

Abstract
Phytochromes are red-light photoreceptor proteins that regulate a variety of responses and cellular processes in plants, bacteria, and fungi. The phytochrome light activation mechanism involves isomerization around the C15═C16 double bond of an open-chain tetrapyrrole chromophore, resulting in a flip of its D-ring. In an important new development, bacteriophytochrome (Bph) has been engineered for use as a fluorescent marker in mammalian tissues. Here we report that an unusual Bph, RpBphP3 from Rhodopseudomonas palustris, denoted P3, is fluorescent. This Bph modulates synthesis of light-harvesting complex in combination with a second Bph exhibiting classical photochemistry, RpBphP2, denoted P2. We identify the factors that determine the fluorescence and isomerization quantum yields through the application of ultrafast spectroscopy to wild-type and mutants of P2 and P3. The excited-state lifetime of the biliverdin chromophore in P3 was significantly longer at 330–500 ps than in P2 and other classical phytochromes and accompanied by a significantly reduced isomerization quantum yield. H/D exchange reduces the rate of decay from the excited state of biliverdin by a factor of 1.4 and increases the isomerization quantum yield. Comparison of the properties of the P2 and P3 variants shows that the quantum yields of fluorescence and isomerization are determined by excited-state deprotonation of biliverdin at the pyrrole rings, in competition with hydrogen-bond rupture between the D-ring and the apoprotein. This work provides a basis for structure-based conversion of Bph into an efficient near-IR fluorescent marker.
Keywords: hydrogen bonds, near-IR fluorescent protein, reaction mechanism, kinetic isotope effect, biophotonics
Light is used as a switch to regulate various physiological processes in eukaryotes and prokaryotes through one or more photochromic proteins. Phytochromes constitute such a family of light sensors in plants, fungi, and bacteria (1, 2). Their light-sensing module comprises PAS (Per-ARNT-Sim), GAF (cGMP phosphodiesterase/adenylyl cyclase/FhlA), and PHY (phytochrome) domains and covalently binds a linear tetrapyrrole, phytochromobilin (PΦB), and phycocyanobilin (PCB) in plant and cyanobacterial phytochromes and biliverdin (BV) in bacteriophytochromes (BPhs). In the dark, most phytochromes adopt a red-absorbing state known as Pr and, upon light absorption, convert to a far-red-absorbing state known as Pfr.
The recent determination of crystal structures of various Bphs and the cyanobacterial phytochrome Cph1 has explored the light-activated function of phytochromes (3–7). The linear tetrapyrrole chromophore is bound to the PAS (Bph) or GAF (Cph1) domain at ring A through covalent linkage to a conserved cysteine. The overall structures of Bph and Cph1 are quite similar with most of the chromophore-protein contacts residing in the GAF domain. In the Pr state the chromophore assumes a ZZZssa configuration and is positioned in its binding pocket through steric interactions and hydrogen bonds from protein residues to the pyrrole rings and propionate side chains (Fig. 1 and Fig. S1). Recent studies have indicated that 15Za to 15Ea isomerization of the chromophore at the C15═C16 double bond, which causes a flip of pyrrole ring D, accompanies formation of the Pfr state (6). The primary photoproduct, denoted Lumi-R, is formed on the 10- to 100-ps time scale and adopts the 15Ea configuration (8–12).
Fig. 1.

The BV binding site in the x-ray structure of R. palustris P3 (monomer II). Weak interactions that fall out of range for H-bond distances are indicated with crosses.
In an important recent development, BPh from Deinococcus radiodurans (DrBph) has been engineered for use as a fluorescent marker in mammalian tissues (13). Fluorescent phytochromes were first reported by Fischer and Lagarias (14) and Vierstra and co-workers (15). DrBph fluoresces in the near-IR at ∼720 nm, a wavelength less prone to scattering that can penetrate more deeply into tissue than light emitted by GFP-derived fluorescent proteins. BV is a naturally occurring cofactor in mammalian tissue that covalently binds to a conserved cysteine in the Bph, and hence Bphs can readily be genetically encoded. Bph photochemistry is thus of considerable significance for biomedical research and technology. Because the natural function of Bphs is as light-dependent signaling molecules, they have evolved to maximize the quantum yield for entry into the photocycle and signaling and concomitantly to minimize the quantum yield for fluorescence. Hence, their fluorescence properties and in particular the fluorescence quantum yield need to be improved if they are to find routine application as fluorescent markers.
Like plant and cyanobacterial phytochromes, Bph proceeds through a photocycle involving Lumi-R and Meta-R intermediates that finally results in formation of Pfr that absorbs near 750 nm (1, 16). Little is known regarding the early Bph photochemistry, and many questions remain on the molecular mechanisms that govern BV excited-state dynamics, deactivation, and isomerization. What is the rate-limiting step in the isomerization process? What is the role of specific chromophore–protein interactions? Why is the quantum yield of Pfr and Lumi-R formation so low at 0.1–0.2 (10, 12, 17, 18)? More generally, what processes determine the fluorescence quantum yield in Bph?
RpBphP3 (hereafter referred to as P3) from Rhodopseudomonas palustris is a Bph with unusual photochemical properties. Its dark state absorbs at 705 nm and is similar to that of the Pr state of Bphs exhibiting classical Pr/Pfr photoconversion, but upon light activation P3 forms a state with a blueshifted absorption near 645 nm, a state referred to as Pnr (2). The x-ray structure of P3 in the Pr state revealed interesting differences from classical Bph and Cph1 in its BV binding pocket, in particular around ring D. In addition to a conserved histidine residue found in both DrBph and P3, two additional polar residues, Lys-183 and Ser-297, are located in P3 within hydrogen-bonding distance of the carbonyl group of ring D of the bilin chromophore (Fig. 1) (5).
