

Abstract
The amyloid-β peptide (Aβ) plays a central role in the pathogenesis of Alzheimer's disease (AD). The fibrillar form of Aβ (fAβ) exerts toxic effects on neurons through mechanisms not well understood. We have shown that the aged primate cortex is selectively vulnerable to fAβ toxicity at low concentrations. In addition to neuronal loss, fAβ induced massive activation of microglia in the aged rhesus cortex. We now demonstrate that inhibition of microglia activation abolishes fAβ toxicity. Injection or pump delivery of macrophage/microglia inhibitory factor (MIF) significantly reduced activation of microglia and the volume of damage caused by fAβ. Microglia isolated from aged rhesus cortex produced substantial reactive oxygen species when stimulated by fAβ, which was inhibited by MIF in a dose-dependent manner. This is the first definitive in vivo demonstration that the fAβ-induced microglia activation and inflammation mediate, at least in part, its toxic effects on neurons. Combined with our earlier observations, these findings suggest that aged primate microglia may display an exaggerated inflammatory response to fAβ when compared with young microglia.
Keywords: Amyloid toxicity, Microglia, Fibrillar, Neuronal loss, Non-human primate
1. Introduction
A large body of evidence supports a central role for the amyloid-β peptide (Aβ) in the pathogenesis of Alzheimer's disease (AD) (Selkoe, 2000). Aβ is deposited in the cerebral cortex in AD brains and deposits containing fibrillar Aβ (fAβ) are associated with dystrophic neurites and are used for pathological diagnosis of AD (Mirra et al., 1991; Terry and Wisniewski, 1972). The neuritic pathology associated with fAβ is suggested as indicative of the toxicity of this peptide.
Various conformations of Aβ are toxic to neurons (Lorenzo and Yankner, 1994; Hartley et al., 1999). However, the mechanisms which may mediate this toxicity are not well understood. We have shown that the aged primate cerebral cortex is selectively vulnerable to fAβ toxicity (Geula et al., 1998). Injections of plaque equivalent concentrations (200 pg) of fAβ into the cerebral cortex of aged rhesus or marmoset monkeys produced significant neuronal loss and induced hyperphosphorylation of tau, both features of the AD brain. Similar injections in young primates or aged rats were without effect. Injections of freshly prepared soluble Aβ did not result in toxicity.
Injections of fAβ in the aged primate brain also resulted in the recruitment and activation of a large population of microglia (Geula et al., 1998). In vitro evidence has clearly demonstrated the ability of fAβ to activate microglia and result in the production of reactive oxygen species (ROS), chemokines such as MCP-1 and cytotoxic cytokines such as interleukin-1 (IL-1) and tumor necrosis factor-α (TNFα) (El Khoury et al., 1996; Coraci et al., 2002; Meda et al., 1999). Therefore, it is likely that the inflammatory reaction precipitated by fAβ and mediated through activated microglia is responsible, at least in part, for neuronal loss seen in AD. However, in vivo evidence for the ability of fAβ activated microglia to mediate toxic effects on neurons is lacking. This issue is of particular relevance because fAβ deposits in the AD brain invariably contain activated microglia (Mochizuki et al., 1996; Sheng et al., 1997). Significantly, long-term use of non-steroidal anti-inflammatory drugs (NSAID) is associated with substantially reduced risk of AD (Etminan et al., 2003; Zandi et al., 2002), further strengthening the suggestion that inflammation contributes to AD pathogenesis. It must be noted that some anti-inflammatory agents inhibit γ-secretase activity required for the production of Aβ (Weggen et al., 2003a, 2003b). Thus, it is possible that the reduced risk of AD conferred by long-term use of NSAIDs is mediated, at least in part, through direct interference with Aβ production.
In the present set of experiments, we sought to determine the contribution of activated microglia to fAβ toxicity in the aged primate model in vivo. We used a fragment of tuftsin for this purpose. Tuftsin, also known as macrophage/microglia stimulating peptide (MSP), is a naturally occurring tetrapeptide with the sequence Thr-Lys-Pro-Arg. It is liberated from gamma-globulin (IgG) with the aid of two enzymes, one produced in the spleen and the other on plasma membranes (Najjar, 1983). Tuftsin has been found to stimulate all functions of phagocytic cells, including phagocytosis, pinocytosis, motility, immunogenic activity, release of potentially toxic substances such as TNF-α, ROS and nitric oxide (NO) and bactericidal activity (Najjar, 1983; Cillari et al., 1994). Consistent with its function, tuftsin deficiency in many disease states is associated with an increased incidence of infections (Corazza et al., 1991; Szkaradkiewicz, 1992; Trevisani et al., 2002). A tripeptide fragment of tuftsin (Thr-Lys-Pro, macrophage/microglia inhibitory factor [MIF]), acts as an immunosuppressant, inhibits microglial activation, abolishes the above effects (Siemion et al., 1994; Wieczorek et al., 1994) and thereby protects neurons in vivo and in vitro from several modes of damage (Kaul and Lipton, 1999; Rogove and Tsirka, 1998; Thanos et al., 1993).
2. Methods
2.1. Animals and surgery
Fibrillar Aβ40 and Aβ42 were freshly prepared and evaluated prior to surgery, as described previously (Lorenzo and Yankner, 1994). Seven specific pathogen free female rhesus monkeys 28–31 years of age were used in the present study. Following inhalation-induced anesthesia, each animal was mounted on a Kopf stereotaxic head-holder. Under aseptic conditions, the scalp was opened, a bone flap was removed and the dura was resected. Phosphate buffered saline (PBS), fAβ, MIF or fAβ in combination with MIF were injected in a volume of 1 μl over 5 min and at a depth of 1.5–2.5 mm into anatomically distinct gyri in the frontal lobe. The principalis sulcus and the upper and lower rami of the arcuate sulcus were used as landmarks to identify 4 gyri in each frontal lobe. At least five injections of one compound or a combination of compounds separated by a distance of approximately 5 mm were made in each gyrus. Injections in different gyri were separated by at least 1 cm. The gyri into which specific compounds were administered were randomized. In one animal, an Alzet osmotic minipump (#2M2L) with the capacity of 2 ml and flow rate of 5 μl/h was implanted such that the flow needle, affixed with dental cement and jewelry screws to the skull, was 0.5 mm above the fontal cortex. The anterior–posterior and medial–lateral coordinates were visually determined such that the tip of the needle was at the confluence of the upper and lower rami of arcuate sulcus. The reservoir and tubing were installed below the posterior scalp. The pump was filled with a 1 mM solution of MIF and delivery was ascertained by an empty pump at the conclusion of the experiment. Fifteen days after surgery, each animal was deeply anesthetized with sodium pentobarbital, the brain was removed, separated into two coronal halves through a cut at the mid-parietal region and placed in cold 4% paraformaldehyde in 0.1 M phosphate buffer at 4 °C for 36 h and then taken through graded sucrose solutions (10–30%) at 4 °C for cryoprotection.
2.2. Identification of injection sites
Forty micrometers thick sections of each brain were obtained on a freezing microtome. A series of one in four sections was stained with Cresyl violet. Anatomical landmarks used during surgery were utilized to identify each injection type. For each individual injection site, the sections in which tissue damage was observed and the location of damage in each section were marked following microscopic examination. Only injection sites in which the needle track could be identified were used for further analysis.
