

Abstract
Defects in the enamelin gene (ENAM) cause amelogenesis imperfecta (AI). Our objective was to identify the genetic etiology of enamel hypoplasia in a Caucasian proband. Our hypothesis was that ENAM was defective. The proband and his father have an AG insertion (g.13185_13186insAG; p.422FsX448) in ENAM previously identified in AI kindreds from Slovenia and Turkey. The proband, his brother, and his mother have a novel missense mutation (g.12573C>T) that substitutes leucine for a phosphorylated serine (p.S216L) in the 32-kDa enamelin cleavage product. In this family, a defect in one ENAM allele caused minor pitting or localized enamel hypoplasia, whereas defects in both alleles caused severe enamel malformations, with little or no mineral covering dentin. Ser216 is one of two serines on the 32-kDa enamelin that is phosphorylated by Golgi casein kinase and is thought to mediate calcium binding. We propose that phosphorylation of enamelin is critical for its function.
Keywords: inherited diseases, mutations, enamel, tooth, amelogenesis imperfecta
Introduction
Enamel formation is a complex process regulated by ameloblasts that requires the secretion of amelogenin (Snead et al., 1985), ameloblastin (Krebsbach et al., 1996; Hu et al., 1997a), and enamelin and their proteolytic processing by enamelysin (Mmp-20) (Bartlett et al., 1996). These genes have been individually disrupted by gene targeting, and in each case, severe enamel defects were observed in the null condition (Gibson et al., 2001; Caterina et al., 2002; Fukumoto et al., 2004; Hu et al., 2008). Enamelin functions at the mineralization front where the enamel mineral ribbons initiate and lengthen, which is along the outer surface of the ameloblast distal membrane where enamel proteins are secreted. In the absence of enamelin, the mineralization front fails, and enamel crystals do not form. Instead, mineral deposits slowly, within a layer of accumulated enamel protein and in the intercellular spaces between secretory ameloblasts, resulting in a crusty material covering the coronal dentin that cannot support mastication and readily abrades away (Hu et al., 2008).
Enamelin is the largest enamel protein, having an apparent molecular mass of 186 kDa on SDS-PAGE (Hu et al., 1997b). The C-terminal region of enamelin is short-lived. Antibodies that exclusively bind to the enamelin C-terminus specifically immunolabel the mineralization front at the enamel surface (Hu et al., 1997b), suggesting that after enamelin serves its primary function, the C-terminal domain is excised by proteolysis, re-absorbed into the ameloblast, and degraded, which precludes its accumulation in the deeper enamel. An interesting feature of the enamelin C-terminal domain is the presence of 6 highly conserved cysteines that are presumed to form disulfide bridges, but there is currently no direct evidence to support this (Hu et al., 1998; Hu et al., 2007). Most of enamelin is rapidly degraded, with the notable exception of a 32-kDa cleavage product that accumulates throughout the developing enamel layer (Uchida et al., 1991) and constitutes 1% of total protein in porcine secretory-stage enamel (Tanabe et al., 1990). The 32-kDa enamelin is the most highly conserved part of enamelin (Al-Hashimi et al., in press). The 32-kDa enamelin cleavage product can be isolated from developing porcine enamel, which has facilitated its structural and functional characterization. The porcine 32-kDa enamelin has 3 N-linked glycosylations (Yamakoshi, 1995; Yamakoshi et al., 1998) that protect the protein from further proteolytic processing by Mmp-20 (Yamakoshi et al., 2006). In vitro, the 32-kDa enamelin has properties consistent with roles in crystal nucleation and regulating crystal habit (Bouropoulos and Moradian-Oldak, 2004; Fan et al., 2008; Hu et al., 2008).
Enamelin is a member of the secretory calcium-binding phosphoprotein (SCPP) gene family. In humans, the SCPP family includes amelogenin and 21 genes clustered on chromosome 4 (Kawasaki and Weiss, 2003, 2006; Kawasaki et al., 2004). All SCPP proteins have at least one, and usually many, SXE/S(p) Golgi casein kinase target motifs (Brunati et al., 2000; Kawasaki et al., 2004). The 32-kDa enamelin has two such motifs, and these motifs are conserved in all of the 36 known mammalian enamelin sequences (Al-Hashimi et al., in press). By identifying ENAM mutations underlying defined enamel defects, we gain insight into the structural elements that are necessary for its function.