Here we report a femtosecond transient absorption study of the photochemistry of P3, both wild type and variants in which we replaced Lys-183 and Ser-297 by corresponding residues found in a closely related Bph from the same organism, RpBphP2 (hereafter referred to as P2), which shows classical Pr-Pfr photochemistry. We show that P3 has an unusually long excited-state lifetime, which translates into a significantly increased fluorescence quantum yield as compared to a classical Bph such as P2 or DrBph. Thus P3 constitutes an attractive starting point for engineering a highly fluorescent Bph. The P3 photochemistry differs significantly from the models proposed for plant and cyanobacterial phytochromes and provides a basis for rational design of Bphs into efficient near-IR fluorescent markers.
Results and Discussion
Photochemistry of P3: H/D Exchange Effects Reveal Proton-Transfer Dynamics.
Femtosecond time-resolved absorption spectroscopy was performed on P3 wild-type (PAS-GAF-PHY) to assess its primary photochemistry. The excitation wavelength was 680 nm, and the time-resolved absorbance changes were monitored over a wavelength range from 550 to 780 nm. The data were globally analyzed in terms of a kinetic scheme with sequentially interconverting species, i.e., 1 → 2 → 3 → ⋯, where each species is characterized by an evolution-associated difference spectrum (EADS), which indicates the difference spectrum with respect to the Pr ground state. Five components were required for an adequate description of the time-resolved data, with lifetimes of 0.5, 4.7, 45, and 330 ps and a nondecaying component. The resulting EADS are shown in Fig. 2A. As discussed in SI Text (Fig. S1), the 0.5-ps process has an overall low amplitude and will not be further discussed here. The processes that occur on the 4.7- and 45-ps time scales likely involve structural evolution of excited BV on the excited-state potential energy surface. Importantly, the 4.7- and 45-ps decay-associated difference spectra do not show any loss of ground-state bleach or stimulated emission features and consequently do not involve loss of excited-state population, as discussed in the legend of Fig. S1. Time-resolved fluorescence experiments indicated a single exponential decay of 330 ps (Fig. S1), indicating that only the 330-ps component corresponds to decay of the BV excited state. This excited-state lifetime is significantly longer than that of classical (bacterio)phytochromes and implies an increase of the fluorescence quantum yield. Indeed, the P3 fluorescence quantum yield amounts to 0.045, which is substantially higher than that of P2 (< 0.01) and plant phytochrome (0.005) (8).
Fig. 2.

Time-resolved spectroscopy of Bphs. (A) EADS and their corresponding lifetimes resulting from global analysis of ultrafast transient absorption experiments on wild-type R. palustris P3 (PAS-GAF-PHY) upon excitation at 680 nm; kinetic traces at 709 (B) and 730 nm (C) of P3 in H2O (Black Circles) and D2O (Red Circles) and the P3MA mutant (Blue Circles); (D) EADS for wild-type R. palustris P2 (PAS-GAF-PHY); kinetic traces at 711 (E) and 733 nm (F) of P2 in H2O (Black Circles) and D2O (Red Circles) and the P2KS mutant (Blue Circles). The solid lines in B, C, E, and F denote the result of global fitting.
The EADS with a lifetime of 330 ps (Fig. 2A, Black Line) shows a major ground-state bleach at 710 nm, a bleach of the vibronic band at 640 nm, and excited-state absorption (ESA) at 750 nm. We assign it to a vibrationally relaxed and structurally distorted BV in the excited state. This state decays to the nondecaying component, which we assign to the primary photoproduct Lumi-R (Fig. 2A, Red Line). Lumi-R shows a ground-state bleach at 700 nm and an induced absorption at 730 nm. The spectral shape of Lumi-R indicates that, as in phytochromes with classical photochemistry, the primary photoproduct of P3 is redshifted with respect to the Pr ground state. The quantum yield of Lumi-R formation is estimated at 0.06 in P3 (Fig. S2), significantly lower than that observed in classical phytochromes such as P2 with yields of 0.10–0.20 [(8, 17, 18) and see below].
The primary photochemistry of P3 exhibits pronounced H/D exchange effects. Fig. 2B shows kinetic traces that represent excited-state decay, at the ground-state bleach maximum, for P3 in H2O (black circles) and D2O (red circles). The D2O dataset was multiplied with a scaling number so that its signal amplitude after photon absorption was the same as in the H2O dataset. Strikingly, the kinetics are significantly slower in D2O (450 ps) as compared to H2O (330 ps) by a factor of 1.4. Fig. 2C shows kinetic traces at 730 nm. An initial decay of BV ESA (0–100 ps) is followed by the rise of Lumi-R (100 ps–2 ns). Remarkably, the final amplitude of Lumi-R absorption is ∼20% higher in D2O (red circles) compared with H2O (black circles). It thus appears that in D2O, the quantum yield for formation of Lumi-R exceeds that in H2O by 20%.