2.3. Identification of microglia
A one in four series of sections through each injection site were processed immunohistochemically using the Vectastain Elite ABC kit. Microglia were identified using a monoclonal antibody (1/500; Dako) to class II major histocompatibility glycoprotein HLA-DR, a marker of microglia activation. The presence of microglia was confirmed using a monoclonal antibody to CD68 (1/500; Dako).
2.4. Quantitative analysis
Visual examination of sections was performed in each injection site to determine the extent of damage. All injection sites displayed clear neuronal loss and gliosis. The border of the area displaying neuronal loss and gliosis was sharply defined. The area inside this border displayed visible neuronal loss and gliosis. No observable disturbance in the cortex, the density of neurons or gliosis was observable beyond this border. Two types of analysis were performed to quantitatively determine the extent of toxicity. First, the area of neurotoxic damage in mm2 was estimated in all the sections which passed through an injection site using methods described previously (Geula et al., 1998). The mean area of damage from multiple injections of the same compound was determined in each animal. As a second method, the volume of damage in each injection site was estimated using unbiased stereological techniques utilizing the Stereologer (Systems Planning) software. The two methods resulted in an identical magnitude of change (data not shown). In this report we present the data from the more accurate unbiased stereological determinations.
The number of microglia in each injection site was estimated using the fractionator unbiased stereological method, employing the Stereologer software and a one in four series of sections stained for HLA-DR. We have described the details of this method elsewhere (Geula et al., 2003). Coefficients of error for the estimations of volume of damage and number of microglia were less than 0.1.
2.5. Aβ blots
To determine whether MIF obscures fAβ or otherwise changes its fibrillar conformation, 20 μl samples of fAβ were blotted onto glass slides and allowed to air dry overnight. Then each slide was incubated overnight in PBS or PBS containing 1 mM MIF. The blots were then stained with thioflavin-S to visualize the β-pleated sheet conformation of fAβ. The presence of fAβ in the blots was determined using epifluorescence microscopy.
2.6. Rhesus microglia cultures
Blocks of frontal cortex from three aged rhesus monkeys (29–30 years old) were cut into small (1–2 mm) pieces and triturated by vigorous pipetting in RPMI to break up cell clumps and incubated in a mixture of collagenase and dispase for 1 h. The suspension was then passed through a 70 μm nitex filter and resuspended in isotonic percoll. In order to separate cells from debris and myelin, the suspension was centrifuged for 30 min at 500 × g, the pellet containing cells was resuspended in RPMI containing 10% FBS and the cells allowed to adhere to tissue culture plates for 1 h. Nonadherent cells were then washed away and the adherent cells were found to be more than 95% microglia as evidenced by CD11b staining and their ability to take up DiI AcLDL.
2.7. Detection of reactive oxygen species
Aged rhesus microglia isolated as described above were plated onto multispot glass slides (6 mm diameter spots) and incubated with 10 μg/spot fAβ, or control peptide with identical amino acid composition but reverse sequence (revAβ), in the presence of 1 mg/ml nitroblue tetrazolium (NBT) to measure reactive oxygen species (Coraci et al., 2002). ROS production leads to deposition of insoluble dark colored formazan clearly visible in the cells. To quantify ROS precipitated formazan was solubilized in a mixture of DMSO and KOH and read in a multiwell plate reader at 650 nm. Alternatively, the number of microglia containing formazan was counted in each spot. Experiments were performed in the presence or absence of various concentrations of MIF.
2.8. Effects of MIF on human neuronal cultures
SH-SY5Y neuronal cell line was maintained in Dulbecco's modified Eagle's medium (DMEM)/F12 50/50 (Fisher). Prior to Aβ exposure, 1.25 × 104 cells were seeded in 24-well culture plates for 24 h. Cells were exposed to 10 μM Aβ42 or PBS in serum free medium for 24 h in the presence or absence of MIF. Degenerating neurons were detected through uptake of Trypan blue. Neuronal loss was determined in unstained cultures. Neurons were counted in five random fields in each culture in five experiments.
2.9. Statistical analysis
When data were normally distributed, statistical analysis of the results was performed using ANOVA followed by the Neuman–Keuls post-hoc tests or t-tests. When data failed the test of normality, the non-parametric versions of the above tests were used. Relationships were determined using Student's moment correlation. Statistical analysis was carried out using the GraphPad Instat software.
3. Results
3.1. Inhibition of fAβ activated microglia
We used MIF to investigate the effects of inhibition of microglia activation on fAβ toxicity. We first determined the best doses of MIF which when injected along with fAβ would result in decreased toxicity. For this purpose, PBS vehicle, 200 pg of fAβ 40, or fAβ 42 alone or with various concentrations of MIF were injected in distinct anatomical sites within the frontal cortex of one aged rhesus monkey. Five injections of each compound or combination of compounds were made in anatomically distinct gyri and each injection was used as a data point for statistical analysis. Consistent with our previous study (Geula et al., 1998), fAβ injections resulted in a significant volume of damage as determined using unbiased stereological methods (Fig. 1). Co-injection of fAβ with MIF caused a dose-dependent decrease in the volume of damage. Concentrations of 500 μM or higher of MIF resulted in significant reduction in the fAβ induced volume of damage. Concentrations of 500 μM and 1 mM of MIF were used in the remainder of the experiments.
Fig. 1.
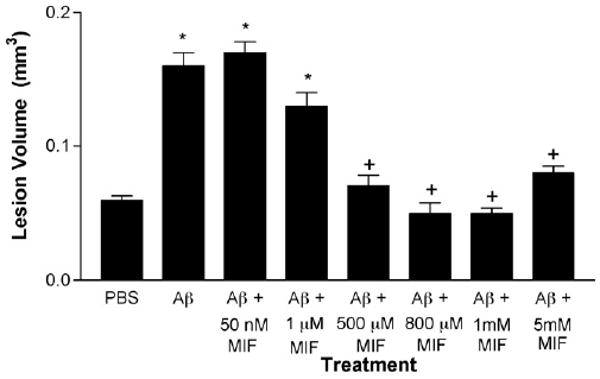
MIF exerts a dose-dependent effect on fAβ toxicity in the aged rhesus cortex. Injections of fAβ caused significantly larger volume of damage when compared with control injections of phosphate buffered saline (PBS). Co-injection of various concentrations of MIF with fAβ resulted in a dose-dependent reduction in the volume of damage. Concentrations of 50 nM and 1 μM MIF did not result in significant reduction in the volume of damage. The volume of damage was significantly larger than that in PBS injection sites. In contrast, concentrations between 500 μM and 5 mM MIF resulted in reduced volume of damage which was significantly smaller than that induced by fAβ alone and nearly identical to the damage caused by PBS control. Concentrations of 500 μM and 1 mM of MIF were used in the remainder of the experiments. *p < 0.001 compared to PBS. +p < 0.001 compared to fAβ alone and fAβ with 50 nM or 1 μM MIF and p > 0.05 compared with PBS.