Materials & Methods
Protocol Approval
The study protocol and patient consent forms were reviewed and approved by the Institutional Review Board at the University of Michigan.
Participant Recruitment and Examination
The proband was a 19-year-old Caucasian male of Eastern European descent (Czech Republic) who presented to the General Dentistry Clinic at the University of Michigan, School of Dentistry. Oral photographs were taken of the proband, mother, and brother. The father’s oral photograph and the dental radiographs of all the participants were obtained from their dental care providers. Blood samples (5 cc) were obtained from the proband and his father, mother, and brother. Genomic DNA purification was performed with the QIA amp DNA Blood Midi Kit (Qiagen Inc., Valencia, CA, USA). The quality and quantity of the isolated DNA were determined by spectrophotometry at OD260 and OD280.
Polymerase Chain-reaction and DNA Sequence Analyses
All ENAM coding exons (exons 3 through 10), along with adjoining intron sequences, were amplified by polymerase chain-reaction (PCR). The PCR products were purified with the QIAquick PCR Purification Kit (Qiagen). The sizes and quantities of the product were analyzed on 1% agarose gels stained with ethidium bromide. The University of Michigan Sequencing Core determined the DNA sequences of the PCR products. The chromatograms were analyzed by means of FinchTV (Geospiza, Seattle, WA, USA) software. The resulting sequences were compared with the ENAM cDNA (NM_031889.2) and genomic (NC_000004.11) reference sequences and the dbSNPs database (http://www.ncbi.nlm.nih.gov/projects/SNP/).
The PCR reactions were carried out with 290-470 ng of genomic DNA and Supermix (Invitrogen, Carlsbad, CA, USA). All reactions had a denaturation at 94°C, followed by 40 cycles each with denaturation at 94°C for 30 sec, primer annealing at 55°C to 61°C for 60 sec, and product extension at 72°C for 90 sec. The final product extension was for 7 min. The exon-specific PCR amplification primer pairs (F = forward and R = reverse) and their annealing temperatures were: (exon 3) F-TGTCAACAT CGCCCTAGAA and R-GGATGACTGAGATCCTTCC at 55°C; (exon 4/5) F-GCCCCATCCATTTCCATACT and R-TGATGGC TGGGGAAATTACT at 55°C; (exon 6) F-TGTTCTCAAAT CCTGAACTCAAA and R-TGTGAGAGG ATAGGGGCAAT at 55°C; (exon 7) F-TGCCTTTTGGTTTGTTTTGG and R-AG TCGACTTGCCATGCAGTT at 55°C; (exon 8) F-GATTGGAA TCCTGGCTTCAG and R-TGCACTGGTT TTGTTTCATACC at 61°C; (exon 9) F-TGGTAAGGAGGAT TGCCAAC and R-ATCTTTGGACCACTGGCTTG at 55°C; (exon 10a) F-AC CTCGTGATCCACCTGTCT and R-CCAGTGGGGCAACAG TAGTT at 55°C; (exon 10b) F-CAGGAAACA GTACCCAGGA and R-GGGAAGGATGGGGTAAATGT at 57°C; (exon 10c) F-AGAATTTGCCCAAAGGGATT and R-GGGCTGGTTA CCTTTCTGGT at 57°C; (exon 10d) F-CAAAGGAGGCCCAA CAGTTA and R-TTGTTCCCATCTCCTCCA AG at 55°C; and (exon 10e) F-CCCTAACTTCATCCCACCAA and R-TCA TGGCCTTCTTGCTTTTT at 55°C.
Scanning Electron Microscopy (SEM)
Scanning electron microscopy was performed in the Microscopy and Image-analysis Laboratory (MIL) at the University of Michigan, Department of Cell and Developmental Biology, as described previously (Simmer et al., 2009).
