

Abstract
A dramatic stage-migration in diagnosis of prostate cancer has led to earlier detection of clinically localized carcinoma and an increased use of radiation therapy. The p53 protein responds to irradiation-induced DNA damage by removing critically damaged cells from the proliferative pool. This review will focus on the dominant role that p53-dependent cellular senescence, rather than cell death, plays in determining the radiosensitivity of human prostate cancer cells in vitro. The finding that senescence is a primary mechanism of tumor regression indicates that p53 activators or downstream effectors may prove effective in radiosensitizing some carcinoma of the prostate.
Keywords: radiosensitization, prostate cancer, p53, nutlin, Akt, senescence, PTEN, MDM2
RADIOSENSITIZATION OF PROSTATE CANCER
In the last 15 years, active screening for prostate cancer has lead to declining rates of mortality in the United States and a significantly decreased proportion of men presenting with advanced, high risk, disease. A majority of prostate carcinomas (CaP) are now detected while the disease is still clinically localized and with intermediate-to-low risk factor characteristics.1 However, the incidence of CaP remains high and optimal treatment of men with intermediate risk CaP continues to be controversial.2,3 Conventional-dose (72 Gy) external beam radiation is elected as a preferred treatment modality by many of these patients.1 However, there is evidence that conventional-dose radiation often does not provide complete tumor eradication with a resultant radiorecurrent prostate cancer and five year distant metastasis-free survival of less than 80%.4,5 Given this problem, it is not surprising that radiotherapy dose escalation has been recommended for patients with poor prognostic features (e.g., positive surgical margins);6 but the toxicity associated with dose-escalated therapy is not trivial and it is recognized that 5-10% of all radiotherapy patients develop severe acute or late effects.7 Accordingly, there is intense interest in understanding critical determinants in the efficacy of irradiation (IR)-induced CaP cell killing and developing targeted therapeutic strategies for radiosensitization.
Curative radiation therapy aims at preventing tumor regrowth by inducing tumor cell death and loss of reproductive integrity. These processes together are referred to as clonogenic death. Clonogenic cells are defined as tumor cells possessing the capacity to produce of colony of progeny and therefore contribute to the regrowth of the tumor. Radiation therapy creates substantial DNA damage that is recognized by the tumor suppressor p53 and forces most cells to either undergo programmed cell death (e.g., apoptosis), necrosis or enter a state of reproductive death (i.e., cellular senescence). Radiation therapy is conventionally delivered in fractions of 1.8-2.0 Gy doses for five days a week until a total of 60-80 Gy is reached. After sequential doses, more than 99% of the malignant cells are typically killed. However, the surviving fraction may amount to more than a million cells per gram of tumor volume.8 These clonogenic survivors retain the potential to actively contribute to radiorecurrent prostate cancer and are a major target for radiosensitization. However, the cellular and molecular heterogeneity of CaP make this subpopulation of radioresistant cells a difficult target to strike using a single modality of adjunctive therapy.
The effectiveness of radiotherapy in treating prostate cancer varies significantly and inherently depends on the molecular context of the tumor. Numerous proteins are often mutated, resulting in activation of oncogenes and losses of tumor suppressor proteins. There is no dominant molecular profile that initiates or maintains prostatic carcinogenesis, but rather a complex mixture of mutations that disrupt cell cycle control and cell death pathways. Cell cycle proteins commonly mutated at early to late stages of prostate cancer progression include Rb, p53, p14, p16 and p27.9-13 Mutations within these proteins result in defective cell cycle checkpoint control, leading to further chromosomal instability and inactivation of the protective reproductive death pathway termed, cellular senescence. The MAPK and PI3K-AKT pathways are also commonly the sight of mutations that promote cell proliferation and block apoptosis. Combined mutations within and among these pathways may limit the success of radiation therapy. Knowledge of the pathways altered, prior to initiating radiation therapy, would provide a sort of manual for clinicians to optimize radiosensitization strategies based on the individual's profile. The use of small-molecule inhibitors aimed at mutated pathways in combination with radiation may improve the success of treatment and prolong survival of the patient.
p53
Although the frequency of p53 mutations in early prostate cancer is low, heterozygous loss of function mutations often accompany late stage carcinoma.14,15 Current models of the molecular network coordinating responses to DNA damage place p53 at the crossroads of several stress response pathways critical for maintaining genome integrity; including cell cycle arrest, DNA repair, mitotic catastrophe, apoptosis and cellular senescence (Fig. 1). How p53 directs cells down each of these alternative avenues of cell fate determination appears to differ among cells of various origins.16-20 It is important that each of these distinct forms of p53-dependent clonogenic cell death be collectively evaluated when investigating new therapeutic strategies for increasing the radiosensitivity of CaP cells.
Figure 1.