3.2. MIF inhibits fAβ-induced activation of microglia in aged rhesus cortex
We determined the ability of MIF to inhibit fAβ induced activation of microglia. In six aged rhesus, we stereologically determined the number of HLA-DR immunoreactive, activated microglia in PBS, fAβ and fAβ plus MIF injection sites. Injections of 200 pg of fAβ alone resulted in activation of a significant number of microglia when compared with injections of vehicle (Fig. 2A, B and D). Co-injection of 500 μM MIF with fAβ resulted in substantial reduction in the number of activated microglia which was not significantly different from that observed in PBS injection sites (Fig. 2). Similar results where obtained using the 40 or 42 amino acid sequence of Aβ. Thus, at the concentrations used, co-injection of MIF results in significant inhibition of fAβ induced activation of microglia in aged rhesus cortex.
Fig. 2.
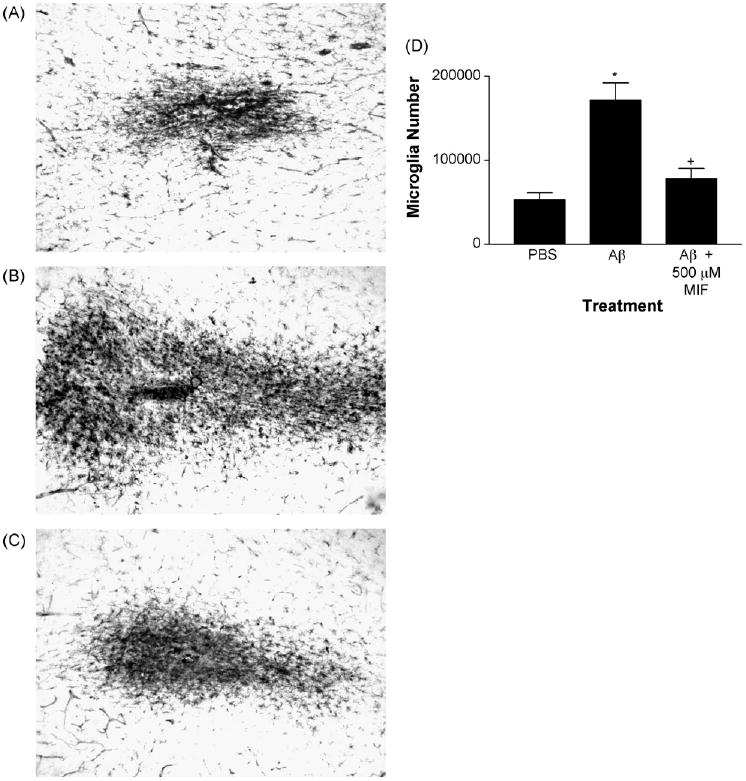
Injections of MIF reduce fAβ induced activation of microglia in aged rhesus cortex. (A) Injection of PBS control resulted in activation of a small number of microglia as indicated by the presence of HLA-DR immunoreactivity. (B) In contrast, injections of fAβ (200 pg) resulted in activation of a substantial number of microglia. (C) Co-injection of MIF (500 μM) with fAβ resulted in a substantial reduction in the number of activated microglia. (D) Quantitative analysis using unbiased stereological counting techniques in five aged rhesus monkeys demonstrated a substantial and significant increase in the number of HLA-DR-positive activated microglia in fAβ injection sites and a decrease in sites co-injected with MIF. *p < 0.05 compared with PBS. +p > 0.05 compared with PBS. Magnification in A–C is 100×.
3.3. Inhibition of microglia activation reduces fAβ toxicity in aged rhesus cortex
In six aged rhesus monkeys, 1 μl injections of 50 nM fAβ42 or fAβ40 (200 pg) resulted in a substantial volume of damage which was significantly larger than the volume of damage produced by injections of the PBS vehicle (Fig. 3A, B and D). The effects of Aβ42 and Aβ40 were similar. Injections of higher concentrations of Aβ (2.5 μM and 25 μM) resulted in slight but non-significant increases in the volume of damage, indicating that the damage induced by fAβ had peaked at 50 nM.
Fig. 3.
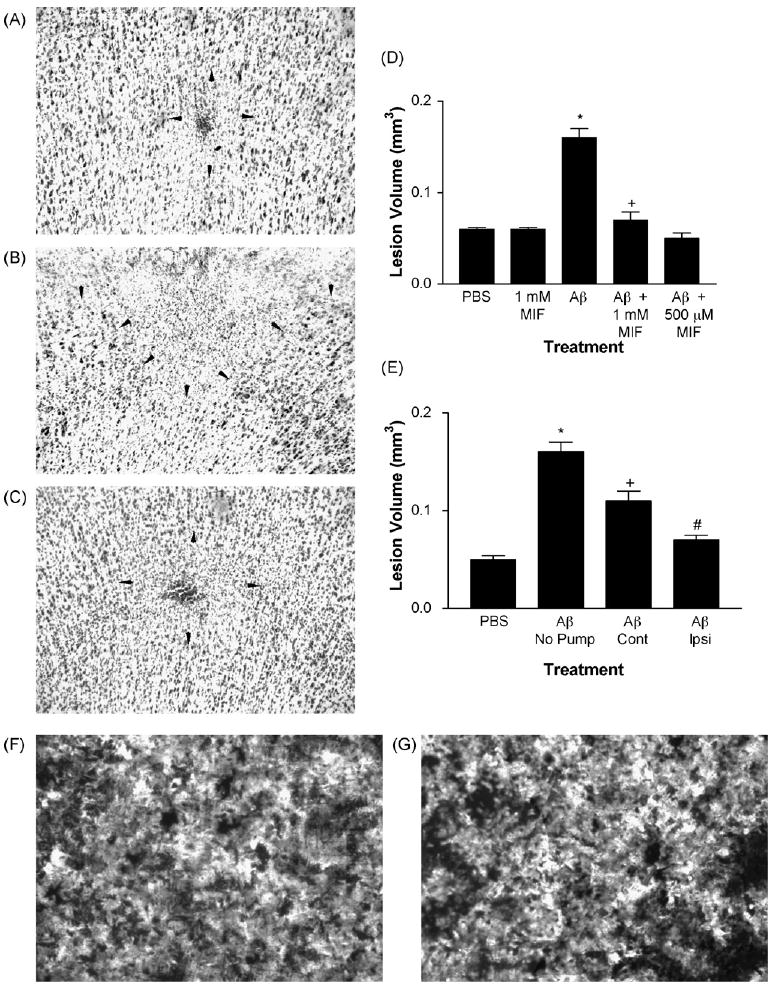
Injection or pump delivery of MIF in aged rhesus cortex significantly reduces the volume of damage induced by fAβ. (A) Injections of PBS resulted in a small area of gliosis and neuronal loss at the center of the injection (depicted by arrowheads). (B) In contrast, injections of fAβ resulted in a large area of damage (extending to the cortical surface towards the top). (C) Co-injection of MIF with fAβ resulted in a significant reduction in the area of damage. (D) In five aged rhesus monkeys, injections of PBS vehicle or MIF alone (1 mM) resulted in a small and comparable volume of damage determined using unbiased stereological methods. Injections of fAβ induced a significantly larger volume of damage when compared with PBS or MIF alone. Co-injections of MIF (500 μM or 1 mM) with fAβ resulted in a significant reduction in the volume of damage. *p < 0.001 compared to PBS and MIF. +p < 0.001 when compared with fAβ and p > 0.05 when compared with PBS or MIF alone. (E) Injections of fAβ in the hemisphere contralateral (Cont) to pump delivery of MIF displayed significantly smaller volume of damage when compared with similar injections in animals without pump delivery. However, this volume of damage was significantly larger than the volume of damage caused by injections of PBS in the ipsilateral hemisphere. Injections of fAβ ipsilateral (Ipsi) to pump delivery of MIF resulted in a significant reduction in the volume of damage which was not different from that in PBS injection sites. *p < 0.001 when compared with PBS. +p < 0.05 when compared with fAβ in animals with no pump and p < 0.01 compared to PBS. #p < 0.001 compared to fAβ in animals with no pump, p < 0.05 compared with fAβ contralateral to the pump, and p > 0.05 compared to PBS. (F) Staining of fAβ blots with thioflavin-S, which binds to the β-pleated sheet conformation of fAβ, resulted in robust epifluorescence (light areas), indicating the presence of fAβ. (G) Incubation of fAβ blots in 1 mM MIF did not alter thioflavin-S epifluorescence, suggesting that MIF does not bind fAβ or otherwise alter its fibrillar conformation. Magnification in A–C is 100× and in F and G 400×.