To investigate the H/D exchange effects quantitatively and understand the implications for the P3 reaction dynamics in terms of microscopic rate constants, we performed target analysis by using a kinetic model shown in Fig. 3. The model involves five molecular states: three BV excited states denoted as B′′, B′, and B*, Lumi-R, and the Pr ground state. The initial excited state B′′ evolves to B′ with rate constant k1, which further evolves to B* with rate constant k2. In this case k1 and k2 correspond to the ∼4 and ∼40 ps relaxation processes, respectively. Excited-state population loss takes place only from B*. The decay of B* is determined by three competing reaction pathways: Lumi-R formation with rate constant k3, relaxation to the Pr ground state with rate constant k4, and radiative decay to the ground state with rate constant kR, which takes fluorescence into account. The latter is fixed at (6.6 ns)-1 to produce the observed fluorescence quantum yield of 0.045. The choice of model is supported by the fact that BV shows a strictly monoexponential excited-state decay, in transient absorption as well as time-resolved fluorescence.
Fig. 3.

Kinetic scheme to describe the excited-state dynamics of R. palustris P3. The time constants estimated from the target analysis of P3 in H2O are indicated in parentheses.
The time-resolved data of P3 in H2O and D2O are simultaneously analyzed, in which the species-associated difference spectra (SADS) are required to remain essentially identical in H2O and D2O but the rate constants k1–4 (which take different values in H2O and D2O) are allowed to vary freely. The lifetime of B* is ((k3 + k4 + kR)-1, and consequently, from the kinetic model alone, k3 and k4 are indeterminate. However, they can be distinguished with additional data on the Lumi-R quantum yield, which in the context of the kinetic model is given by k3/(k3 + k4 + kR) and has values of 0.06 in H2O and 0.072 in D2O (see above). Thus the value for k4 in D2O is fixed in the target analysis to produce the correct BV lifetime and Lumi-R quantum yields. The SADS that result from the target analysis are shown in Fig. S3; the rate constants k1–4 are indicated in Fig. 3 and summarized in Table S1.
The kinetic isotope effect (KIE) is defined as kH2O/kD2O for rate constant k determined in H2O and D2O. For k1, k2, and k3, the KIE is close to unity, but the KIE for k4 is significantly larger at 1.43. Thus, the slowdown of excited-state decay in P3 upon H/D exchange (Fig. 2B) is primarily because of a KIE on the population loss channel to the Pr ground state. We arrive at the important conclusion that, in P3, the BV excited-state deactivation to Pr is rate-limited by a proton-transfer process, which explains why the Lumi-R quantum yield is higher in D2O: If the decay-to-Pr rate constant k4 is reduced with respect to the Lumi-R formation rate constant k3 (Fig. 3), the yield of the latter process will increase.
The Photochemistry of P2.
Femtosecond time-resolved absorption spectroscopy was performed on P2 wild-type (PAS-GAF-PHY) to allow comparison with P3 and other phytochromes with classical photochemistry. The Pr state of P2 absorbs at 710 nm (5). The global analysis procedure indicated that five components were required for an adequate description of the time-resolved data, with time constants of 0.4, 4, 43, and 170 ps, and a nondecaying component. Fig. 2D shows the EADS that result. The 0.4- and 4-ps components (blue and green dotted lines) likely represent structural wevolution on the excited-state potential energy surface (see Fig. S4).
The EADS with a lifetime of 43 ps (black line, Fig. 2D) shows a major ground-state bleach/stimulated emission band at 720 nm, a bleach of the vibronic band at 640 nm, and ESA between 650–700 nm. We assign it to a vibrationally relaxed and structurally distorted BV in the excited state. This state decays in 43 ps to the EADS with a lifetime of 170 ps (red line). As compared to the black EADS, its overall amplitude is decreased, which indicates decay from the excited state of BV with a time constant of 43 ps. As we demonstrate below with target analysis, the EADS with a 170-ps lifetime corresponds to a mixture of the BV excited state and the primary photoproduct Lumi-R. The evolution to the final, nondecaying EADS (cyan line) takes place in 170 ps. The final EADS shows a bleach at 706 nm and an induced absorption at 734 nm, which we assign to Lumi-R. The excited-state decay times (43 and 170 ps) agree well with those observed in time-resolved fluorescence experiments (Fig. S4) and fall within the range of time constants observed in other Bph and Cph1 (9, 10, 12, 19). The Lumi-R quantum yield is estimated at 0.13 (Fig. S2). This value is comparable with those of many other phytochromes with classical photochemistry (8, 17–19) but is significantly higher than in P3 by a factor of ∼2.
Although the P2 primary photochemistry exhibits a significant H/D exchange effect, this is not as striking as in P3. The kinetics that probe BV excited-state decay (712 nm) are slower in D2O (Fig. 2E, red trace) than in H2O (black trace) with fitted time constants of 50 and 250 ps in D2O. Time-resolved fluorescence experiments show a similar trend (Fig. S4). The Lumi-R quantum yield is about 8% higher in D2O (Fig. 2F, red trace) than in H2O (black trace).