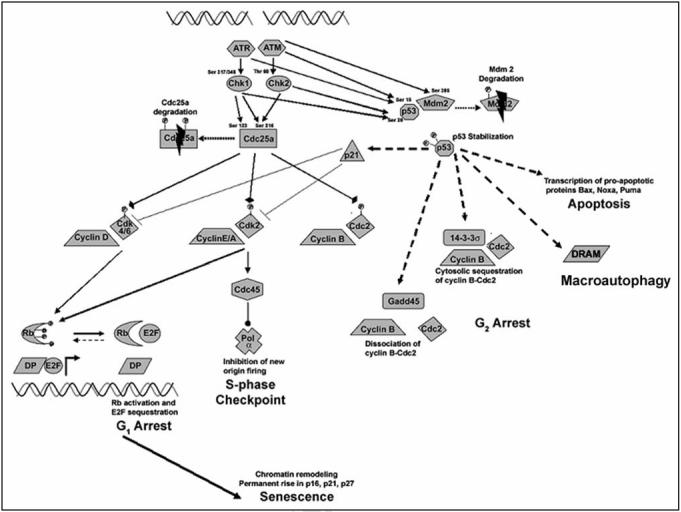
Response of p53 to DSBs generated by ionizing radiation. Activated p53 can induce cell cycle arrest at the G1, S and G2 phases, trigger apoptosis or force a cell into cellular senescence. Stabilization of p53 protein is mediated by multiple phosphorylations on p53 and Mdm2 via the ATM/ATR and Chk1/2 kinases. p53-mediated transcription of p21 inhibits both cyclin D-cdk4/6 (G1 arrest) and cyclin E/A-cdk2 (S-phase arrest). Cyclin-cdk inhibition allows unphosporylated Rb to sequester E2F necessary for transition into S phase. Prolonged p53 activation may lead to chromatin remodeling and permanent cell cycle arrest (senescence). p53-mediated transcription of 14-3-3s and Gadd45 inhibit cyclin B-cdc2 (G2 arrest). Prolonged p53 activation can also trigger apoptosis through transcription of proapoptic proteins bax, noxa and puma. p53-mediated transription of DRAM can lead to cell macroautophagy.
P53 ACTIVATION
Since p53 function is crucial to coordinating the DNA damage response, multiple upstream pathways control p53 activation. While the list of stress signals that activate p53 continues to grow, including hypoxia, ribosomal stress and loss of cell-cell contacts, the primary pathways appear to be oncogenic stress and DNA damage. Interestingly, different stress signals use different mechanisms for p53 activation (Fig. 2). Under resting conditions, the ubiquitin degradation pathway maintains a high rate of p53 turnover that is dependent on the E3 ubiquitin ligase, murine double-minute 2 (MDM2).21 Hypophosphorylated MDM2 binds the p53 C-terminus and targets p53 for degradation.22 Moreover, the p53 binding domain of MDM2 overlaps with the DNA binding domain of p53, effectively impairing p53 functionality.23 The p53 protein is also regulated by MDMX, a homologue of MDM2, that binds MDM2 through it's C-terminal RING domain and stimulates the MDM2-dependent ubiquitination of p53.21,24 IR-induced DNA damage triggers a phosphorylation cascade involving ataxia telangiectasia mutated (ATM) and ATM-related (ATR) proteins that prevent p53 degradation through multiple phosphorylations on MDM2, MDMX and p53.25-28
Figure 2.
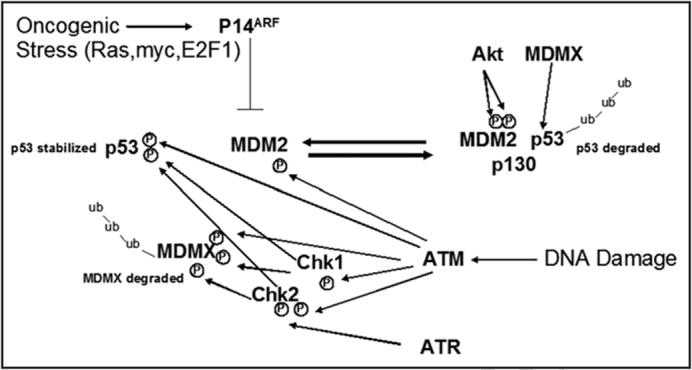
Mechanisms of p53 stabilization. Two primary pathways serve to activate p53: oncogneic stress and DNA damage. Without stimulus, p53 is ubiquitinated and targeted for proteosomal degradation with the help of mdm2, mdmx and p300. Oncogenic stress triggers an up-regulation of p14, which can stabilize p53 by binding to mdm2 and preventing p53 degradation. DNA damage activates ATM/ATR which can phosphorylate p53, mdm2 and mdmx directly or indirectly through chk1/2.
Active p53 functions as a transcriptional regulator within a complex network involving the selective transactivation and transrepression of multiple genes involved in cell-cycle control and apoptosis. p53 functions as a tetrameric transcription factor and the hetero-oligomerization between a heterozygous mutant allele and wild-type p53 proteins may be sufficient to alter the function of p53 tetramers. The dominant negative actions of missense p53 mutations were first demonstrated in experiments with ectopic expression of wild-type and mutant p53 proteins17,29 and more recently in thymocytes expressing p53 point mutations at single-copy levels.30 Alternatively, mutant p53 alleles may impart a gain of function (GOF) that could also impact tumor development and the progression of prostate cancer.31-33
A DOMINANT ROLE FOR P53-DEPENDENT CELLULAR SENESCENCE IN RADIOSENSITIZATION OF PROSTATE CANCER CELLS
Many studies of in vitro radiosensitivity have used apoptosis assays to assess the efficacy of cell killing and have been limited to a few CaP cell lines that were derived from metastatic lesions containing p53 mutations rarely observed in organ-confined tumors.34-38 The best characterized lines include PC-3 and DU145 cells that harbor pairs of deleted or inactivated p53 alleles and display a complete loss of p53 function, a condition often associated with decreased radiosensitivity7 (Table 1). However, in cultures of CaP cells retaining a functional p53 protein (LNCaP and 22Rv1) cellular senescence is the dominant form of clonogenic death after a single exposure to IR. Alternative stress response pathways controlled by this tumor suppressor, including cell cycle arrest, DNA damage repair, mitotic catastrophe and apoptosis, contributed significantly less to radiation-induced clonogenic death.39 Although autophagy is an additional form of cell death that has recently been reported to enhance the radiosensitivity of PC-3 and DU145 cells,37 neither of these cell lines express a functional p53. Thus, in the contemporary setting of earlier detection where complete p53 inactivation is rare, the induction of terminal growth arrest may be a primary mode of clonogenic death in radiation therapy of prostate cancer. This would be consistent with the observation that in some patients complete regression of prostatic tumors can take more than a year after completing radiation therapy.
