

Abstract
Molecular dynamics (MD) simulations provide essential information about the thermodynamics and dynamics of proteins. To construct the free-energy surface from equilibrium trajectories, it is necessary to group the individual snapshots in a meaningful way. The inherent structures (IS) are shown to provide an appropriate discretization of the trajectory and to avoid problems that can arise in clustering algorithms that have been employed previously. The IS-based approach is illustrated with a 30-ns room temperature “native” state MD simulation of a 10-residue peptide in a β-hairpin conformation. The transitions between the IS are used to construct a configuration space network from which a one-dimensional free-energy profile is obtained with the mincut method. The results demonstrate that the IS approach is useful and that even for this simple system, there exists a nontrivial organization of the native state into several valleys separated by barriers as high as 3 kcal/mol. Further, by introducing a coarse-grained network, it is demonstrated that there are multiple pathways connecting the valleys. This scenario is hidden when the snapshots of the trajectory are used directly with rmsd clustering to compute the free-energy profile. Application of the IS approach to the native state of the PDZ2 signaling domain indicates its utility for the study of biologically relevant systems.
Keywords: complex networks, conformational dynamics, energy landscapes, molecular dynamics simulations
Protein function depends critically on the synergy between structure and dynamics. The dynamics often involves the interconversion of conformational states on a complex multidimensional free-energy surface. It is very difficult to study such conformational transitions experimentally at an atomic level of detail, although techniques such as single-molecule FRET (1), x-ray crystallography (2), and NMR (3) supply useful, but limited, information. Consequently, molecular dynamics simulations are playing an increasing role in determining the free-energy surface, as a supplement to the experimental studies. The energy surface is made up of many deep valleys connected by saddles (4), suggesting that protein dynamics can be divided into intravalley and intervalley motions (5). The former represent the oscillations around local minima, while the latter involve barrier crossings from one minimum to another (6). Most descriptions of the surface have been rather qualitative because computations to sample the underlying surface for such multidimensional systems have not been possible. Recently, particularly for peptides and even a few small proteins (7–9), molecular dynamics (MD) simulations that extend into the microsecond range are providing the information required for a more quantitative analysis (10). Because of the large number of degrees of freedom involved, analysis of the results based on graph theory have been found to be useful (11, 12). The essential idea is to map the calculated trajectory on a conformation space network (CSN), whose nodes represent the different conformations visited during the simulation and whose links correspond to direct transitions between the nodes (13). This approach has been successfully applied to obtain an understanding of peptide folding and biomolecular structural transitions (11, 12, 14–17), as well as to interpret electron transfer experiments (18) and time-resolved IR measurements (19, 20). Alternative methods for determining the presence of metastable states and transition pathways combine many short trajectories to determine the kinetic connectivity (21, 22).
It has long been realized that in liquids at room temperature, thermal fluctuation can hide the underlying architecture of the energy surface. To deal with this problem, Stillinger and Weber (23) introduced the concept of “inherent structures (IS),” defined as the local minima on the potential energy surface. They are determined by calculating an MD trajectory at a given temperature and quenching the system by gentle energy minimization. All conformations that under this mapping go to the same IS define the basin (of attraction) of the IS. Such a partitioning has been used to study the thermodynamics of liquids, and for supercooled liquids to obtain insight into the dynamics (24–26). Early studies of proteins based on the IS concept (6, 27) demonstrated the multiminimum nature of the potential surface but were limited by the short trajectories that were accessible. Thermodynamic aspects of coarse-grained models of protein folding have been analyzed more recently (28–31) following the original prescription of Stillinger and Weber (23, 24); see also ref. 32.
In this paper, we show how the IS can be used to facilitate the analysis of MD trajectories that sample the conformation space under equilibrium conditions. The decomposition of the conformation space in terms of the IS provides a natural and simple description of a dynamical system when the timescales of the motions in a minimum and between minima are well separated. Once the IS have been determined, the IS make the mapping of the energy landscape onto a CSN essentially unique and avoid the uncertainties introduced by a purely geometrical clustering of the trajectory to define the network nodes (11, 12). Although the choice of the rmsd cutoffs for clustering, for example, is less important for the large conformational changes that occur in the transition between the unfolded and folded states of proteins (11, 33), for cases where the conformation is more restricted, as it is in the folded (native) state, the IS appear ideal for clustering the snapshots and determining the free-energy surface and associated CSN.