We performed a target analysis to obtain microscopic rate constants of the photoreaction. The kinetic model is shown in Fig. S5 and is more complex than the model for P3 because of the biexponential excited-state decay. To account for this feature, two components denoted  and
and  that represent the fast (43 ps) and slow (170 ps) reacting species were introduced. The biexponentiality possibly follows from structural heterogeneity of the chromophore in the Pr ground state, which may result in a distinct isomerization reaction rate for each conformer. Such conformational heterogeneity in Pr was reported in DrBph and Agp1 by resonance Raman spectroscopy (15, 20) and in the Pfr ground state of Pseudomonas aeruginosa Bph by crystallography (6). It should be noted, however, that in Cph1, which also shows multiphasic excited-state dynamics (9), a homogeneous Pr ground state was observed (21).
that represent the fast (43 ps) and slow (170 ps) reacting species were introduced. The biexponentiality possibly follows from structural heterogeneity of the chromophore in the Pr ground state, which may result in a distinct isomerization reaction rate for each conformer. Such conformational heterogeneity in Pr was reported in DrBph and Agp1 by resonance Raman spectroscopy (15, 20) and in the Pfr ground state of Pseudomonas aeruginosa Bph by crystallography (6). It should be noted, however, that in Cph1, which also shows multiphasic excited-state dynamics (9), a homogeneous Pr ground state was observed (21).
Target analysis shows that Lumi-R formation proceeds from both  and
and  , indicating that both “fast” and “slow” decay components of P2 are productive. The pattern of KIEs is more complicated than in P3. The rate constants that make up the fast channel (43 ps) show a KIE close to unity, whereas for the deactivation channel in the
, indicating that both “fast” and “slow” decay components of P2 are productive. The pattern of KIEs is more complicated than in P3. The rate constants that make up the fast channel (43 ps) show a KIE close to unity, whereas for the deactivation channel in the  branch, this loss channel has a sizable KIE of 1.45. Thus, only for the slow
branch, this loss channel has a sizable KIE of 1.45. Thus, only for the slow 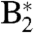 branch do we have clear evidence that in P2, the BV excited-state decay to Pr is rate-limited by a proton-transfer process. This phenomenon effectively raises the Lumi-R quantum yield for P2 in D2O with respect to H2O but to a smaller extent than in P3. (Further details are in SI Text.)
branch do we have clear evidence that in P2, the BV excited-state decay to Pr is rate-limited by a proton-transfer process. This phenomenon effectively raises the Lumi-R quantum yield for P2 in D2O with respect to H2O but to a smaller extent than in P3. (Further details are in SI Text.)
Hydrogen Bonds to Ring D Are Rate-Limiting for Isomerization and Excited-State Lifetime.
P3 differs from P2, Cph1, Agp1, and plant phytochromes with classical photochemistry in two major respects: Its excited-state lifetime is considerably longer (330 ps), and its quantum yield for formation of Lumi-R is lower (0.06). These differences are reflected in the rate constants derived from the kinetic modeling: The Lumi-R formation rate in P3, (5,400 ps)-1, is an order of magnitude smaller than in P2 [main component of (220 ps)-1] (Table S1). In addition, the decay rate to the Pr ground state, (350 ps)-1, is considerably smaller in P3 than in P2 [main component of (52 ps)-1] (Table S1). It thus appears that in P3 both the productive (to Lumi-R) and the nonproductive (to Pr) photochemistry are restricted.
If we assume that P2 structurally resembles DrBph, we can compare the sequences and x-ray structures in the Pr state of the BV binding pockets of P2 and P3. Reexamination of the P3 crystal structure (Protein Data Bank ID code 2OOL) revealed that the two monomers in one asymmetric unit, I and II, show structural heterogeneity. In monomer II, three side chains compete for hydrogen bonding to the carbonyl of ring D: His-299, Lys-183, and Ser-297 (Fig. 1; ref. 5). In monomer I, Lys-183 has slightly rotated with respect to that in monomer II and falls out of H-bond range with the ring D carbonyl, implying that only His-299 and Ser-297 hydrogen bond with ring D (Fig. S1). The sequence of P2 suggests that, as in the structure of DrBph, only a single hydrogen bond is formed between this carbonyl and a conserved histidine (3, 4). Thus, hydrogen-bond interactions with the ring D carbonyl are likely to be significantly stronger in P3 as compared with P2 and will restrict ring D motion upon photoexcitation.
If isomerization about the BV C15═C16 double bond is to occur, the hydrogen bonds to ring D must be broken. Our experiments strongly suggest that in P3 the rate-limiting process in isomerization is a rupture of the hydrogen bonds from these three side chains to the ring D carbonyl. To test this hypothesis, we performed transient absorption experiments on a P3 mutant denoted P3MA in which Lys-183 and Ser-297 are replaced by Met and Ala, respectively (the amino acids at the equivalent positions of P2) and on the corresponding P2 mutant denoted P2KS in which Met-169 and Ala-382 are replaced by Lys and Ser, respectively (the amino acids at the equivalent positions of P3). Absorption maxima in the Pr and Pfr/Pnr states of the P2KS and P3MA mutants occur at wavelengths similar to their respective wild types (5). Fig. 2E shows the kinetics of excited-state decay in the P2KS mutant (blue circles). Fig. S6 shows the result of a global analysis of the data. The BV excited-state lifetime is increased to 300 ps in the P2KS mutant, a value almost identical to that of P3 wild type. The Lumi-R quantum yield in P2KS (Fig. 2F, blue circles) is significantly lower than that of P2 wild type and estimated at 0.07 (Fig. S6), essentially identical to that of P3 wild type. Fig. 2B shows the kinetics for the excited-state lifetime in the P3MA mutant (blue circles). The lifetime in the P3MA mutant is decreased with respect to wild-type P3, i.e., 200 vs. 330 ps. The quantum yield for formation of Lumi-R in P3MA (Fig. 2C, blue circles) is considerably higher than in wild-type P3 and estimated at 0.11 (Fig. S6), very similar to that of P2 wild type. Thus, replacement of Ser and Lys in P3 by the equivalent Met and Ala residues in P2 lowers the BV excited-state lifetime and increases the Lumi-R quantum yield. Conversely, replacement of Met and Ala in P2 by the equivalent Ser and Lys residues in P3 increases the BV excited-state lifetime and decreases the Lumi-R quantum yield. We conclude that constraints on BV ring D rotation through hydrogen bonds to the ring D carbonyl indeed determine the excited-state lifetime and Lumi-R yield. The differences in hydrogen bonding to the ring D carbonyl between P3 and P2 are maintained in the excited state as reflected in the excited-state difference spectra (Fig. S7).
