7, 119.7, 126.1, 128.5, 129.3, 133.6, 137.8, 139.4, 140.2, 145.5, 148.4, 150.1, 168.8 ppm; ESI-MS obsd 2412.5; HRMS (ESI+ of [(MH+ + Na)/2], CH3CN): m/z calcd for C104H106N16O44S4: 1228.2608, found: 1228.2601; HPLC tR = 20.76 min (using a gradient of 60% to 0% of buffer A over 25 min).
5,10,15,20-Tetrakis-[N-(PEG)2methyleneamidophenyl]porphyrin (4-mPEG)
A solution of porphyrin 4 (20 mg, 0.03 mmol) in anhydrous DMF (3 mL) was treated with freshly powdered anhydrous K2CO3 (33 mg, 0.24 mmol, 8 equiv) and PEG3-bromoacetate (0.10 g, 0.36 mmol, 12 equiv). The reaction mixture was subjected to microwave irradiation with the following settings: T = 70 °C, t = 25 min, Pmax = off. The reaction was monitored by LCMS analysis. Upon completion, reaction mixture was diluted with ethyl acetate and water. The organic layer was separated, washed twice with water, dried over anhydr MgSO4, filtered and evaporated to dryness. The resulting oily residue was dissolved in CH2Cl2 and precipitated with hexanes. The crude solid was then purified by preparative HPLC (using a gradient of 60 to 0 of buffer A with flow rate 21 mL/min; buffer A = 100:0.1 water/TFA, buffer B = 90:10:0.1 CH3CN/water/TFA) and all fractions containing the product were combined and evaporated to dryness to afford porphyrin 4-mPEG as a dark green solid (40 mg, 89%): UV/Vis (DMF) λmax (log ε) 437 (5.30), 528 (4.02), 574 (4.05), 664 nm; λem (DMF) 686 (3.98) nm; 1H NMR (500 MHz, CDCl3/[D4]MeOH, 9:1) δ 3.45 (m, 12H), 3.80 (br m, 50H), 4.34 (s, 3H), 4.56 (m, 8H), 7.25 (m, 5H), 8.48 (br m, 14H) ppm; 13C NMR (125 MHz, CDCl3/[D4]MeOH, 9:1) δ 29.7, 45.4, 53.4, 59.1, 64.5, 64.7, 66.6, 68.5, 69.0, 70.6, 72.0, 112.2, 112.7, 127.1, 131.2, 146.3, 170.7 ppm; ESI-MS obsd 1492; HRMS (ESI+ of [MH+], CH3CN): m/z calcd for C80H98N8O20: 1491.6970, found: 1491.6977; HPLC tR = 8.71 min (using a gradient of 60% to 0% of buffer A over 25 min).
Supplementary Material
Acknowledgment
We thank Dr. Scott A. Hilderbrand for his helpful discussions. This work was supported by an NIH grant U24-CA092782 (RW), U01-HL080731 (JM, RW), NIH grants U54-CA119349 (RW), U54-CA126515 (RW). NMR data were obtained at the Department of Chemistry, University of Connecticut, with great thanks to Dr. Martha Morton.
Footnotes
Supporting Information Available. General synthetic and photophysical characterization procedures, NMR (1H and 13C) of all compounds synthesized and HPLC traces of relevant compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).Dolmans DE, Fukumura D, Jain RK. Nat Rev Cancer. 2003;3:380–387. doi: 10.1038/nrc1071. [DOI] [PubMed] [Google Scholar]
- (2).Pandey RK, Zheng G. The Porphyrin Handbook. 2000:157–230. [Google Scholar]
- (3).Sternberg ED, Dolphin D, Brückner C. Tetrahedron. 1998;54:4151–4202. [Google Scholar]
- (4).Rockson SG, Kramer P, Razavi M, Szuba A, Filardo S, Fitzgerald P, Cooke JP, Yousuf S, DeVault AR, Renschler MF, Adelman DC. Circulation. 2000;102:2322–2324. doi: 10.1161/01.cir.102.19.2322. [DOI] [PubMed] [Google Scholar]
- (5).Rockson SG, Lorenz DP, Cheong WF, Woodburn KW. Circulation. 2000;102:591–596. doi: 10.1161/01.cir.102.5.591. [DOI] [PubMed] [Google Scholar]
- (6).Bisland SK, Singh D, Gariepy J. Bioconjug Chem. 1999;10:982–992. doi: 10.1021/bc990020u. [DOI] [PubMed] [Google Scholar]
- (7).Chaloin L, Bigey P, Loup C, Marin M, Galeotti N, Piechaczyk M, Heitz F, Meunier B. Bioconjug Chem. 2001;12:691–700. doi: 10.1021/bc000125t. [DOI] [PubMed] [Google Scholar]