Table 1.
Relationship between p53 status and radiosensitivity of human prostate cancer cells.
| Line | p53 Allele | p53 Status | Radiosensitivity (Sf2) |
|---|---|---|---|
| Abbreviations: Wt = wild-type, ND = Not determined, SF2 = Surviving fraction after a 2 Gy dose of irradiation. | |||
| LNCaP | Wt/Wt69 | Wt | 32.6 |
| 22Rv1 | Q331R/Wt70 | Partial function | 30.7 |
| DU145 | P223L/V274F69 | Dominant negative | 80.2 |
| PC-3 | Deleted | Null | 52.2 |
| Patient Q | R282Q | Gain of function33 | ND |
Recent evidence indicates that tumor regression after radiation therapy might involve the phagocytosis of senescent cells by macrophages and the autophagy of senescent cells themselves. Pronounced alterations in the endosomal/lysosomal pathway are evident in senescent cells. Lipofuscin accumulation at the level of autophagic vacuoles accounts for the increased granularity observed in the side-scatter profile of senescent cells and is a hallmark of cellular senescence. Autophagy is an ancient pathway for homeostatic turnover of long-lived intracellular components and for nutrient acquisition during starvation and stress. Recently, p53 has been shown to positively modulate damage-regulated autophagy modulator (DRAM)-dependent autophagy.40 Thus, IR-induced activation of p53, cellular senescence and autophagy may contribute to the tumor regression observed in vivo when investigators have experimentally “turned on” the p53 gene in tumor-bearing mice.41,42
INITIATION AND MAINTENANCE OF CELLULAR SENESCENCE
The p53, p21 and Rb tumor suppressors are important senescence regulators. p53, p21 and Rb are transiently activated in response to stressors that induce ‘premature’ senescence in tumor cells. Transactivation of the cyclin-dependent kinase (CDK) inhibitor p21 appears most relevant as a downstream mediator of p53-dependent terminal growth arrest.43 How p21 and other CDK inhibitors (e.g., p16Ink4a) promote senescence is not precisely known, but one well-studied mechanism for p21-induced senescence involves the activation of Rb. Rb family members (Rb, p107, p130) are corepressors of the E2F transcription factors required for cell proliferation. With an accumulation of CDK inhibitors and the onset of senescence arrest, Rb is converted to an active hypophosphorylated form that stably interacts with, and sequesters, E2F and other proteins that influence gene expression and promote cell cycle progression.44 Notably, the cellular levels of p53, p21 and active Rb do not remain elevated after the onset of senescence, suggesting that molecular interactions required to initiate the senescent state may be dispensable for its maintenance. For example, conditional expression of p53, p21, or p16 causes cellular senescence in many settings, and the cells remain in a senescent state after promoter shutoff.45-47 Because expression of another CDK inhibitor, p16, rises as p21 goes down, it has been suggested that p16 may be responsible for maintaining a stable growth arrest, while others contend that both p53 and p16 function in the maintenance of growth arrest in senescent cells.48
Another recent explanation for the permanence of cellular senescence is that p16 enables Rb to establish heterochromatin changes that maintain E2F-responsive promoters in an irreversibly silenced state and no longer dependent on the presence of p16 or Rb.44 These unusual foci of heterochromatin are physically associated with tightly packed and trimethylated histone proteins (e.g., H3K9) and the histone methyltransferase Suv39h1.49,50 Whether these p16/Rb-initiated modifications of heterochromatin are truly irreversible remains to be determined. Moreover, there is evidence that the premature senescence induced by drugs and IR in tumor cells can occur in the absence of p53, p21 as well as p16, indicating that additional genes and pathways may also mediate damage-induced senescence of tumor cells.51
PRIMING THE P53 PATHWAY TO RESPOND TO RADIATION THERAPY
p53 activity is regulated by the E3 ubiquitin ligase MDM2, that binds and targets p53 for degradation.52 Overexpression of MDM2 has been reported in prostate cancer cells and may serve to protect those cells from p53-mediated cellular senescence in response to irradiation.53 Thus, targeting MDM2 seems to be a reasonable target for radiosensitization. Investigators have down-regulated MDM2 using antisense and successfully radiosensitized LNCaP cells.54,55 While antisense raised against MDM2 demonstrated that regulation of p53 is critical for radiosensitization, the use of antisense may prove to be difficult in the clinical setting.