BV Pyrrole Ring Deprotonation Competes with C15═C16 Isomerization.
A key conclusion arises from a significant H/D exchange effect on the primary photochemistry of Bph. These observations contrast with those on plant phytochrome, where no effect of H/D exchange on the excited-state lifetime was observed (22). The target analysis of P3 indicates that BV excited-state deactivation to the Pr ground state is subject to a KIE of ∼1.4, which implies that this process is rate-limited by a proton-transfer event. What proton-transfer process may efficiently deactivate the BV excited state? We note that all four pyrrole nitrogens are protonated in the Pr state of plant phytochrome, Cph1 and Bph (16, 23–25). Recent experimental and computational evidence suggests that pyrroles can efficiently deprotonate in the excited state and deactivate to the ground state through a conical intersection (26, 27). Thus, a candidate mechanism for BV excited-state deactivation is a light-induced deprotonation reaction of a BV pyrrole nitrogen through excited-state proton transfer (ESPT).
The x-ray structures of DrBph, P3, and Cph1 indicated that the pyrrole nitrogens of rings A, B, and C form an intricate hydrogen-bonding network with a bound internal water, a highly conserved histidine, and the main-chain carbonyl of a highly conserved Asp in the PASDIP sequence motif (3, 5, 7). In the P3 structure, monomer I contains a water molecule that hydrogen bonds to the chromophore and Asp-216 in a way similar to that in DrBph and Cph1 (Fig. S1; refs. 3 and 7). However, monomer II does not bind a water near the BV pyrrole rings, and the pyrrole nitrogens hydrogen bond only to Asp-216 (ref. 5; Fig. 1). The KIE observed here for BV deprotonation is modest at 1.43. KIEs in such a range correspond to the fully adiabatic limit for proton transfer and reflect strong hydrogen-bond interactions between donor and acceptor groups and shallow barriers where the dominating effect of the KIE is reflected in isotope-dependent splitting in the crossing region of the potential surfaces in question (28). The structures shown in Fig. 1 and Fig. S1 readily provide a proton-transfer network with the strong hydrogen-bond interactions required for fully adiabatic proton transfer. The idea that proton transfer occurs at this particular site is supported by the results on the P3 D216A and Y272F mutants, which show a significantly increased excited-state lifetime of 500 ps (Fig. S8). These results are consistent with a decrease of the BV deprotonation rate, presumably resulting from a locally perturbed hydrogen-bonding structure around the BV pyrrole rings. We propose that BV decays from the excited state to the Pr ground state through proton transfer from the ring A, B, or C pyrrole nitrogen to the main-chain carbonyl of Asp-216 or to the bound water and ensuing rapid internal conversion to the molecular ground state.
The proton-transfer rate is considerably smaller in P3 (350 ps-1) than in P2 [main component of (52 ps)-1]. The results on the P2KS and P3MA mutants (Fig. 2) indicate that this decreased proton-transfer rate is a consequence of the strong hydrogen bonding to the ring D carbonyl in the Pr state, which restricts rotational motion of ring D upon photoexcitation. We conclude that a certain degree of twisting around the C15═C16 double bond is required to activate the BV deprotonation channel. Quantum chemical calculations on the related PΦB chromophore indicated that the acidity of pyrrole rings B and C does not change appreciably in the excited state (29), which is consistent with the idea that an additional structural distortion is required. Further support is provided by recent experiments on pyridyl indoles with an intramolecular hydrogen bond showing that light-induced intramolecular proton transfer from the indole to pyridyl moiety along the hydrogen-bond coordinate was coupled to twisting of the bond angle between pyridyl and indole moieties, leading to rapid excited-state deactivation. A hydrogen/deuterium KIE of 1.4 was measured for this reaction (30).
The ring D pyrrole nitrogen may hydrogen bond with a bound water (4), which points at an additional potential proton-transfer pathway for excited-state deactivation. Thus, a light-driven deprotonation reaction at ring D cannot be excluded.
The Mechanism of Bph Isomerization and Excited-State Deactivation.
We arrive at the following physical-chemical picture of BV Z/E isomerization about the C15═C16 double bond in P3, as depicted schematically in Fig. 4.† In the Pr state, the BV chromophore assumes the ZZZssa configuration (Fig. 4A). Upon excitation with red light, the B′′ state is formed that relaxes to B′ in 4 ps and in turn to B* in 40 ps. Because of the π-π∗ character of the BV excited state, the structural distortion of BV will mainly involve a twisting around the double bonds of the methine bridges, to which the C15═C16 double bond is most susceptible (Fig. 4B). The twisting about the C15═C16 double bond will strain the hydrogen bonds between His-299, Ser-297, and Lys-183 and the ring D carbonyl. Decay from the conformationally distorted, excited-state B* occurs via three distinct, competing processes:
Rupture of the hydrogen bonds to the ring D carbonyl occurs with a rate constant of (5,400 ps)-1 (Fig. 4C) This event releases ring D, allowing one-bond isomerization about C15═C16 and concomitant rapid flipping of ring D and formation of Lumi-R (Fig. 4D). This pathway accounts for only 6% of excited-state decay, in keeping with the low quantum yield for formation of Lumi-R.