Co-injection of MIF with fAβ resulted in significantly reduced volume of damage (Fig. 3A-D). This effect was seen at all concentrations of fAβ. Both 500 μM and 1 mM concentrations of MIF resulted in significant decreases in the volume of damage. The volume of damage displayed a significant positive correlation with the number of activated microglia in all injection sites (r = 0.78; p < 0.0002). Therefore, reductions in fAβ-induced activation of microglia result in a significant decrease in the toxic effects of fAβ on cortical neurons.
Next, we determined whether the results obtained are due to binding and occlusion of fAβ by MIF, preventing exposure of microglia and neurons to the β-pleated sheets of the peptide rather than due to inhibition of microglia. Dried blots of various concentrations of fAβ on glass slides were incubated in the presence or absence of 1 mM MIF and stained with thioflavin-S to detect fAβ. Robust and identical thioflavin-S fluorescence was observed under the two conditions (Fig. 3F and G), indicating that MIF does not occlude fAβ nor does it alter the fibrillar conformation of the peptide.
3.4. Continuous delivery of macrophage/microglia inhibitory factor reduces fAβ toxicity
The co-injection results demonstrate that inhibition of microglia activation using a bolus of MIF results in significant reduction of fAβ toxicity. To investigate the relevance of the results to therapeutic treatments where lower concentration of drug is available over longer periods, we utilized an Alzet pump for delivery of MIF. A 2 ml pump filled with a 1 mM solution of MIF was implanted in one animal with the flow directed towards the frontal cortex in which injections of fAβ were made. The pump delivered MIF solution at a rate of 5 μl/h over the two week survival time. Pump delivery of MIF resulted in a small and non-significant decrease in the volume of damage in fAβ injection sites contralateral to the implanted pump when compared with similar injections in animals without pumps. In contrast, the fAβ induced volume of damage ipsilateral to the implanted pump was significantly smaller when compared with those in animals without pumps or in the contralateral hemisphere (Fig. 3E). Therefore, continuous presence of MIF at low concentrations effectively inhibits fAβ toxicity in the aged rhesus cortex.
3.5. Protective effects of macrophage/microglia inhibitory factor are not due to direct interaction with neurons
To determine whether inhibition of fAβ toxicity by MIF is due, at least in part, to direct interaction of MIF with neurons, we exposed cultured human-derived neuroblastoma cells (SH-SY5Y) to fAβ in the presence of MIF. This cell line has been shown to be sensitive to direct toxic effects of fAβ. Exposure of SH-SY5Y cells to fAβ (10 μM) resulted in a significant increase in the percentage of degenerating cells as indicated by uptake of Trypan blue (Supplementary Fig. 1), and a significant decrease in the number of surviving cells (p < 0.05). Co-administration of MIF (10–100 μM) had no effect on the toxic effects of fAβ on SH-SY5Y cells (p > 0.05). These results demonstrate that the protective effects of MIF against fAβ toxicity are not due to direct interaction with neurons.
As a further test for the possibility that MIF may bind to fAβ and thus cover the toxic moiety of the peptide, we pre-incubated fAβ with 100 μM MIF and then exposed SH-SY5Y cells to the resulting mixture. Pre-incubation with MIF exerted no influence on the toxic effects of fAβ as determined by the percentage of neurons taking up Trypan blue or the number of surviving neurons (p > 0.05), indicating that MIF does not bind to or alter the β-pleated sheet structure of fAβ, allowing the latter to manifest its full toxic effects.
3.6. Aged rhesus microglia produce reactive oxygen species in response to fAβ
To determine the potential mechanism through which aged rhesus microglia mediate fAβ toxicity, we isolated microglia from the frontal cortex of three aged rhesus monkeys and exposed the cultured microglia to fAβ. Cultures consisted of more than 98% microglia as indicated by uptake of DiI AcLDL, a ligand for scavenger receptor activity and a marker of microglia (Fig. 4A). Cultured microglia exposed to fAβ displayed a significant increase in production of ROS, as determined by oxidation of nitroblue tetrazolium into the dark blue formazan salt, when compared with control cultures exposed to Aβ with the reverse sequence (Fig. 4B–D).
Fig. 4.
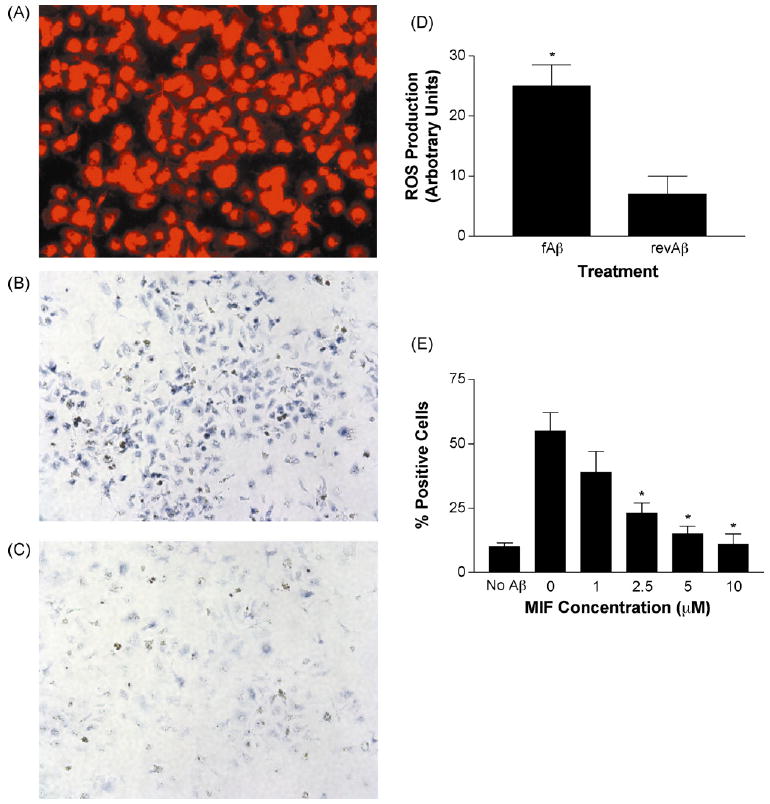
MIF inhibits fAβ-induced production of reactive oxygen species by cultured aged rhesus microglia. (A) Microglia isolated from aged rhesus frontal cortex take up DiI (red color) conjugated AcLDL, a ligand of microglia scavenger receptors. (B) Exposure of cultured aged rhesus microglia to fAβ results in production of reactive oxygen species (ROS) (dark blue reaction product). (C) In contrast, a peptide with the reverse sequence of Aβ does not induce production of ROS in cultured microglia from the aged rhesus cortex. (D) Solubilization and quantitation of ROS reaction product demonstrated significantly increased production of ROS in aged rhesus microglia exposed to fAβ when compared with microglia exposed to peptide with the reverse sequence. *p < 0.0001. (E) The presence of MIF resulted in a dose-dependent inhibition of fAβ-induced production of ROS by aged rhesus microglia. *p < 0.05 compared to 0 and 1 μM concentration.