Recently, a selective small-molecule inhibitor of MDM2-p53 binding, nutlin-3 has been identified and characterized. Nutlin-3 disrupts MDM2 binding and activates p53 in cell culture at a concentration of 5-10 μM or inhibits tumor growth when given orally at 200 mg/kg.56 Nutlin-3 specifically activated the p53 pathway in numerous cancer cells possessing wild-type p53, inducing cell cycle arrest and apoptosis to varying extents.57 Since nutlin-3 uncouples p53 from MDM2-mediated degradation, treatment with the drug prior to radiation treatment may increase steady-state levels of p53, effectively priming the cellular ability to respond maximally to radiation (Fig. 3). Investigators first showed that administration of nutlin-3 prior to irradiation can effectively radiosensitize wild-type p53 lung cancer cells.35 We have recently shown that disruption of p53-MDM2 interactions by the small-molecule MDM2 antagonist nutlin-3 can effectively sensitize prostate cancer cells to a clinically-relevant dose (2 Gy) of IR, provided these CaP cells express wild-type p53.39 Moreover, the increase in clonogenic cell death was entirely attributable to an increased induction of p53-dependent cellular senescence.
Figure 3.
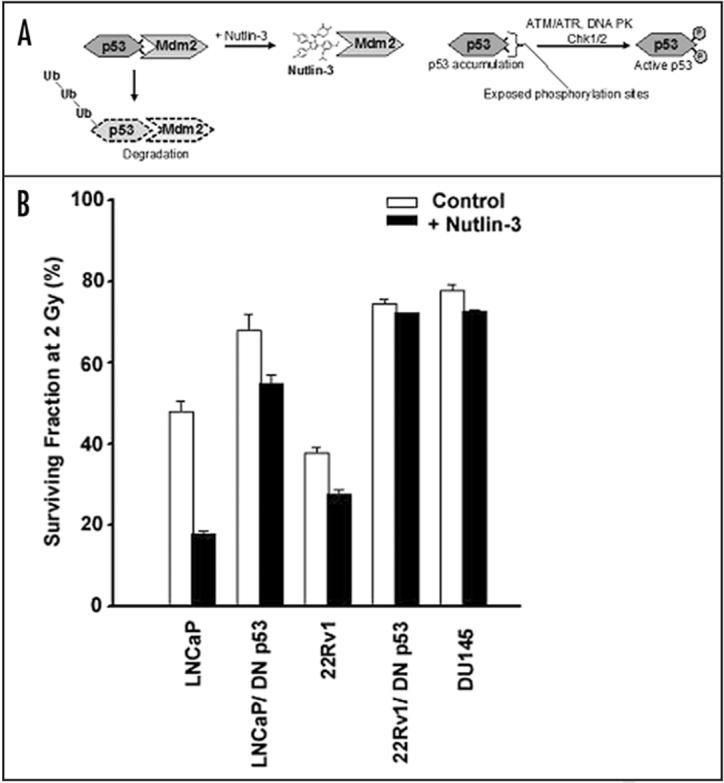
Uncoupling of p53 from MDM2-mediated degradation with the small molecule inhibitor, nutlin-3 radiosensitizes prostate cancer cells. (A) MDM2 binds and targets p53 to the proteosome pathway. The small molecule inhibitor, nutlin-3 specifically binds MDM2 and prevents its association with p53. As a result, steady-state levels of p53 rise and are available for immediate phosphorylation from the DNA damage response pathway. (B) Surviving fraction of LNCaP and 22Rv1 prostate cancer cells possessing functional p53 or transfected with a dominant negative construct (DN p53) were pre-treated with nutlin-3 prior to a 2 Gy dose of irradiation. DU145 prostate cancer cells, lacking a functional p53 was treated in a similar fashion.
While nutlin-3 may radiosensitize some tumors by “priming” the p53 pathway, it seems clear that this strategy will not apply to all prostate tumors. p53 activation requires both MDM2 degradation and MDMX inactivation. Nutlin-3 does not disrupt HDMX binding of MDM2 and MDMX overexpression prevents p53 activation by nutlin-3.58 The observations suggest that further development of small-molecule inhibitors of MDMX, or alternative molecular targets, may be useful in combination with nutlin-3 to provide a clinically effective radioresponse. Moreover, p53 activators could have an adverse outcome in patients with GOF p53 mutant alleles and it will be essential to determine their p53 status prior to initiating treatment. Because prostate cancer is slow growing and is now often detected while still localized, the clinician may some day have the opportunity to individualize treatment based on knowledge of the patient's p53 status.
GENETIC INTERACTIONS BYPASSING CELLULAR SENESCENCE IN PROSTATE CANCER PROGRESSION
Rb inactivation is common in prostate cancer and generally precedes somatic alterations affecting the tumor suppressor genes p53 and phosphatase and tensin homologue (PTEN).12,14,59 Interactions among these three major tumor suppressor genes appear to directly influence tumor development in the mouse prostate.60,61 For instance, conditional deletion of an Rb allele specifically in the mouse prostate epithelium results in the development of focal hyperplasia but not tumorigenesis,62 while acute inactivation of PTEN alone induces a p53-dependent cellular senescence.60 In contrast, when combined with inactivation of the Rb family proteins (Rb/p107/p130) or p53, a loss of PTEN either elicits an accelerated rate of tumor progression or development of invasive adenocarcinoma in mice, respectively.60,61 These transgenic studies establish that a p53-mediated senescence response restricts cell proliferation after loss of PTEN in the mouse prostate in vivo and that a concomitant loss of either Rb or p53 is sufficient to bypass this impediment to tumor growth.