Fluorescence to the Pr ground state occurs through radiative decay at a rate of (6.6 ns)-1, accounting for ∼5% of excited-state decay (Fig. 4E). Note that the radiative lifetime is an intrinsic property of the cationic BV chromophore.
ESPT takes place from the pyrrole nitrogen of ring A, B, or C to Asp-216 in (350 ps)-1 resulting in neutral BV and protonation of the Asp-216 backbone to form a transient R-C+-OH species (Fig. 4F). This process is the dominant pathway of excited-state deactivation, accounting for 89% of the excited population. Proton transfer from ring C is shown in Fig. 4F, but it cannot be excluded that it occurs from ring A or B. The twist in the C15═C16 double bond that evolves in 4–40 ps (Fig. 4B) activates the proton-transfer channel. Upon ESPT a rapid deactivation to the ground state takes place, possibly via a conical intersection. Once the system is in the electronic ground state, the proton is rapidly shuttled back to the BV chromophore because the protonated backbone of Asp-216 is not a stable chemical species. This process results in the reformation of Pr (Fig. 4E). Because the proton back-shuttle process is much faster than the BV excited-state decay (330 ps), a deprotonated BV ground-state species does not accumulate and cannot be directly observed.
Fig. 4.

The reaction mechanism for Lumi-R formation, excited-state deactivation to Pr through ESPT, and fluorescence in R. palustris P3 bacteriophytochrome.
Although this model favors deprotonation at ring A, B, or C, we note that deprotonation at ring D cannot be excluded. However, given the effects of D216A and Y272F mutations on the BV excited-state lifetime (Fig. S8), we consider deprotonation at ring A, B, or C most likely. This isomerization mechanism is distinctly different from that recently proposed for plant and cyanobacterial phytochromes (11, 19). Excited-state decay in plant phytochrome is significantly faster than in P3, with single decay times of 30–50 ps (8, 19) that do not display an H/D exchange effect (22). Ultrafast Raman spectroscopy on Cph1 reveals signs of initial isomerization of its PCB chromophore about its C15═C16 double bond at times as short as 3 ps. This observation was interpreted as an isomerization in the excited state of PCB, followed by excited-state decay to Lumi-R on a 30-ps time scale (11). In the context of our data, BV isomerization in the excited state would require that excited-state 15Ea BV has absorption and emission properties identical to those of excited-state 15Za BV. Such a situation cannot a priori be excluded, given the insensitivity of UV-visible spectroscopy to local structure. However, any chromophore isomerization in the excited state requires breaking of the hydrogen bonds to ring D prior to excited-state decay. The results on P3, P2KS, and P3MA strongly suggest that hydrogen-bond rupture between ring D and the apoprotein is rate-limiting to isomerization, which is difficult to reconcile with isomerization in the excited state. A recent study also suggested that the isomerization reaction proceeds along distinctly different pathways in plant and cyanobacterial phytochromes on the one hand and Bph on the other (31).
In P2, H/D exchange effects similar to those on P3 were observed for the slow phase in the excited-state decay (170 ps with a 40% amplitude), which indicates that BV excited-state deprotonation competes with Lumi-R formation for this fraction. The fast phase in excited-state decay (43 ps, 60% amplitude) did not exhibit an H/D exchange effect. We note that the absence of a KIE does not necessarily imply the absence of a proton-transfer event (28). Thus, the fast 43-ps decay phase in P2 is either associated with an ESPT process at the BV pyrrole nitrogens similar to that of P3 or is rate-limited by a different excited-state deactivation process.
The relaxation processes in the BV excited state in P3 (the 4.7- and/or 40-ps components) were assigned to structural dynamics of ring D. Another possible origin involves formation of a transition state that precedes ESPT. A clear example of such a proton-transfer transition state in ESPT is given by recent ultrafast stimulated Raman study on GFP, where initial chromophore wagging motions on a subpicosecond time scale poised the system for ESPT in picoseconds (32). However, these motions could be revealed only with stimulated Raman spectroscopy and were spectrally silent in visible spectroscopy (see, e.g., ref. 33). Thus, whereas the (sub)picosecond processes observed in this study may be related to structural evolution of ring D, it remains unclear whether they are mechanistically connected to the ESPT process through a local structural evolution at the pyrrole nitrogens.