3.7. Macrophage/microglia inhibitory factor dose dependently inhibits fAβ induced ROS production
Next, we determined whether MIF inhibits fAβ-induced production of ROS by aged rhesus microglia. Exposure of cultured aged rhesus microglia to fAβ in the presence of various concentrations of MIF resulted in a dose-dependent decrease in ROS production when compared with control cultures as determined by the number of microglia which displayed the dark blue formazan salt (Fig. 4E).
4. Discussion
The results summarized here clearly demonstrate that fAβ toxicity in the aged rhesus cortex is mediated through activation of microglia. Both injected and deposited fAβ result in activation of a substantial number of microglia. The presence of MIF leads to inhibition of fAβ induced microglia activation and toxicity. Continuous presence of low concentrations of MIF is sufficient for inhibition of microglia activation, leading to protection of neurons against fAβ toxicity. Significantly, MIF did not occlude the β-pleated sheets of fAβ or otherwise change its conformation nor did it protect cultured neurons against direct toxic effects of fAβ. Therefore, the protective effects of MIF were not due to direct interactions with fAβ or with neurons.
Recent evidence suggests that peripheral cells of monocytic origin migrate into the central nervous system and may continuously replenish the pool of microglia (Davoust et al., 2008; Simard and Rivest, 2004; Kokovay and Cunningham, 2005). However, most such studies have used irradiated animals, leading to the possibility that the migration of the bone marrow chimeras used into the central nervous system may be the result of damage to the blood-brain barrier caused by irradiation rather than to a natural tendency of blood monocytes to enter the brain (Davoust et al., 2008). It is possible that the activated microglia observed in the brain in the present experiment originate in the blood, particularly since the resection of dura during the craniotomy procedure and the needle penetrations used for injections are likely to disrupt the blood-brain barrier. This possibility does not alter the conclusions of the present experiments, particularly because monocyte lineage cells which have been shown to migrate into the brain display identical characteristics of microglia once they are differentiated in the brain (Simard and Rivest, 2004).
Exposure of microglia to fAβ in vitro leads to activation and production of chemokines and potentially cytotoxic cytokines (El Khoury et al., 1996, 2003; Meda et al., 1999). Production of ROS by fAβ stimulated microglia is a consistent finding of in vitro models (Coraci et al., 2002; El Khoury et al., 2003; Moore et al., 2002). Production of the above substances by microglia exposed to fAβ has been shown to lead to neuronal toxicity and degeneration (Guilian et al., 1996). We found that cultured microglia from aged rhesus cortex substantially increased production of ROS when exposed to fAβ. The presence of MIF resulted in significant and dose-dependent reduction of ROS production. Thus the effects of MIF on fAβ toxicity are mediated, at least in part, through oxidative damage and release of other toxic substances by activated microglia.
At least one mechanism through which Aβ-induced toxicity is mediated involves activation of the n-methyl-d-aspartate (NMDA) subtype of glutamate receptors (Inoue et al., 2008). Damaging effects of microglia activation may also be mediated through NMDA receptor activity (Rosi et al., 2006), and this process appears to require generation of reactive oxygen species (Barger et al., 2007). Thus it is potentially possible that the reactive oxygen species generated by fAβ stimulated microglia in the present study operate through NMDA receptor activation to induce neuronal degeneration.
4.1. Influence of Aβ conformation on toxicity
Aβ exists in various physical conformations which include soluble oligomers, protofibrils and fibrillar forms (Lorenzo and Yankner, 1994; Walsh et al., 1999; Hartley et al., 1999). The initial stages of Aβ polymerization and fibrillogenesis have been shown to give rise to intermediate species of small soluble Aβ oligomers and larger oligomers of so-called Aβ protofibrils which are smaller than 200 nm in length and are likely precursors of full Aβ fibrils (Walsh et al., 1999; Harper et al., 1999). Soluble Aβ oligomers and protofibrils alter neuronal activity (Hartley et al., 1999; Walsh et al., 2002; Lambert et al., 1998), result in intracellular calcium dysregulation and membrane disruption (Demuro et al., 2005; Kayed et al., 2004) and may cause degeneration of neurons and other cells in vitro (Chromy et al., 2003; Lambert et al., 2001). It has been reported that soluble Aβ oligomers exert toxic effects on cultured cells in the absence of protofibrils or fibrils (Chromy et al., 2003). Moreover, at least in some in vitro systems, soluble Aβ oligomers are more toxic to neurons than Aβ fibrils, although both conformations of the peptide produce toxic effects (Manelli et al., 2006). These findings have led some to conclude that the neuronal degeneration caused by Aβ is mostly due to the early appearance of oligomers with a minor contribution of the later appearing fAβ.
The above conclusion, based entirely on in vitro systems, runs counter to a host of in vitro and in vivo studies which indicate that fAβ exerts powerful toxic effects on neurons (Yankner, 1996; Emre et al., 1992; Yankner et al., 1990). Our earlier findings indicated that the aged primate brain is selectively vulnerable to the toxic effects of fAβ (Geula et al., 1998), with the implication that the use of lower species and in vitro models may not be appropriate for the determination of the full range of Aβ pathology. We had used relatively fresh preparations of soluble Aβ (hours prior to injection), which likely contained soluble oligomers, as a control. Injections of such Aβ preparations resulted in no toxic effects (Geula et al., 1998). In preliminary work (unpublished observations), we have also made injections of insoluble aggregated but nonfibrillar Aβ in the aged rhesus. Such preparations, which most likely contain soluble oligomers, did not result in toxicity. Thus, while it is likely that soluble Aβ oligomers interfere with synaptic function, the convincing demonstration of neuronal degeneration caused by this Aβ conformation must await future in vivo studies, preferably using non-human primate models. In contrast, all available evidence points to substantial and perhaps species-dependent neuronal killing by fAβ. We have also isolated fAβ by centrifugation of our preparations and then used the freshly reconstituted fAβ pellet and the supernatant which is likely to contain soluble Aβ oligomers to stimulate human microglia isolated from postmortem brains. We found robust production of ROS by human microglia only following stimulation by fAβ (data not shown). Thus, the effects observed in the above experiments are due to fAβ rather than contamination by soluble Aβ oligomers.