AKT AS AN ADJUNCTIVE TARGET To P53 IN RADIOSENSITIZATION OF PROSTATE CANCER
Small molecule inhibitors of PI3K such as LY294002 have been reported to effectively radiosensitize LNCaP cells.63 Thus, inactivating mutations of PTEN may limit IR-induced cell killing in prostate tumors due to unrestrained Akt activity. This may be related to the observation that the PI3K-Akt pathway effectively inhibits p53 by increasing its degradation via the MDM2 pathway (Fig. 4).64-66 More recently, however, it has been reported that activation of PI3K signaling results in the downstream activation of p53 and the p53-dependent senescence pathway.67,68 This discrepancy may be the result of oncogenic stress stimulating p14, which would inactivate Mdm2 regardless of the increased shuttling into the nucleus via Akt phosphorylation. Thus it is unclear whether targeting the Akt pathway in conjunction with p53 activation will synergize or antagonize the radioresponsiveness of prostate cancer cells. Future studies will be directed at resolving this controversy in order to understand how and when these tumor suppressor pathways interact to influence the radiosensitivity of CaP cells.
Figure 4.
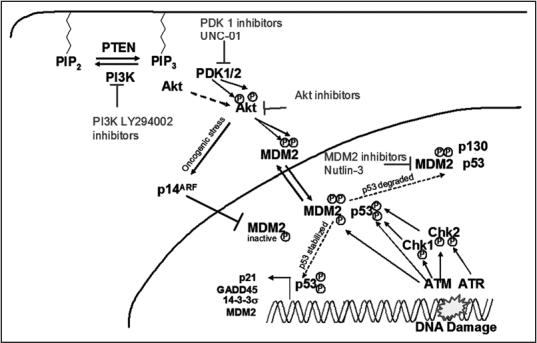
Crosstalk between the PI3K/AKT pathway and p53 activation: Potential stratagies for combined radiosensitization. Gain of function mutations in PI3K or loss of function mutations in PTEN result in recruitment of Akt and PDK1/2 to the membrane where Akt is phosphorylated. Active Akt can phosphorylate MDM2 increasing cytoplasmic to nuclear shuttling and effectively dampening the p53 pathway. The effectiveness of the p53 pathway can be augmented by increasing p53 stability (nutlin-3), or targeting PI3K (LY294002), PDK1 (UNC-01) or Akt simultaneously.
ACKNOWLEDGEMENTS
Supported in part by grants from the NIH (R01098195) and the Brody Brothers Foundation Endowment (#997729).
ABBREVIATIONS
- CaP
prostate carcinoma
- IR
ionizing radiation
- MDM2
murine double-minute
- ATM
ataxia teleangetasia mutated
- ATR
ATM-related
- DSB
double strand break
- CDK
cyclin-dependent kinase
- PTEN
phosphatase and tensin homologue
References
- 1.Cooperberg MR, Broering JM, Litwin MS, Lubeck DP, Mehta SS, Henning JM, Carroll PR. The contemporary management of prostate cancer in the United States: Lessons from the cancer of the prostate strategic urologic research endeavor (CapSURE), a national disease registry. J Urol 2004. 171:1393–401. doi: 10.1097/01.ju.0000107247.81471.06. [DOI] [PubMed] [Google Scholar]
- 2.Nichol AM, Warde P, Bristow RG. Optimal treatment of intermediate-risk prostate carcinoma with radiotherapy: Clinical and translational issues. Cancer. 2005;104:891–905. doi: 10.1002/cncr.21257. [DOI] [PubMed] [Google Scholar]
- 3.Ward JF, Blute ML. Use and timing of radiotherapy in high-risk prostate cancer. Jama. 2004;291:2817. doi: 10.1001/jama.291.23.2817-a. author reply 2817-8. [DOI] [PubMed] [Google Scholar]
- 4.Kupelian PA, Potters L, Khuntia D, Ciezki JP, Reddy CA, Reuther AM, Carlson TP, Klein EA. Radical prostatectomy, external beam radiotherapy <72 Gy, external beam radiotherapy > or =72 Gy, permanent seed implantation, or combined seeds/external beam radiotherapy for stage T1-T2 prostate cancer. Int J Radiat Oncol Biol Phys. 2004;58:25–33. doi: 10.1016/s0360-3016(03)00784-3. [DOI] [PubMed] [Google Scholar]
- 5.Zelefsky MJ, Ben-Porat L, Chan HM, Fearn PA, Venkatraman ES. Evaluation of postradio-therapy PSA patterns and correlation with 10-year disease free survival outcomes for prostate cancer. Int J Radiat Oncol Biol Phys. 2006;66:382–8. doi: 10.1016/j.ijrobp.2006.05.012. [DOI] [PubMed] [Google Scholar]
- 6.Pollack A, Zagars GK, Starkschall G, Antolak JA, Lee JJ, Huang E, von Eschenbach AC, Kuban DA, Rosen I. Prostate cancer radiation dose response: Results of the M. D. Anderson phase III randomized trial. Int J Radiat Oncol Biol Phys. 2002;53:1097–105. doi: 10.1016/s0360-3016(02)02829-8. [DOI] [PubMed] [Google Scholar]
- 7.Rosen EM, Fan S, Rockwell S, Goldberg ID. The molecular and cellular basis of radiosensitivity: Implications for understanding how normal tissues and tumors respond to therapeutic radiation. Cancer Invest. 1999;17:56–72. [PubMed] [Google Scholar]
- 8.Steel GG. The case against apoptosis. Acta Oncol. 2001;40:968–75. doi: 10.1080/02841860152708251. [DOI] [PubMed] [Google Scholar]
- 9.Feilotter HE, Nagai MA, Boag AH, Eng C, Mulligan LM. Analysis of PTEN and the 10q23 region in primary prostate carcinomas. Oncogene. 1998;16:1743–8. doi: 10.1038/sj.onc.1200205. [DOI] [PubMed] [Google Scholar]