Results
The proband’s teeth appeared discolored and small, with generalized spacing, consistent with hypoplastic amelogenesis imperfecta (Fig. 1). His chief complaints were esthetics and thermal sensitivity. The exposed crowns were the color of dentin, with no evidence of enamel being present. Gingival inflammation and a slightly increased overbite were also observed. Before oral photographs were taken, but after the full-mouth series of radiographs was taken, the proband had undergone gingival surgery for crown lengthening, and the maxillary posterior teeth were covered with temporary crowns. The radiographs showed no evidence of an enamel layer (Appendix). The dentin and root morphologies were within normal limits.
Figure 1.
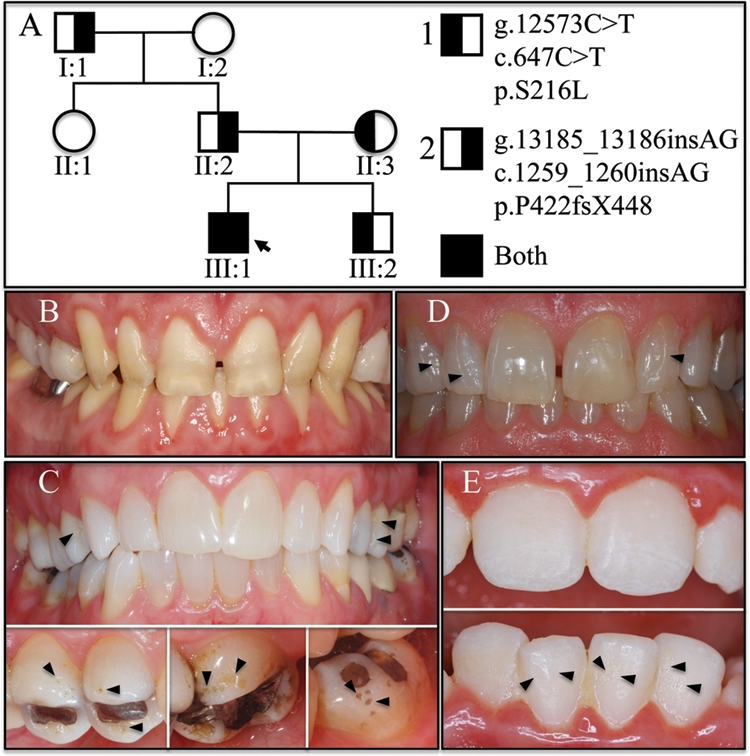
Genotypes and phenotypes of the kindred. (A) Pedigree of the family with autosomal-dominant amelogenesis imperfecta. Two mutant ENAM alleles were identified in this family: mutation 1 (g.12573C>T; c.647C>T; p.S216L) and mutation 2 (g.13185_13186insAG; c.1259_1260insAG). The proband (III:I; arrow) is represented by a completely shaded square because he is a compound heterozygote for both mutations. The proband’s mother (II:3) and younger brother (III:2) are heterozygous for mutation 1, while the father (II:2) is heterozygous for mutation 2. (B) Oral photograph of the proband showing generalized severe enamel hypoplasia. (C) Oral photographs of the mother (p.S216L), showing highly polished enamel with localized pitting (arrowheads). (D) Oral photograph of the father (p.P422fsX448), showing polished enamel with localized pitting defects (arrowheads). (E) Oral photographs of the younger sibling (p.S216L), showing chalky-white enamel resembling fluorosis with localized pitting defects (arrowheads).
Mutation analyses identified 2 disease-associated mutations. The first was a C to T transition (g.12573C>T; c.647C>T) that replaces Ser216 with Leu216 (p.S216L). This is a novel mutation that has never before been reported and is not listed in the single-nucleotide-polymorphism database. The second ENAM mutation is an AG insertion (g.13185_13186insAG; c.1259_1260 insAG) that leads to the translation of 421 normal amino acids (including the 39-amino-acid signal peptide), followed by 26 extraneous amino acids (VPNLALLFAMKKSKIQRRSPWVQ KNK), and then terminates after codon 448 (p.P422fsX448). This frameshift deletes 721 amino acids from the enamelin C-terminus (Pro422 to Gln1142), but does not alter the 32-kDa cleavage product. This frameshift mutation has previously been reported to cause amelogenesis imperfecta (Ozdemir et al., 2005; Pavlic et al., 2007; Kang et al., 2009).