ESPT is a key feature of the fluorescence properties of GFP and its derived proteins. However, the role of ESPT in GFP is fundamentally different from that in Bph. In GFP, ESPT results in a redshift of the fluorescence, but it does not lead to excited-state decay (34). The question is what function ESPT has in Bph. It is important to realize that the Bphs that undergo this type of ESPT in fact do not proceed through the photocycle. In P3, ESPT underlies the low Pnr quantum yield of 0.06; in a classical Bph such as P2, ESPT partly contributes to the low Pfr quantum yield of ∼0.15. Notably, the low fluorescence quantum yield of Bph is also largely determined by the ESPT process and hardly by BV isomerization. ESPT may arise from a specific structural feature of BV and the surrounding protein in functional Bphs: the conserved hydrogen-bond network between the pyrrole nitrogens, the bound water, and the main-chain carbonyl of the conserved Asp. In addition, ring D is required to twist in the excited state for isomerization to occur (Fig. 4B). These structural requirements have the inevitable side effect that in excess of 85% of molecules that have absorbed a photon decay through ESPT before ring D has been stabilized in the Lumi-R intermediate. Hence the quantum yield for Lumi-R formation and entry into the productive, signaling pathway is low, 0.15. Likewise, strategies for enhancing the fluorescence properties of Bph through genetic engineering may prove successful when aimed at minimizing the ESPT process.
Materials and Methods
Ultrafast transient absorption spectroscopy with a time resolution of 120 fs was carried out with a setup on the basis of an amplified Ti:sapphire laser system described earlier (35). Data were analyzed as described in SI Text. Samples were prepared as described in ref. 5.
Supplementary Material
Acknowledgments.
K.C.T. and J.T.M.K. were supported by Nederlandse Organisatie voor Wetenschappelijk Onderzoek – Aard- en Levenswetenschappen through a VIDI grant (to J.T.M.K.). E.A.S and K.M. were supported by National Institutes of Health Grant GM036452 (to K.M.).
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/cgi/content/full/0911535107/DCSupplemental.
†In this work, we have observed strictly homogeneous excited-state kinetics for P3, which strongly suggests that P3 assumes a single conformation in solution. The following discussion is based on the structure of P3 monomer II; for monomer I, a qualitatively similar picture can be drawn with either Asp-216 or bound water as transient proton acceptor.
References
- 1.Rockwell NC, Su YS, Lagarias JC. Phytochrome structure and signaling mechanisms. Annu Rev Plant Biol. 2006;57:837–858. doi: 10.1146/annurev.arplant.56.032604.144208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Giraud E, et al. A new type of bacteriophytochrome acts in tandem with a classical bacteriophytochrome to control the antennae synthesis in Rhodopseudomonas palustris. J Biol Chem. 2005;280:32389–32397. doi: 10.1074/jbc.M506890200. [DOI] [PubMed] [Google Scholar]
- 3.Wagner JR, Brunzelle JS, Forest KT, Vierstra RD. A light-sensing knot revealed by the structure of the chromophore-binding domain of phytochrome. Nature. 2005;438:325–331. doi: 10.1038/nature04118. [DOI] [PubMed] [Google Scholar]
- 4.Wagner JR, et al. High resolution structure of Deinococcus bacteriophytochrome yields new insights into phytochrome architecture and evolution. J Biol Chem. 2007;282:12298–12309. doi: 10.1074/jbc.M611824200. [DOI] [PubMed] [Google Scholar]
- 5.Yang X, Stojkovic EA, Kuk J, Moffat K. Crystal structure of the chromophore binding domain of an unusual bacteriophytochrome, RpBphP3, reveals residues that modulate photoconversion. Proc Natl Acad Sci USA. 2007;104:12571–12576. doi: 10.1073/pnas.0701737104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yang X, Kuk J, Moffat K. Crystal structure of Pseudomonas aeruginosa bacteriophytochrome: photoconversion and signal transduction. Proc Natl Acad Sci USA. 2008;105:14715–14720. doi: 10.1073/pnas.0806718105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Essen LO, Mailliet J, Hughes J. The structure of a complete phytochrome sensory module in the Pr ground state. Proc Natl Acad Sci USA. 2008;105:14709–14714. doi: 10.1073/pnas.0806477105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Andel F., III, et al. Femtosecond time-resolved spectroscopy of the primary photochemistry of phytochrome. Biospectroscopy. 1997;3:421–433. [Google Scholar]
- 9.Heyne K, et al. Ultrafast dynamics of phytochrome from the cyanobacterium synechocystis, reconstituted with phycocyanobilin and phycoerythrobilin. Biophys J. 2002;82:1004–1016. doi: 10.1016/S0006-3495(02)75460-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van Thor JJ, Ronayne KL, Towrie M. Formation of the early photoproduct lumi-R of cyanobacterial phytochrome cph1 observed by ultrafast mid-infrared spectroscopy. J Am Chem Soc. 2007;129:126–132. doi: 10.1021/ja0660709. [DOI] [PubMed] [Google Scholar]
- 11.Dasgupta J, et al. Ultrafast excited-state isomerization in phytochrome revealed by femtosecond stimulated Raman spectroscopy. Proc Natl Acad Sci USA. 2009;106:1784–1789. doi: 10.1073/pnas.0812056106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schumann C, et al. Sub-picosecond mid-infrared spectroscopy of phytochrome Agp1 from Agrobacterium tumefaciens. ChemPhysChem. 2007;8:1657–1663. doi: 10.1002/cphc.200700210. [DOI] [PubMed] [Google Scholar]