We study a model system, a 10-residue peptide in a β-hairpin conformation that is simple enough so that a full description of the dynamics with the IS approach can be achieved, while the system has a large enough number of degrees of freedom to be interesting. A 30-ns MD simulation at 300 K of the peptide in its folded (“native”) state provides a full sampling of the conformation space accessible at equilibrium. Mapping the potential energy onto the free-energy surface by means of the IS-based CSN and mincut free-energy profiles (34) demonstrates the correspondence between conformational changes, energy barriers, and transition kinetics. Comparison with the standard CSN analysis based on clustering of the 300 K MD trajectory shows certain limitations of the latter. An illustrative application of the IS analysis to a PDZ2 domain demonstrates the utility of the method for studying the native state dynamics of biologically relevant systems.
Results and Discussion
The Conformation Space Network and Free-Energy Profile.
Inherent structures and their transitions.
The application of the mapping into IS of the 30-ns long (1.5 × 107 snapshots) MD trajectory of the GS10 peptide results in 1,561 IS, as described in Methods (see Fig. S1). The set of IS defines an ensemble of microstates that can be used to characterize the dynamics of the peptide. Two short segments of the time series of the IS are shown in Fig. 1A. It is evident that different long-lived regions (called valleys) are sampled. The time series of the conformations at room temperature does not show this organization (gray lines in Fig. 1A). The valleys, which are described in more detail in terms of the free-energy profile (see below), include many IS basins and are characterized by different values of the potential energy. In Fig. 1A Left, a transition between two valleys (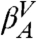 and
and  in Fig. 1C) is shown at t ≈ 22 ps. When the system is sampling one valley, it rapidly interconverts among a small number of minima with similar energies but slightly different conformations (e.g., all-atom rmsd between 0.3 and 0.6 Å) while, rarely, there is a transition to another valley. Fig. 1A Right shows a series of transitions between several valleys over the time scale of 600 ps. The fast transitions within a valley and the slow transitions between different ones recall the classification used in the field of supercooled liquids between type β- and α-transitions, respectively (see figure 3 of ref. 35 for more details).
in Fig. 1C) is shown at t ≈ 22 ps. When the system is sampling one valley, it rapidly interconverts among a small number of minima with similar energies but slightly different conformations (e.g., all-atom rmsd between 0.3 and 0.6 Å) while, rarely, there is a transition to another valley. Fig. 1A Right shows a series of transitions between several valleys over the time scale of 600 ps. The fast transitions within a valley and the slow transitions between different ones recall the classification used in the field of supercooled liquids between type β- and α-transitions, respectively (see figure 3 of ref. 35 for more details).
Fig. 1.

(A) IS trajectory. Two sample windows of the IS energies of the GS10 peptide in kcal/mol and the room temperature potential energy (in arbitrary units for comparison) time series are shown in red and gray, respectively. In the left part a transition between the 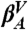 and
and 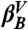 valley is depicted, whereas in the right part a series of transitions between multiple valleys is shown. (B) CSN of the GS10 peptide. Nodes and links represent IS and MD transitions, respectively; the color code corresponds to that in D, except that white nodes are characterized by Z > 0.97. The size of the nodes and links is proportional to their populations. For clarity, only nodes that have been visited more than 200 times are shown; there are a total of 571 nodes. (C) IS CFEP of the GS10 peptide relative to the most populated microstate, βA. The four most populated valleys are shown in dashed lines, and the corresponding lowest energy microstate structures are schematically sketched; only the atoms involved in the relevant hydrogen bonding and SER orientations are displayed. (D) Reaction pathways between the most important IS of the system displayed as a CCSN. IS populations and average number of transitions are shown both by the numbers and by the size of the nodes and the thickness of the links, respectively (see text for comments).
valley is depicted, whereas in the right part a series of transitions between multiple valleys is shown. (B) CSN of the GS10 peptide. Nodes and links represent IS and MD transitions, respectively; the color code corresponds to that in D, except that white nodes are characterized by Z > 0.97. The size of the nodes and links is proportional to their populations. For clarity, only nodes that have been visited more than 200 times are shown; there are a total of 571 nodes. (C) IS CFEP of the GS10 peptide relative to the most populated microstate, βA. The four most populated valleys are shown in dashed lines, and the corresponding lowest energy microstate structures are schematically sketched; only the atoms involved in the relevant hydrogen bonding and SER orientations are displayed. (D) Reaction pathways between the most important IS of the system displayed as a CCSN. IS populations and average number of transitions are shown both by the numbers and by the size of the nodes and the thickness of the links, respectively (see text for comments).