- (8).Choi Y, McCarthy JR, Weissleder R, Tung CH. ChemMedChem. 2006;1:458–463. doi: 10.1002/cmdc.200500036. [DOI] [PubMed] [Google Scholar]
- (9).Del Governatore M, Hamblin MR, Shea CR, Rizvi I, Molpus KG, Tanabe KK, Hasan T. Cancer Res. 2000;60:4200–4205. [PubMed] [Google Scholar]
- (10).Hudson R, Carcenac M, Smith K, Madden L, Clarke OJ, Pelegrin A, Greenman J, Boyle RW. Br. J. Cancer. 2005;92:1442–1449. doi: 10.1038/sj.bjc.6602517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hudson R, Boyle RW. J. Porphyrins Phthalocyanines. 2004;8:954–975. [Google Scholar]
- (12).McCarthy JR, Jaffer FA, Weissleder R. Small. 2006;2:983–987. doi: 10.1002/smll.200600139. [DOI] [PubMed] [Google Scholar]
- (13).McCarthy JR, Perez JM, Bruckner C, Weissleder R. Nano Lett. 2005;5:2552–2556. doi: 10.1021/nl0519229. [DOI] [PubMed] [Google Scholar]
- (14).McCarthy JR, Weissleder R. ChemMedChem. 2007;2:360–365. doi: 10.1002/cmdc.200600244. [DOI] [PubMed] [Google Scholar]
- (15).Stefflova K, Chen J, Zheng G. Curr Med Chem. 2007;14:2110–2125. doi: 10.2174/092986707781389655. [DOI] [PubMed] [Google Scholar]
- (16).Stefflova K, Li H, Chen J, Zheng G. Bioconjug Chem. 2007;18:379–388. doi: 10.1021/bc0602578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Zheng G, Chen J, Stefflova K, Jarvi M, Li H, Wilson BC. Proc. Natl. Acad. Sci. U. S. A. 2007;104:8989–8994. doi: 10.1073/pnas.0611142104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Choi Y, Weissleder R, Tung CH. Cancer Res. 2006;66:7225–7229. doi: 10.1158/0008-5472.CAN-06-0448. [DOI] [PubMed] [Google Scholar]
- (19).Bouffard J, Kim Y, Swager TM, Weissleder R, Hilderbrand SA. Org Lett. 2008;10:37–40. doi: 10.1021/ol702539v. [DOI] [PubMed] [Google Scholar]
- (20).Jiang W, Fu Q, Fan H, Ho J, Wang W. Angew Chem Int Ed Engl. 2007;46:8445–8448. doi: 10.1002/anie.200702271. [DOI] [PubMed] [Google Scholar]
- (21).Shibata A, Furukawa K, Abe H, Tsuneda S, Ito Y. Bioorg. Med. Chem. Lett. 2008;18:2246–2249. doi: 10.1016/j.bmcl.2008.03.014. [DOI] [PubMed] [Google Scholar]
- (22).Heafield MT, Fearn S, Steventon GB, Waring RH, Williams AC, Sturman SG. Neurosci. Lett. 1990;110:216–220. doi: 10.1016/0304-3940(90)90814-p. [DOI] [PubMed] [Google Scholar]
- (23).Nekrassova O, Lawrence NS, Compton RG. Talanta. 2003;60:1085–1095. doi: 10.1016/S0039-9140(03)00173-5. [DOI] [PubMed] [Google Scholar]
- (24).Nygard O, Nordrehaug JE, Refsum H, Ueland PM, Farstad M, Vollset SE. N Engl J Med. 1997;337:230–236. doi: 10.1056/NEJM199707243370403. [DOI] [PubMed] [Google Scholar]
- (25).Refsum H, Ueland PM, Nygard O, Vollset SE. Annu Rev Med. 1998;49:31–62. doi: 10.1146/annurev.med.49.1.31. [DOI] [PubMed] [Google Scholar]
- (26).Luguya R, Jaquinod L, Fronczek FR, Vicente MGH, Smith KM. Tetrahedron. 2004;60:2757–2763. [Google Scholar]
- (27).Dean ML, Schmink JR, Leadbeater NE, Bruckner C. Dalton Trans. 2008:1341–1345. doi: 10.1039/b716181f. [DOI] [PubMed] [Google Scholar]
- (28).Causgrove TP, Cheng P, Brune DC, Blankenship RE. J. Phys. Chem. 1993;97:5519–5524. doi: 10.1021/j100123a011. [DOI] [PubMed] [Google Scholar]
- (29).Dal Pozzo A, Acquasaliente M, Donzelli G, Delor F, Ferruti P. Farmaco [Sci] 1986;41:622–629. [PubMed] [Google Scholar]
- (30).Townsend DM, Tew KD, Tapiero H. Biomed Pharmacother. 2003;57:145–155. doi: 10.1016/s0753-3322(03)00043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Yuasa M, Oyaizu K, Yamaguchi A, Kuwakado M. J. Am. Chem. Soc. 2004;126:11128–11129. doi: 10.1021/ja0486216. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.