Although both ENAM mutations were in the exon 10 coding region, they were on different PCR amplification products, so it was not immediately apparent if the 2 mutations were on the same or different alleles. According to the family history, the dental enamel of the mother, brother, and father was normal. The younger brother’s panorex, taken at age 7, showed enamel in good contrast with dentin (Appendix). An oral exam revealed that the enamel was chalky-white, with patches of surface roughness (Fig. 1E) and enamel pits, particularly on the cusp tips. Genetic analysis demonstrated that the younger brother had the first mutation (g.12573C>T), which substituted leucine for the phosphoserine at position 216, but did not have the second mutation, the AG insertion that shifted the reading frame. The mother had the same ENAM genotype, with one allele that substituted a leucine for Ser216, along with a normal wild-type ENAM allele. The mother’s teeth appeared to be highly polished, especially her anterior teeth (Fig. 1C). Minor pitting was evident, which was most obvious on the mesial-buccal area of the maxillary first bicuspid (#5). The father’s anterior teeth also exhibited a polished appearance (Fig. 1D). Genetic analysis of the father showed that he had a wild-type ENAM allele and a mutant allele with the AG frameshift. Chromatograms showing the mutations identified in each person and additional oral photographs and dental radiographs are provided in the Appendix.
Naturally avulsed primary maxillary right central incisors that had been saved by the proband and his younger brother were analyzed by scanning electron microscopy and compared (Fig. 2). The mineral covering dentin in the proband was only 4% as thick as in his brother’s tooth (22.5 µm vs. 512 µm) and had disappeared from the incisal edge and lingual surfaces. These findings support the conclusion that, in this family, a subtle AI phenotype (localized hypoplastic AI) is caused by defects in a single ENAM allele, and that severe enamel hypoplasia is the phenotype when both ENAM alleles are defective.
Figure 2.

SEM evaluation of primary incisors from the proband and his brother. (Top row) Scanning electron micrographs of maxillary right primary central incisor (tooth E) from the proband (III:I; p.S216L and p.P422fsX448). A thin layer of mineral measuring 22.5 µm thick covers dentin on the facial surface (arrowheads), but is absent from the cusp tip. (Bottom row) SEM of maxillary right primary central incisor from the proband’s younger brother (III:2; p.S216L). Bars = 100 µm.
Discussion
The range of enamel phenotypes associated with ENAM defects is becoming increasingly well-characterized. The first-reported AI-causing mutation in ENAM was a heterozygous G to A transition in the first nucleotide of intron 8 that was predicted to cause a deletion of exon 8 (p.A158-Q178del) during RNA processing (Rajpar et al., 2001). The crowns of the teeth were small, thin, and yellow, with little or no enamel. The teeth looked similar to those of our proband, who had defects in both ENAM alleles. In another case, a heterozygous missense mutation truncated the enamelin protein after only 53 amino acids (p.K53X) (Mårdh et al., 2002). The resulting localized hypoplastic enamel showed prominent horizontal grooves and pits, especially in the cervical third of the crown (Kim et al., 2006). A heterozygous deletion of 1 G within a run of 7 Gs at the end of exon 9 and the beginning of intron 9 was shown to cause severe generalized enamel hypoplasia (Kida et al., 2002). The defect was predicted to cause a frameshift that would have expressed only the first 197 amino acids of enamelin (p.N197fsX277). This same mutation was found in several other families with generalized and severe thinning of the enamel (PS Hart et al., 2003; Kim et al., 2005; Pavlic et al., 2007). A heterozygous ENAM missense mutation that truncated the protein after 246 amino acids (p.S246X) caused autosomal-dominant localized enamel hypoplasia (Ozdemir et al., 2005). Radiographs showed decreased contrast between enamel and dentin, and affected members had extensive dental restorations to decrease sensitivity and improve esthetics. These early reports clearly indicated that heterozygous ENAM mutations could cause severe enamel hypoplasia.