Like Aβ, abnormally phosphorylated tau, which is the main constituent of tangles in AD, also occurs in unaggregated, soluble form in neurons (pre-tangles) prior to tangle formation (Lauckner et al., 2003). Such pre-tangles appear to be functionally compromised as indicated by reduced expression of the presynaptic protein synaptophysin (Callahan et al., 2002).
Based on available evidence, we propose a two-component model of AD pathology. The first and early component consists of production and accumulation of soluble Aβ and phosphorylated tau in neurons, leading to neuronal (and particularly synaptic) dysfunction. The second and later component involves deposition of fAβ in plaques and aggregated tau in tangles, which exert toxic effects on neurons and result in neuronal loss. Significantly, the presence and density of neuritic plaques rich in fAβ and of neurofibrillary tangles are used for the pathological diagnosis of AD (Mirra et al., 1991; Hyman and Trojanowski, 1997). Thus, when clinical symptoms of dementia are manifest, fAβ and aggregated phosphorylated tau are present in abundance and are likely to exert powerful toxic effects on neurons.
Our recent observations in relation to microglia are consistent with the two-component model of AD pathology proposed above. Absence of the chemokine receptor Ccr2 resulted in markedly decreased microglia recruitment and led to earlier Aβ accumulation and premature death in β-amyloid precursor protein (β-APP) transgenic mice known to display increased levels of Aβ and plaque formation as they age (El Khoury et al., 2007). Thus, absence of early microglial accumulation leads to decreased Aβ clearance and increased mortality. Microglia express pattern recognition receptors including the class A scavenger receptor (SR-A) (El Khoury et al., 1996; Paresce et al., 1996) and the class B scavenger receptor type 2 (SR-B2), or CD36 (Coraci et al., 2002). Microglia have been shown to scavenge and degrade Aβ (Paresce et al., 1996; Rogers and Lue, 2001; Webster et al., 2001) and this function appears to be mediated by the SR-A receptor. SR-A promotes microglial endocytosis of fAβ but it is not clear if it is required for fAβ-induced microglial activation (Paresce et al., 1996; Antic et al., 2000). The CD36 receptor, on the other hand, appears to mediate the inflammatory and potentially neurotoxic effects of fAβ stimulated microglia (El Khoury et al., 1996; Coraci et al., 2002). Microglia and macrophages isolated from CD36 knockout mice (CD36−/−) unlike those isolated from wild-type animals, displayed marked reductions in fAβ induced secretion of cytokines, chemokines and ROS (El Khoury et al., 2003). Thus, in the first proposed component of AD pathology, microglia are likely to exert a beneficial influence by clearing soluble Aβ oligomers from the brain, while in the second component, they appear to be activated by fAβ and are likely to mediate neuronal degeneration. Furthermore, it is likely that each component is mediated through distinct scavenger receptors.
It must be noted that the results obtained from Ccr2 knockout βAPP transgenic animals were derived from a model of relatively chronic and prolonged Aβ production, whereas the results of the fAβ injection studies in the rhesus represent an acute model of Aβ pathology, and thus the two sets of findings cannot be directly compared. However, recent preliminary observations from our laboratory (Geula, C., Shah, P., unpublished observations) have indicated robust activation of microglia, loss of neurons and degeneration of cholinergic axons observed exclusively in the immediate vicinity of thioflavin-S positive, fAβ containing plaques in the aged rhesus and human cortex. This finding indicates that the pathology of acute injections of fAβ we describe in the present set of experiments is also associated with naturally deposited fAβ in plaques formed slowly over many years.
4.2. Therapeutic implications
With very few exceptions, a large number of epidemiological studies has indicated that use of NSAID lowers the risk for AD (Etminan et al., 2003; Landi et al., 2003; Zandi et al., 2002). These results suggest that preventing the inflammatory response, which is mediated in great part by microglia, is likely to be beneficial in AD, and probably influences the pathophysiology of the disease process. It is also possible that NSAIDs directly lower Aβ production by inhibiting γ-secretase activity (Weggen et al., 2003a, 2003b). However, clinical trials of a few NSAIDs in AD have shown no significant effects on symptoms (Reines et al., 2004; Aisen et al., 2003).
Participation of inflammatory processes in the pathophysiology of AD is likely to produce its effects over very long periods (perhaps decades). This is indicated by the fact that disease pathology in AD appears to originate decades before the clinical symptoms of the disease become apparent (Ohm et al., 1995). Therefore, the relatively short periods of treatment in clinical trials (Reines et al., 2004; Aisen et al., 2003) are unlikely to result in enough of an alteration in pathology to produce disease modification. In fact, many epidemiological studies have indicated that the effect of NSAID use on risk of AD is time-dependent and a substantial effect is obtained primarily following long-term use (Landi et al., 2003; Zandi et al., 2002). Furthermore, as indicated above, Aβ interaction with microglia is likely to stimulate different scavenger receptors, giving rise to very different outcomes. Therefore, NSAID treatments which reduce the general inflammatory response are likely to affect both the beneficial and detrimental results of microglia activation, giving rise to no net gain. For anti-inflammatory treatments to be effective, agents must be identified which interact with particular receptors and pathways which are identified as participants in this process in AD. Furthermore, it will be necessary to enhance the beneficial effects of microglia activation and diminish the detrimental effects of the inflammatory response.
Supplementary Material
Acknowledgments
We thank Naima Gedi and Nicholas Nagykery for expert technical assistance. This work was supported in part by grants from the Alzheimer's Association, the Dana Foundation Neuroimmunology Program and the Illinois Department of Public Health Alzheimer's Disease Fund and the Northwestern University Alzheimer's Disease Center (P30 AG013854).
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at dx.doi.org/10.1016/j.neurobiolaging.2009.02.025.
Footnotes
Disclosure statement: There are no actual or potential conflicts of interest. The procedures used in the animal experiments reported here were approved by an institutional animal care and use committee.