- 10.Guo Y, Sklar GN, Borkowski A, Kyprianou N. Loss of the cyclin-dependent kinase inhibitor p27(Kip1) protein in human prostate cancer correlates with tumor grade. Clin Cancer Res. 1997;3:2269–74. [PubMed] [Google Scholar]
- 11.Navone NM, Troncoso P, Pisters LL, Goodrow TL, Palmer JL, Nichols WW, von Eschenbach AC, Conti CJ. p53 protein accumulation and gene mutation in the progression of human prostate carcinoma. J Natl Cancer Inst. 1993;85:1657–69. doi: 10.1093/jnci/85.20.1657. [DOI] [PubMed] [Google Scholar]
- 12.Phillips SM, Barton CM, Lee SJ, Morton DG, Wallace DM, Lemoine NR, Neoptolemos JP. Loss of the retinoblastoma susceptibility gene (RB1) is a frequent and early event in prostatic tumorigenesis. Br J Cancer. 1994;70:1252–7. doi: 10.1038/bjc.1994.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Konishi N, Nakamura M, Kishi M, Nishimine M, Ishida E, Shimada K. Heterogeneous methylation and deletion patterns of the INK4a/ARF locus within prostate carcinomas. Am J Pathol. 2002;160:1207–14. doi: 10.1016/S0002-9440(10)62547-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bookstein R, MacGrogan D, Hilsenbeck SG, Sharkey F, Allred DC. p53 is mutated in a subset of advanced-stage prostate cancers. Cancer Res. 1993;53:3369–73. [PubMed] [Google Scholar]
- 15.Voeller HJ, Sugars LY, Pretlow T, Gelmann EP. p53 oncogene mutations in human prostate cancer specimens. J Urol. 1994;151:492–5. doi: 10.1016/s0022-5347(17)35000-0. [DOI] [PubMed] [Google Scholar]
- 16.Subler MA, Martin DW, Deb S. Overlapping domains on the p53 protein regulate its transcriptional activation and repression functions. Oncogene. 1994;9:1351–9. [PubMed] [Google Scholar]
- 17.Kern SE, Pietenpol JA, Thiagalingam S, Seymour A, Kinzler KW, Vogelstein B. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science. 1992;256:827–30. doi: 10.1126/science.1589764. [DOI] [PubMed] [Google Scholar]
- 18.Scott SL, Earle JD, Gumerlock PH. Functional p53 increases prostate cancer cell survival after exposure to fractionated doses of ionizing radiation. Cancer Res. 2003;63:7190–6. [PubMed] [Google Scholar]
- 19.Chan WM, Siu WY, Lau A, Poon RY. How many mutant p53 molecules are needed to inactivate a tetramer? Mol Cell Biol. 2004;24:3536–51. doi: 10.1128/MCB.24.8.3536-3551.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohn KW, Pommier Y. Molecular interaction map of the p53 and Mdm2 logic elements, which control the Off-On switch of p53 in response to DNA damage. Biochem Biophys Res Commun. 2005;331:816–27. doi: 10.1016/j.bbrc.2005.03.186. [DOI] [PubMed] [Google Scholar]
- 21.Linares LK, Hengstermann A, Ciechanover A, Muller S, Scheffner M. HdmX stimulates Hdm2-mediated ubiquitination and degradation of p53. Proc Natl Acad Sci USA. 2003;100:12009–14. doi: 10.1073/pnas.2030930100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fuchs SY, Adler V, Buschmann T, Wu X, Ronai Z. Mdm2 association with p53 targets its ubiquitination. Oncogene. 1998;17:2543–7. doi: 10.1038/sj.onc.1202200. [DOI] [PubMed] [Google Scholar]
- 23.Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–45. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- 24.Gu J, Kawai H, Nie L, Kitao H, Wiederschain D, Jochemsen AG, Parant J, Lozano G, Yuan ZM. Mutual dependence of MDM2 and MDMX in their functional inactivation of p53. J Biol Chem. 2002;277:19251–4. doi: 10.1074/jbc.C200150200. [DOI] [PubMed] [Google Scholar]
- 25.Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Natl Acad Sci USA. 1999;96:13777–82. doi: 10.1073/pnas.96.24.13777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tibbetts RS, Brumbaugh KM, Williams JM, Sarkaria JN, Cliby WA, Shieh SY, Taya Y, Prives C, Abraham RT. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev. 1999;13:152–7. doi: 10.1101/gad.13.2.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuerbitz SJ, Plunkett BS, Walsh WV, Kastan MB. Wild-type p53 is a cell cycle checkpoint determinant following irradiation. Proc Natl Acad Sci USA. 1992;89:7491–5. doi: 10.1073/pnas.89.16.7491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Helt CE, Cliby WA, Keng PC, Bambara RA, O'Reilly MA. Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage. J Biol Chem. 2005;280:1186–92. doi: 10.1074/jbc.M410873200. [DOI] [PubMed] [Google Scholar]