- 13.Shu XK, et al. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science. 2009;324:804–807. doi: 10.1126/science.1168683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer AJ, Lagarias JC. Harnessing phytochrome’s glowing potential. Proc Natl Acad Sci USA. 2004;101:17334–17339. doi: 10.1073/pnas.0407645101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wagner JR, et al. Mutational analysis of Deinococcus radiodurans bacteriophytochrome reveals key amino acids necessary for the photochromicity and proton exchange cycle of phytochromes. J Biol Chem. 2008;283:12212–12226. doi: 10.1074/jbc.M709355200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Borucki B, et al. Light-induced proton release of phytochrome is coupled to the transient deprotonation of the tetrapyrrole chromophore. J Biol Chem. 2005;280:34358–34364. doi: 10.1074/jbc.M505493200. [DOI] [PubMed] [Google Scholar]
- 17.Gensch T, Churio MS, Braslavsky SE, Schaffner K. Primary quantum yield and volume change of phytochrome—A phototransformation determined by laser-induced optoacoustic spectroscopy. Photochem Photobiol. 1996;63:719–725. [Google Scholar]
- 18.Lamparter T, et al. Characterization of recombinant phytochrome from the cyanobacterium Synechocystis. Proc Natl Acad Sci USA. 1997;94:11792–11797. doi: 10.1073/pnas.94.22.11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muller MG, et al. Femtosecond kinetics of photoconversion of the higher plant photoreceptor phytochrome carrying native and modified chromophores. Biophys J. 2008;94:4370–4382. doi: 10.1529/biophysj.106.091652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.von Stetten D, et al. Chromophore heterogeneity and photoconversion in phytochrome crystals and solution studied by resonance Raman spectroscopy. Angew Chem Int Ed Engl. 2008;47:4753–4755. doi: 10.1002/anie.200705716. [DOI] [PubMed] [Google Scholar]
- 21.Spillane KM, Dasgupta J, Lagarias JC, Mathies RA. Homogeneity of phytochrome Cph1 vibronic absorption revealed by resonance Raman intensity analysis. J Am Chem Soc. 2009;131:13946–13948. doi: 10.1021/ja905822m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brock H, et al. Fluorescence lifetimes and relative quantum yields of 124-kilodalton oat phytochrome in H2O and D2O solutions. Biochemistry. 1987;26:1412–1420. [Google Scholar]
- 23.Strauss HM, Hughes J, Schmieder P. Heteronuclear solution-state NMR studies of the chromophore in cyanobacterial phytochrome Cph1. Biochemistry. 2005;44:8244–8250. doi: 10.1021/bi050457r. [DOI] [PubMed] [Google Scholar]
- 24.Rohmer T, et al. Light-induced chromophore activity and signal transduction in phytochromes observed by C-13 and N-15 magic-angle spinning NMR. Proc Natl Acad Sci USA. 2008;105:15229–15234. doi: 10.1073/pnas.0805696105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kneip C, et al. Protonation state and structural changes of the tetrapyrrole chromophore during the P-r → P-fr phototransformation of phytochrome: A resonance Raman spectroscopic study. Biochemistry. 1999;38:15185–15192. doi: 10.1021/bi990688w. [DOI] [PubMed] [Google Scholar]
- 26.Lan ZG, Frutos LM, Sobolewski AL, Domcke W. Photochemistry of hydrogen-bonded aromatic pairs: Quantum dynamical calculations for the pyrrole-pyridine complex. Proc Natl Acad Sci USA. 2008;105:12707–12712. doi: 10.1073/pnas.0801062105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.David O, et al. Hydrogen transfer in excited pyrrole-ammonia clusters. J Chem Phys. 2004;120:10101–10110. doi: 10.1063/1.1704639. [DOI] [PubMed] [Google Scholar]
- 28.Kuznetsov AM, Ulstrup J. Proton and hydrogen atom tunnelling in hydrolytic and redox enzyme catalysis. Can J Chem. 1999;77:1085–1096. [Google Scholar]
- 29.Borg OA, Durbeej B. Relative ground and excited-state pK(a) values of phytochromobilin in the photoactivation of phytochrome: A computational study. J Phys Chem B. 2007;111:11554–11565. doi: 10.1021/jp0727953. [DOI] [PubMed] [Google Scholar]
- 30.Nosenko Y, et al. Proton transfer with a twist? Femtosecond dynamics of 7-(2-pyridyl)-indole in condensed phase and in supersonic jets. Angew Chem Int Ed Engl. 2008;47:6037–6040. doi: 10.1002/anie.200801350. [DOI] [PubMed] [Google Scholar]
- 31.Rockwell NC, Shang L, Martin SS, Lagarias JC. Distinct classes of red/far-red photochemistry within the phytochrome superfamily. Proc Natl Acad Sci USA. 2009;106:6123–6127. doi: 10.1073/pnas.0902370106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fang C, Frontiera RR, Tran R, Mathies RA. Mapping GFP structure evolution during proton transfer with femtosecond Raman spectroscopy. Nature. 2009;462:200–U274. doi: 10.1038/nature08527. [DOI] [PubMed] [Google Scholar]
- 33.Kennis JTM, et al. Uncovering the hidden ground state of green fluorescent protein. Proc Natl Acad Sci USA. 2004;101:17988–17993. doi: 10.1073/pnas.0404262102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chattoraj M, King BA, Bublitz GU, Boxer SG. Ultra-fast excited state dynamics in green fluorescent protein: Multiple states and proton transfer. Proc Natl Acad Sci USA. 1996;93:8362–8367. doi: 10.1073/pnas.93.16.8362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Berera R, van Grondelle R, Kennis JTM. Ultrafast transient absorption spectroscopy: Principles and application to photosynthetic systems. Photosynth Res. 2009;101:105–118. doi: 10.1007/s11120-009-9454-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.