References
- Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ. Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA. 2003;289:2819–2826. doi: 10.1001/jama.289.21.2819. [DOI] [PubMed] [Google Scholar]
- Antic A, Dzenko KA, Pachter JS. Engagement of the scavenger receptor is not responsible for beta-amyloid stimulation of monocytes to a neurocytopathic state. Exp Neurol. 2000;161:96–101. doi: 10.1006/exnr.1999.7265. [DOI] [PubMed] [Google Scholar]
- Barger SW, Goodwin ME, Porter MM, Beggs ML. Glutamate release from activated microglia requires the oxidative burst and lipid peroxidation. J Neurochem. 2007;101:1205–1213. doi: 10.1111/j.1471-4159.2007.04487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan LM, Vaules WA, Coleman PD. Progressive reduction of synaptophysin message in single neurons in Alzheimer disease. J Neuropathol Exp Neurol. 2002;61:384–395. doi: 10.1093/jnen/61.5.384. [DOI] [PubMed] [Google Scholar]
- Chromy BA, Nowak RJ, Lambert MP, Viola KL, Chang L, Velasco PT, Jones BW, Fernandez SJ, Lacor PN, Horowitz P, Finch CE, Krafft GA, Klein WL. Self-assembly of Abeta(1-42) into globular neurotoxins. Biochemistry. 2003;42:12749–12760. doi: 10.1021/bi030029q. [DOI] [PubMed] [Google Scholar]
- Cillari E, Arcoleo F, Dieli M, D'Agostino R, Gromo G, Leoni F, Milano S. The macrophage-activating tetrapeptide tuftsin induces nitric oxide synthesis and stimulates murine macrophages to kill Leishmania parasites in vitro. Infect Immun. 1994;62:2649–2652. doi: 10.1128/iai.62.6.2649-2652.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El Khoury JB. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160:101–112. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corazza GR, Zoli G, Ginaldi L, Cancellieri C, Profeta V, Gasbarrini G, Quaglino D. Tuftsin deficiency in AIDS. Lancet. 1991;337:12–13. doi: 10.1016/0140-6736(91)93331-3. [DOI] [PubMed] [Google Scholar]
- Davoust N, Vuaillat C, Androdias G, Nataf S. From bone marrow to microglia: barriers and avenues. Trends Immunol. 2008;29:227–234. doi: 10.1016/j.it.2008.01.010. [DOI] [PubMed] [Google Scholar]
- Demuro A, Mina E, Kayed R, Milton SC, Parker I, Glabe CG. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J Biol Chem. 2005;280:17294–17300. doi: 10.1074/jbc.M500997200. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Hickman SE, Thomas CA, Cao L, Silverstein SC, Loike JD. Scavenger receptor-mediated adhesion of microglia to beta-amyloid fibrils. Nature. 1996;382:716–719. doi: 10.1038/382716a0. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Toft M, Hickman SE, Means TK, Terada K, Geula C, Luster AD. Ccr2 deficiency impairs microglial accumulation and accelerates progression of Alzheimer-like disease. Nat Med. 2007;13:432–438. doi: 10.1038/nm1555. [DOI] [PubMed] [Google Scholar]
- El Khoury JB, Moore KJ, Means TK, Leung J, Terada K, Toft M, Freeman MW, Luster AD. CD36 mediates the innate host response to beta-amyloid. J Exp Med. 2003;197:1657–1666. doi: 10.1084/jem.20021546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emre M, Geula C, Ransil BJ, Mesulam MM. The acute neurotoxicity and effects upon cholinergic axons of intracerebrally injected beta-amyloid in the rat brain. Neurobiol Aging. 1992;13:553–559. doi: 10.1016/0197-4580(92)90055-3. [DOI] [PubMed] [Google Scholar]
- Etminan M, Gill S, Samii A. Effect of non-steroidal antiinflammatory drugs on risk of Alzheimer's disease: systematic review and meta-analysis of observational studies. BMJ. 2003;327:128. doi: 10.1136/bmj.327.7407.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geula C, Bu J, Nagykery N, Scinto LF, Chan J, Joseph J, Parker R, Wu CK. Loss of calbindin-D28k from aging human cholinergic basal forebrain: relation to neuronal loss. J Comp Neurol. 2003;455:249–259. doi: 10.1002/cne.10475. [DOI] [PubMed] [Google Scholar]
- Geula C, Wu CK, Saroff D, Lorenzo A, Yuan M, Yankner BA. Aging renders the brain vulnerable to amyloid β-protein neurotoxicity. Nat Med. 1998;4:827–831. doi: 10.1038/nm0798-827. [DOI] [PubMed] [Google Scholar]
- Guilian D, Haverkamp LJ, Yu JH, Karshin W, Tom D, Li J, Kirkpatrick J, Kuo LM, Roher AE. Specific domains of beta-amyloid from Alzheimer's plaque elicit neuron killing in human microglia. J Neurosci. 1996;16:6021–6037. doi: 10.1523/JNEUROSCI.16-19-06021.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JD, Wong SS, Lieber CM, Lansbury PT., Jr Assembly of A beta amyloid protofibrils: an in vitro model for a possible early event in Alzheimer's disease. Biochemistry. 1999;38:8972–8980. doi: 10.1021/bi9904149. [DOI] [PubMed] [Google Scholar]
- Hartley DM, Walsh DM, Ye CP, Diehl T, Vasquez S, Vassilev PM, Teplow DB, Selkoe DJ. Protofibrillar intermediates of amyloid beta-protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci. 1999;19:8876–8884. doi: 10.1523/JNEUROSCI.19-20-08876.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT, Trojanowski JQ. Consensus recommendations for the postmortem diagnosis of Alzheimer disease from the National Institute on Aging and the Reagan Institute Working Group on diagnostic criteria for the neuropathological assessment of Alzheimer disease. J Neuropathol Exp Neurol. 1997;56:1095–1097. doi: 10.1097/00005072-199710000-00002. [DOI] [PubMed] [Google Scholar]
- Inoue R, Hashimoto K, Harai T, Mori H. NMDA- and beta-amyloid 1-42-induced neurotoxicity is attenuated in serine racemase knock-out mice. J Neurosci. 2008;28:14486–14491. doi: 10.1523/JNEUROSCI.5034-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul M, Lipton SA. Chemokines and activated macrophages in HIV gp120-induced neuronal apoptosis. Proc Natl Acad Sci U S A. 1999;96:8212–8216. doi: 10.1073/pnas.96.14.8212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Sokolov Y, Edmonds B, McIntire TM, Milton SC, Hall JE, Glabe CG. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J Biol Chem. 2004;279:46363–46366. doi: 10.1074/jbc.C400260200. [DOI] [PubMed] [Google Scholar]
- Kokovay E, Cunningham LA. Bone marrow-derived microglia contribute to the neuroinflammatory response and express iNOS in the MPTP mouse model of Parkinson's disease. Neurobiol Dis. 2005;19:471–478. doi: 10.1016/j.nbd.2005.01.023. [DOI] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Abeta1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Viola KL, Chromy BA, Chang L, Morgan TE, Yu J, Venton DL, Krafft GA, Finch CE, Klein WL. Vaccination with soluble Abeta oligomers generates toxicity-neutralizing antibodies. J Neurochem. 2001;79:595–605. doi: 10.1046/j.1471-4159.2001.00592.x. [DOI] [PubMed] [Google Scholar]
- Landi F, Cesari M, Onder G, Russo A, Torre S, Bernabei R. Non-steroidal anti-inflammatory drug (NSAID) use and Alzheimer disease in community-dwelling elderly patients. Am J Geriatr Psychiatry. 2003;11:179–185. [PubMed] [Google Scholar]