- 29.Milner J, Medcalf EA, Cook AC. Tumor suppressor p53: Analysis of wild-type and mutant p53 complexes. Mol Cell Biol. 1991;11:12–9. doi: 10.1128/mcb.11.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Vries A, Flores ER, Miranda B, Hsieh HM, van Oostrom CT, Sage J, Jacks T. Targeted point mutations of p53 lead to dominant-negative inhibition of wild-type p53 function. Proc Natl Acad Sci USA. 2002;99:2948–53. doi: 10.1073/pnas.052713099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: Functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–65. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 32.Song H, Hollstein M, Xu Y. p53 gain-of-function cancer mutants induce genetic instability by inactivating ATM. Nat Cell Biol. 2007;9:573–80. doi: 10.1038/ncb1571. [DOI] [PubMed] [Google Scholar]
- 33.Shi XB, Nesslinger NJ, Deitch AD, Gumerlock PH, deVere White RW. Complex functions of mutant p53 alleles from human prostate cancer. Prostate. 2002;51:59–72. doi: 10.1002/pros.10072. [DOI] [PubMed] [Google Scholar]
- 34.Algan O, Stobbe CC, Helt AM, Hanks GE, Chapman JD. Radiation inactivation of human prostate cancer cells: The role of apoptosis. Radiat Res. 1996;146:267–75. [PubMed] [Google Scholar]
- 35.Cao C, Shinohara ET, Subhawong TK, Geng L, Woon Kim K, Albert JM, Hallahan DE, Lu B. Radiosensitization of lung cancer by nutlin, an inhibitor of murine double minute 2. Mol Cancer Ther. 2006;5:411–7. doi: 10.1158/1535-7163.MCT-05-0356. [DOI] [PubMed] [Google Scholar]
- 36.Raffoul JJ, Wang Y, Kucuk O, Forman JD, Sarkar FH, Hillman GG. Genistein inhibits radiation-induced activation of NF-kappaB in prostate cancer cells promoting apoptosis and G2/M cell cycle arrest. BMC Cancer. 2006;6:107. doi: 10.1186/1471-2407-6-107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cao C, Subhawong T, Albert JM, Kim KW, Geng L, Sekhar KR, Gi YJ, Lu B. Inhibition of mammalian target of rapamycin or apoptotic pathway induces autophagy and radiosensitizes PTEN null prostate cancer cells. Cancer Res. 2006;66:10040–7. doi: 10.1158/0008-5472.CAN-06-0802. [DOI] [PubMed] [Google Scholar]
- 38.Nguyen KH, Hachem P, Khor LY, Salem N, Hunt KK, Calkins PR, Pollack A. Adenoviral-E2F-1 radiosensitizes p53wild-type and p53null human prostate cancer cells. Int J Radiat Oncol Biol Phys. 2005;63:238–46. doi: 10.1016/j.ijrobp.2005.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lehmann BD, McCubrey JA, Jefferson HS, Paine MS, Chappell WH, Terrian DM. A dominant role for p53-dependent cellular senescence in radiosensitization of human prostate cancer cells. Cell Cycle. 2007;6:595–605. doi: 10.4161/cc.6.5.3901. [DOI] [PubMed] [Google Scholar]
- 40.Crighton D, Wilkinson S, O'Prey J, Syed N, Smith P, Harrison PR, Gasco M, Garrone O, Crook T, Ryan KM. DRAM, a p53-induced modulator of autophagy, is critical for apoptosis. Cell. 2006;126:121–34. doi: 10.1016/j.cell.2006.05.034. [DOI] [PubMed] [Google Scholar]
- 41.Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, Newman J, Reczek EE, Weissleder R, Jacks T. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661–5. doi: 10.1038/nature05541. [DOI] [PubMed] [Google Scholar]
- 42.Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, Cordon-Cardo C, Lowe SW. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656–60. doi: 10.1038/nature05529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dotto GP. p21(WAF1/Cip1): More than a break to the cell cycle? Biochim Biophys Acta. 2000;1471:M43–56. doi: 10.1016/s0304-419x(00)00019-6. [DOI] [PubMed] [Google Scholar]
- 44.Narita M, Nunez S, Heard E, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703–16. doi: 10.1016/s0092-8674(03)00401-x. [DOI] [PubMed] [Google Scholar]
- 45.Dai CY, Enders GH. p16 INK4a can initiate an autonomous senescence program. Oncogene. 2000;19:1613–22. doi: 10.1038/sj.onc.1203438. [DOI] [PubMed] [Google Scholar]
- 46.Sugrue MM, Shin DY, Lee SW, Aaronson SA. Wild-type p53 triggers a rapid senescence program in human tumor cells lacking functional p53. Proc Natl Acad Sci USA. 1997;94:9648–53. doi: 10.1073/pnas.94.18.9648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fang L, Igarashi M, Leung J, Sugrue MM, Lee SW, Aaronson SA. p21Waf1/Cip1/Sdi1 induces permanent growth arrest with markers of replicative senescence in human tumor cells lacking functional p53. Oncogene. 1999;18:2789–97. doi: 10.1038/sj.onc.1202615. [DOI] [PubMed] [Google Scholar]
- 48.Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: Roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–22. doi: 10.1093/emboj/cdg417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O'Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, Kouzarides T. Rb targets histone H3 methylation and HP1 to promoters. Nature. 2001;412:561–5. doi: 10.1038/35087620. [DOI] [PubMed] [Google Scholar]