The inheritence pattern of enamel defects associated with ENAM mutations blurred after characterization of three kindreds with severe generalized enamel hypoplasia inherited in an autosomal-recessive pattern (TC Hart et al., 2003). All three probands were homozygous for p.P422fsX448 (mutation 2 in this report). Most interestingly, all persons who were hetero-zygous for this mutation had localized hypoplastic enamel pitting. These defects were originally thought to be too minor to classify as AI, but in the light of these and later genetic analyses, they are clearly an enamel phenotype caused by a defective single allele of ENAM. In another AI kindred with severe generalized enamel hypoplasia, the proband was a compound heterozygote, with the p.P422fsX448 defect in one ENAM allele paired with a novel in-frame insertion of 7 amino acids (p.V340_M341 insSQYQYCV) in the other (Ozdemir et al., 2005). In addition, simple heterozygotes with either of these defective ENAM alleles paired with a wild-type allele displayed mild localized hypoplasia consisting mainly of well-circumscribed enamel pits. Characterization of other simple heterozygotes with the p.P422fsX448 defect showed that the enamel phenotype could range from chalky white enamel with only mild local hypoplastic alterations to local hypoplastic AI (Pavlic et al., 2007). Simple heterozyotes for another ENAM frameshift mutation (p.L998fsX1062) showed similar differences in severity (Kang et al., 2009). One affected person had chalky white enamel with localized pitting, while another had prominent horizontal bands of hypoplastic enamel affecting primarily the cervical third of the crowns. In our study, individuals with mutation 1 (p.S216L) in one ENAM allele paired with a wild-type allele had chalky-white enamel with surface roughness or highly polished enamel with some localized surface pitting.
We propose that all inherited enamel defects, including minor pitting and surface roughness, be included under the designation ‘amelogenesis imperfecta’. This is appropriate because the threshold for including or not including a phenotype within the AI designation is arbitrary. Excluding minor phenotypes would sometimes require diagnosing siblings with the same ENAM mutation as having and not having AI when one enamel phenotype is only moderately more severe than the other. If we accept that minor inherited enamel defects are AI, ENAM defects can be said to have an autosomal-dominant pattern of inheritance. Penetrance is variable, since some individuals heterozygous for the p.P422fsX448 mutation showed no detectable enamel phenotype, not even pitting (Kang et al., 2009). Expressivity is also variable, since persons heterozygous for the p.P422fsX448 mutation can exhibit chalky enamel with minor pitting or more severe horizontal grooves of enamel hypoplasia.
Enamelin defects usually show a dose effect: The enamel phenotype is typically much more severe when both ENAM alleles are defective. However, in some cases, a single defective ENAM allele can cause a severe enamel phenotype, which is presumably due to the defective enamelin protein causing a dominant-negative effect on ameloblasts or the extracellular matrix. When a severe enamel phenotype is observed, it is especially important to characterize all of the enamelin coding exons and intron borders to ascertain if the second ENAM allele is also defective.
There are now 10 novel ENAM disease-associated mutations that have been characterized (Appendix) and 2 reports of probands with severe enamel hypoplasia caused by 2 different defects in the 2 ENAM alleles (compound heterozygosity). Replacement of phosphoserine at position 216 in enamelin ablates its function and causes enamel malformations.
Supplementary Material
Acknowledgments
We thank the family who participated in this study, Dr. Kevin Hale for sending patient radiographs and photos and Dorothy Sorenson for her assistance with the SEM analyses. USPHS Research Grants DE015846 and DE011301 (NIDCR/NIH) supported this investigation.
Footnotes
A supplemental appendix to this article is published electronically only at http://jdr.sagepub.com/Appendix
All authors declare that there are no conflicting interests.