- Lauckner J, Frey P, Geula C. Comparative distribution of tau phosphorylated at Ser262 in pre-tangles and tangles. Neurobiol Aging. 2003;24:767–776. doi: 10.1016/s0197-4580(02)00228-2. [DOI] [PubMed] [Google Scholar]
- Lorenzo A, Yankner BA. B-Amyloid neurotoxicity requires fibril formation and is inhibited by Congo red. Proc Natl Acad Sci U S A. 1994;91:12243–12247. doi: 10.1073/pnas.91.25.12243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manelli AM, Bulfinch LC, Sullivan PM, LaDu MJ. Abeta42 neurotoxicity in primary co-cultures: effect of apoE isoform and Abeta conformation. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meda L, Baron P, Prat E, Scarpini E, Scarlato G, Cassatella MA, Rossi F. Proinflammatory profile of cytokine production by human monocytes and murine microglia stimulated with beta-amyloid[25-35] J Neuroimmunol. 1999;93:45–52. doi: 10.1016/s0165-5728(98)00188-x. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Mochizuki A, Peterson JW, Mufson EJ, Trapp BD. Amyloid load and neural elements in Alzheimer's disease and nondemented individuals with high amyloid plaque density. Exp Neurol. 1996;142:89–102. doi: 10.1006/exnr.1996.0181. [DOI] [PubMed] [Google Scholar]
- Moore KJ, El Khoury J, Medeiros LA, Terada K, Geula C, Luster AD, Freeman MW. A CD36-initiated signaling cascade mediates inflammatory effects of beta-amyloid. J Biol Chem. 2002;277:47373–47379. doi: 10.1074/jbc.M208788200. [DOI] [PubMed] [Google Scholar]
- Najjar VA. Tuftsin, a natural activator of phagocyte cells: an overview. Ann N Y Acad Sci. 1983;419:1–11. doi: 10.1111/j.1749-6632.1983.tb37086.x. [DOI] [PubMed] [Google Scholar]
- Ohm TG, Muller H, Braak H, Bohl J. Close-meshed prevalence rates of different stages as a tool to uncover the rate of Alzheimer's disease-related neurofibrillary changes. Neuroscience. 1995;64:209–217. doi: 10.1016/0306-4522(95)90397-p. [DOI] [PubMed] [Google Scholar]
- Paresce DM, Ghosh RN, Maxfield FR. Microglial cells internalize aggregates of the Alzheimer's disease amyloid beta-protein via a scavenger receptor. Neuron. 1996;17:553–565. doi: 10.1016/s0896-6273(00)80187-7. [DOI] [PubMed] [Google Scholar]
- Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, Norman BA, Baranak CC. Rofecoxib: no effect on Alzheimer's disease in a 1-year, randomized, blinded, controlled study. Neurology. 2004;62:66–71. doi: 10.1212/wnl.62.1.66. [DOI] [PubMed] [Google Scholar]
- Rogers J, Lue LF. Microglial chemotaxis, activation, and phagocytosis of amyloid beta-peptide as linked phenomena in Alzheimer's disease. Neurochem Int. 2001;39:333–340. doi: 10.1016/s0197-0186(01)00040-7. [DOI] [PubMed] [Google Scholar]
- Rogove AD, Tsirka SE. Neurotoxic responses by microglia elicited by excitotoxic injury in the mouse hippocampus. Curr Biol. 1998;8:19–25. doi: 10.1016/s0960-9822(98)70016-8. [DOI] [PubMed] [Google Scholar]
- Rosi S, Vazdarjanova A, Ramirez-Amaya V, Worley PF, Barnes CA, Wenk GL. Memantine protects against LPS-induced neuroinflammation, restores behaviorally-induced gene expression and spatial learning in the rat. Neuroscience. 2006;142:1303–1315. doi: 10.1016/j.neuroscience.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Toward a comprehensive theory for Alzheimer's disease. Hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann N Y Acad Sci. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- Sheng JG, Mrak RE, Griffin WS. Neuritic plaque evolution in Alzheimer's disease is accompanied by transition of activated microglia from primed to enlarged to phagocytic forms. Acta Neuropathol. 1997;94:1–5. doi: 10.1007/s004010050664. [DOI] [PubMed] [Google Scholar]
- Siemion IZ, Kluczyk A, Slon JJ, Zimecki M, Wieczorek Z. Immunosuppressive activity of analogs of tripeptide Lys-Arg-Pro with D-amino acid residues. Arch Immunol Ther Exp (Warsz) 1994;42:205–207. [PubMed] [Google Scholar]
- Simard AR, Rivest S. Bone marrow stem cells have the ability to populate the entire central nervous system into fully differentiated parenchymal microglia. FASEB J. 2004;18:998–1000. doi: 10.1096/fj.04-1517fje. [DOI] [PubMed] [Google Scholar]
- Szkaradkiewicz A. Phagocytosis and microbicidal capacity of human monocytes in the course of HIV infection. Immunol Lett. 1992;33:145–150. doi: 10.1016/0165-2478(92)90039-q. [DOI] [PubMed] [Google Scholar]
- Terry RD, Wisniewski HM. Ultrastructure of senile dementia and of experimental analogues. In: Gaitz CM, editor. Aging and the Brain. Plenum Press; New York: 1972. pp. 89–116. [Google Scholar]
- Thanos S, Mey J, Wild M. Treatment of the adult retina with microglia-suppressing factors retards axotomy-induced neuronal degradation and enhances axonal regeneration in vivo and in vitro. J Neurosci. 1993;13:455–466. doi: 10.1523/JNEUROSCI.13-02-00455.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevisani F, Castelli E, Foschi FG, Parazza M, Loggi E, Bertelli M, Melotti C, Domenicali M, Zoli G, Bernardi M. Impaired tuftsin activity in cirrhosis: relationship with splenic function and clinical outcome. Gut. 2002;50:707–712. doi: 10.1136/gut.50.5.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Tseng BP, Rydel RE, Podlisny MB, Selkoe DJ, Hartmann T. The oligomerization of amyloid beta-protein begins intracellularly in cells derived from human brain. Biochem Eur Arch Psychiatr Clin Neurosci. 1999;249:291–298. [Google Scholar]
- Webster SD, Galvan MD, Ferran E, Garzon-Rodriguez W, Glabe CG, Tenner AJ. Antibody-mediated phagocytosis of the amyloid beta-peptide in microglia is differentially modulated by C1q. J Immunol. 2001;166:7496–7503. doi: 10.4049/jimmunol.166.12.7496. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Golde TE, Koo EH. Abeta42-lowering nonsteroidal anti-inflammatory drugs preserve intramembrane cleavage of the amyloid precursor protein (APP) and ErbB-4 receptor and signaling through the APP intracellular domain. J Biol Chem. 2003a;278:30748–30754. doi: 10.1074/jbc.M304824200. [DOI] [PubMed] [Google Scholar]
- Weggen S, Eriksen JL, Sagi SA, Pietrzik CU, Ozols V, Fauq A, Golde TE, Koo EH. Evidence that nonsteroidal anti-inflammatory drugs decrease amyloid beta 42 production by direct modulation of gamma-secretase activity. J Biol Chem. 2003b;278:31831–31837. doi: 10.1074/jbc.M303592200. [DOI] [PubMed] [Google Scholar]
- Wieczorek Z, Zimecki M, Slon JJ, Siemion IZ. The immunomodulatory activity of tetra- and tripeptides of tuftsin-kentsin group. Peptides. 1994;15:215–221. doi: 10.1016/0196-9781(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Yankner BA. Mechanisms of neuronal degeneration in Alzheimer's disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- Zandi PP, Anthony JC, Hayden KM, Mehta K, Mayer L, Breitner JC. Reduced incidence of AD with NSAID but not H2 receptor antagonists: the Cache County Study. Neurology. 2002;59:880–886. doi: 10.1212/wnl.59.6.880. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.