- 50.Vandel L, Nicolas E, Vaute O, Ferreira R, Ait-Si-Ali S, Trouche D. Transcriptional repression by the retinoblastoma protein through the recruitment of a histone methyltransferase. Mol Cell Biol. 2001;21:6484–94. doi: 10.1128/MCB.21.19.6484-6494.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shay JW, Roninson IB. Hallmarks of senescence in carcinogenesis and cancer therapy. Oncogene. 2004;23:2919–33. doi: 10.1038/sj.onc.1207518. [DOI] [PubMed] [Google Scholar]
- 52.Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–7. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- 53.Khor LY, Desilvio M, Al-Saleem T, Hammond ME, Grignon DJ, Sause W, Pilepich M, Okunieff P, Sandler H, Pollack A. MDM2 as a predictor of prostate carcinoma outcome: An analysis of radiation therapy oncology group protocol 8610. Cancer. 2005;104:962–7. doi: 10.1002/cncr.21261. [DOI] [PubMed] [Google Scholar]
- 54.Mu Z, Hachem P, Agrawal S, Pollack A. Antisense MDM2 sensitizes prostate cancer cells to androgen deprivation, radiation, and the combination. Int J Radiat Oncol Biol Phys. 2004;58:336–43. doi: 10.1016/j.ijrobp.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 55.Zhang R, Wang H, Agrawal S. Novel antisense anti-MDM2 mixed-backbone oligonucleotides: Proof of principle, in vitro and in vivo activities, and mechanisms. Curr Cancer Drug Targets. 2005;5:43–9. doi: 10.2174/1568009053332663. [DOI] [PubMed] [Google Scholar]
- 56.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 57.Tovar C, Rosinski J, Filipovic Z, Higgins B, Kolinsky K, Hilton H, Zhao X, Vu BT, Qing W, Packman K, Myklebost O, Heimbrook DC, Vassilev LT. Small-molecule MDM2 antagonists reveal aberrant p53 signaling in cancer: Implications for therapy. Proc Natl Acad Sci USA. 2006;103:1888–93. doi: 10.1073/pnas.0507493103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu B, Gilkes DM, Farooqi B, Sebti SM, Chen J. MDMX overexpression prevents p53 activation by the MDM2 inhibitor Nutlin. J Biol Chem. 2006;281:33030–5. doi: 10.1074/jbc.C600147200. [DOI] [PubMed] [Google Scholar]
- 59.Saric T, Brkanac Z, Troyer DA, Padalecki SS, Sarosdy M, Williams K, Abadesco L, Leach RJ, O'Connell P. Genetic pattern of prostate cancer progression. Int J Cancer. 1999;81:219–24. doi: 10.1002/(sici)1097-0215(19990412)81:2<219::aid-ijc9>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 60.Chen Z, Trotman LC, Shaffer D, Lin HK, Dotan ZA, Niki M, Koutcher JA, Scher HI, Ludwig T, Gerald W, Cordon-Cardo C, Pandolfi PP. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nature. 2005;436:725–30. doi: 10.1038/nature03918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hill R, Song Y, Cardiff RD, Van Dyke T. Heterogeneous tumor evolution initiated by loss of pRb function in a preclinical prostate cancer model. Cancer Res. 2005;65:10243–54. doi: 10.1158/0008-5472.CAN-05-1579. [DOI] [PubMed] [Google Scholar]
- 62.Maddison LA, Sutherland BW, Barrios RJ, Greenberg NM. Conditional deletion of Rb causes early stage prostate cancer. Cancer Res. 2004;64:6018–25. doi: 10.1158/0008-5472.CAN-03-2509. [DOI] [PubMed] [Google Scholar]
- 63.Gottschalk AR, Doan A, Nakamura JL, Stokoe D, Haas-Kogan DA. Inhibition of phosphatidylinositol-3-kinase causes increased sensitivity to radiation through a PKB-dependent mechanism. Int J Radiat Oncol Biol Phys. 2005;63:1221–7. doi: 10.1016/j.ijrobp.2005.08.014. [DOI] [PubMed] [Google Scholar]
- 64.Gottlieb TM, Leal JF, Seger R, Taya Y, Oren M. Cross-talk between Akt, p53 and Mdm2: Possible implications for the regulation of apoptosis. Oncogene. 2002;21:1299–303. doi: 10.1038/sj.onc.1205181. [DOI] [PubMed] [Google Scholar]
- 65.Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc Natl Acad Sci USA. 2001;98:11598–603. doi: 10.1073/pnas.181181198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat Cell Biol. 2001;3:973–82. doi: 10.1038/ncb1101-973. [DOI] [PubMed] [Google Scholar]
- 67.Kim JS, Lee C, Bonifant CL, Ressom H, Waldman T. Activation of p53-dependent growth suppression in human cells by mutations in PTEN or PIK3CA. Mol Cell Biol. 2007;27:662–77. doi: 10.1128/MCB.00537-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lee C, Kim JS, Waldman T. Activated PI3K signaling as an endogenous inducer of p53 in human cancer. Cell Cycle. 2007;6:394–6. doi: 10.4161/cc.6.4.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carroll AG, Voeller HJ, Sugars L, Gelmann EP. p53 oncogene mutations in three human prostate cancer cell lines. Prostate. 1993;23:123–34. doi: 10.1002/pros.2990230206. [DOI] [PubMed] [Google Scholar]
- 70.van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, Nordeen SK, Miller GJ, Lucia MS. Molecular characterization of human prostate carcinoma cell lines. Prostate. 2003;57:205–25. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]