References
- Al-Hashimi N, Sire J-Y, Delgado S. (2009). Evolutionary analysis of mammalian enamelin, the largest enamel protein, supports a crucial role for the 32 kDa peptide and reveals selective adaptation in rodents and primates. J Mol Evol 69:635-656 [DOI] [PubMed] [Google Scholar]
- Bartlett JD, Simmer JP, Xue J, Margolis HC, Moreno EC. (1996). Molecular cloning and mRNA tissue distribution of a novel matrix metalloproteinase isolated from porcine enamel organ. Gene 183:123-128 [DOI] [PubMed] [Google Scholar]
- Bouropoulos N, Moradian-Oldak J. (2004). Induction of apatite by the cooperative effect of amelogenin and the 32-kDa enamelin. J Dent Res 83:278-282 [DOI] [PubMed] [Google Scholar]
- Brunati AM, Marin O, Bisinella A, Salviati A, Pinna LA. (2000). Novel consensus sequence for the Golgi apparatus casein kinase, revealed using proline-rich protein-1 (PRP1)-derived peptide substrates. Biochem J 351(Pt 3):765-768 [PMC free article] [PubMed] [Google Scholar]
- Caterina JJ, Skobe Z, Shi J, Ding Y, Simmer JP, Birkedal-Hansen H, et al. (2002). Enamelysin (matrix metalloproteinase 20)-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem 277:49598-49604 [DOI] [PubMed] [Google Scholar]
- Fan D, Lakshminarayanan R, Moradian-Oldak J. (2008). The 32kDa enamelin undergoes conformational transitions upon calcium binding. J Struct Biol 163:109-115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukumoto S, Kiba T, Hall B, Iehara N, Nakamura T, Longenecker G, et al. (2004). Ameloblastin is a cell adhesion molecule required for maintaining the differentiation state of ameloblasts. J Cell Biol 167:973-983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson CW, Yuan ZA, Hall B, Longenecker G, Chen E, Thyagarajan T, et al. (2001). Amelogenin-deficient mice display an amelogenesis imperfecta phenotype. J Biol Chem 276:31871-31875 [DOI] [PubMed] [Google Scholar]
- Hart PS, Michalec MD, Seow WK, Hart TC, Wright JT. (2003). Identification of the enamelin (g.8344delG) mutation in a new kindred and presentation of a standardized ENAM nomenclature. Arch Oral Biol 48:589-596 [DOI] [PubMed] [Google Scholar]
- Hart TC, Hart PS, Gorry MC, Michalec MD, Ryu OH, Uygur C, et al. (2003). Novel ENAM mutation responsible for autosomal recessive amelogenesis imperfecta and localised enamel defects. J Med Genet 40:900-906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu C-C, Fukae M, Uchida T, Qian Q, Zhang CH, Ryu OH, et al. (1997a). Sheathlin: cloning, cDNA/polypeptide sequences, and immunolocalization of porcine enamel proteins concentrated in the sheath space. J Dent Res 76:648-657 [DOI] [PubMed] [Google Scholar]
- Hu C-C, Fukae M, Uchida T, Qian Q, Zhang CH, Ryu OH, et al. (1997b). Cloning and characterization of porcine enamelin mRNAs. J Dent Res 76:1720-1729 [DOI] [PubMed] [Google Scholar]
- Hu C-C, Simmer JP, Bartlett JD, Qian Q, Zhang C, Ryu OH, et al. (1998). Murine enamelin: cDNA and derived protein sequences. Connect Tissue Res 39:47-61 [DOI] [PubMed] [Google Scholar]
- Hu JC, Chun YH, Al Hazzazzi T, Simmer JP. (2007). Enamel formation and amelogenesis imperfecta. Cells Tissues Organs 186:78-85 [DOI] [PubMed] [Google Scholar]
- Hu JC, Hu Y, Smith CE, McKee MD, Wright JT, Yamakoshi Y, et al. (2008). Enamel defects and ameloblast-specific expression in ENAM knock-out/LacZ knock-in mice. J Biol Chem 283:10858-10871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HY, Seymen F, Lee SK, Yildirim M, Tuna EB, Patir A, et al. (2009). Candidate gene strategy reveals ENAM mutations. J Dent Res 88:266-269 [DOI] [PubMed] [Google Scholar]
- Kawasaki K, Weiss KM. (2003). Mineralized tissue and vertebrate evolution: the secretory calcium-binding phosphoprotein gene cluster. Proc Natl Acad Sci USA 100:4060-4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki K, Weiss KM. (2006). Evolutionary genetics of vertebrate tissue mineralization: the origin and evolution of the secretory calcium-binding phosphoprotein family. J Exp Zoolog B Mol Dev Evol 306:295-316 [DOI] [PubMed] [Google Scholar]
- Kawasaki K, Suzuki T, Weiss KM. (2004). Genetic basis for the evolution of vertebrate mineralized tissue. Proc Natl Acad Sci USA 101:11356-11361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kida M, Ariga T, Shirakawa T, Oguchi H, Sakiyama Y. (2002). Autosomal-dominant hypoplastic form of amelogenesis imperfecta caused by an enamelin gene mutation at the exon-intron boundary. J Dent Res 81:738-742 [DOI] [PubMed] [Google Scholar]
- Kim JW, Seymen F, Lin BP, Kiziltan B, Gencay K, Simmer JP, et al. (2005). ENAM mutations in autosomal-dominant amelogenesis imperfecta. J Dent Res 84:278-282 [DOI] [PubMed] [Google Scholar]
- Kim JW, Simmer JP, Lin BP, Seymen F, Bartlett JD, Hu JC. (2006). Mutational analysis of candidate genes in 24 amelogenesis imperfecta families. Eur J Oral Sci 114(Suppl 1):3-12 [DOI] [PubMed] [Google Scholar]
- Krebsbach PH, Lee SK, Matsuki Y, Kozak CA, Yamada K, Yamada Y. (1996). Full-length sequence, localization, and chromosomal mapping of ameloblastin: a novel tooth-specific gene. J Biol Chem 271:4431-4435 [DOI] [PubMed] [Google Scholar]
- Mårdh CK, Backman B, Holmgren G, Hu JC, Simmer JP, Forsman-Semb K. (2002). A nonsense mutation in the enamelin gene causes local hypoplastic autosomal dominant amelogenesis imperfecta (AIH2). Hum Mol Genet 11:1069-1074 [DOI] [PubMed] [Google Scholar]
- Ozdemir D, Hart PS, Firatli E, Aren G, Ryu OH, Hart TC. (2005). Phenotype of ENAM mutations is dosage-dependent. J Dent Res 84:1036-1041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlic A, Petelin M, Battelino T. (2007). Phenotype and enamel ultrastructure characteristics in patients with ENAM gene mutations g.13185-13186insAG and 8344delG. Arch Oral Biol 52:209-217 [DOI] [PubMed] [Google Scholar]
- Rajpar MH, Harley K, Laing C, Davies RM, Dixon MJ. (2001). Mutation of the gene encoding the enamel-specific protein, enamelin, causes autosomal-dominant amelogenesis imperfecta. Hum Mol Genet 10:1673-1677 [DOI] [PubMed] [Google Scholar]
- Simmer JP, Hu Y, Lertlam R, Yamakoshi Y, Hu JC. (2009). Hypomaturation enamel defects in Klk4 knockout/LacZ knockin mice. J Biol Chem 284:19110-19121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snead ML, Lau EC, Zeichner-David M, Fincham AG, Woo SL, Slavkin HC. (1985). DNA sequence for cloned cDNA for murine amelogenin reveals the amino acid sequence for enamel-specific protein. Biochem Biophys Res Commun 129:812-818 [DOI] [PubMed] [Google Scholar]
- Tanabe T, Aoba T, Moreno EC, Fukae M, Shimizu M. (1990). Properties of phosphorylated 32 kd nonamelogenin proteins isolated from porcine secretory enamel. Calcif Tissue Int 46:205-215 [DOI] [PubMed] [Google Scholar]
- Uchida T, Tanabe T, Fukae M, Shimizu M. (1991). Immunocytochemical and immunochemical detection of a 32 kDa nonamelogenin and related proteins in porcine tooth germs. Arch Histol Cytol 54:527-538 [DOI] [PubMed] [Google Scholar]
- Yamakoshi Y. (1995). Carbohydrate moieties of porcine 32 kDa enamelin. Calcif Tissue Int 56:323-330 [DOI] [PubMed] [Google Scholar]
- Yamakoshi Y, Pinheiro FH, Tanabe T, Fukae M, Shimizu M. (1998). Sites of asparagine-linked oligosaccharides in porcine 32 kDa enamelin. Connect Tissue Res 39:39-46 [DOI] [PubMed] [Google Scholar]
- Yamakoshi Y, Hu JC, Fukae M, Iwata T, Simmer JP. (2006). How do MMP-20 and KLK4 process the 32 kDa enamelin? Eur J Oral Sci 114(Suppl 1):45-51 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.