Given the microstates defined by the IS, we construct a CSN shown in Fig. 1B. Visual inspection of the network indicates the presence of a modular organization, i.e., the presence of different groups of nodes with many links between them and fewer links to other nodes. The weights of the nodes and links have a clear physical meaning representing, respectively, the populations of the basins of attraction of the IS (i.e., proportional to their free energy) and the transition probabilities [i.e., proportional to an effective activation energy, since there could be multiple potential energy barriers between a pair of nodes (36)]. For the detailed analysis of the network, we use the procedure described in Methods to calculate a one-dimensional cut-based free-energy profile (CFEP) (34). In Fig. 1C the CFEP of the GS10 peptide is shown. This profile represents the free-energy surface projected on the partition function-based reaction coordinate Z (see Methods), relative to a given reference microstate, in this case the most populated node (arbitrarily called βA). CFEPs have proven to be a better approach as compared with traditional free-energy profiles (where the landscape is projected onto one or more arbitrarily chosen order parameters (33, 37). The CFEPs provide a correct estimate of the height of the free-energy barriers, as well as their positions on the landscape (34, 38, 39). The cut-based free-energy profile of GS10 has a complex structure with a series of barriers and valleys. This is perhaps a somewhat surprising result for such a small peptide restricted to a well-defined β-hairpin structure. There are four regions that are characterized by broad valleys (dashed lines in the figure). The four valleys are labeled 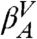 ,
, 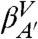 ,
, 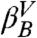 , and
, and  , and the most populated IS in each of them is βA (EIS = -59.5 kcal/mol), βB (EIS = -59.3 kcal/mol), βA′ (EIS = -57.9 kcal/mol), and βB′ (EIS = -57.7 kcal/mol). In addition, the first small valley of the profile located at Z ≈ 0.22 is labeled
, and the most populated IS in each of them is βA (EIS = -59.5 kcal/mol), βB (EIS = -59.3 kcal/mol), βA′ (EIS = -57.9 kcal/mol), and βB′ (EIS = -57.7 kcal/mol). In addition, the first small valley of the profile located at Z ≈ 0.22 is labeled 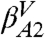 according to the βA2 microstate (EIS = -59.0 kcal/mol).
according to the βA2 microstate (EIS = -59.0 kcal/mol).
Structural analysis.
For this system, it is possible to structurally characterize the differences between the valleys in a straightforward manner. The transition from βA to βA′ is characterized by the rotation of the CBX group about the corresponding ψ dihedral angle, which disrupts the hydrogen bond between NH9 and O2 and forms a hydrogen bond between the O2 and the NH10 group of CBX; see the schematic peptide structures shown in Fig. 1C above the CFEP. The same atomic rearrangement characterizes the transition between βB and βB′. The transition between βA and βB corresponds to the rotation of the serine OH group about the dihedral angles 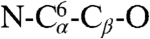 (χ1) and
(χ1) and 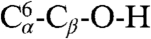 (χ2). This transition is responsible for the highest barrier (≈3 kcal/mol) in the CFEP at position Z ≈ 0.67; it arises primarily from the χ1 dihedral angle barrier (see Fig. S2). The rearrangement between βA and βA2 corresponds to a 120° rotation of the serine OH group about the χ2 dihedral angle. Examination of the high-energy microstates that are accessed within the various valleys (see Fig. 1A) shows that they correspond to a variety of backbone distortions; see Fig. S3.
(χ2). This transition is responsible for the highest barrier (≈3 kcal/mol) in the CFEP at position Z ≈ 0.67; it arises primarily from the χ1 dihedral angle barrier (see Fig. S2). The rearrangement between βA and βA2 corresponds to a 120° rotation of the serine OH group about the χ2 dihedral angle. Examination of the high-energy microstates that are accessed within the various valleys (see Fig. 1A) shows that they correspond to a variety of backbone distortions; see Fig. S3.
Coarse-grained network.
A coarse CSN (CCSN) was built from the original trajectory projected on the microstates by keeping only the five IS: βA, βA2, βA′, βB, βB′; all others were deleted. This was done to have a simple way to capture real transitions between the valleys and eliminate the multiple passes through the transition state. The reaction pathways between the five microstates described above are shown in Fig. 1D. It is clear that there are multiple, not equally probable, pathways between the five IS, instead of a sequential pathway along the Z coordinate. This complexity is not evident from the CFEP by itself. The reaction to go from βB′ to βA can take place by more than one possible route. The most significant pathway proceeds from βB′ to βB, then to βA2, and finally to βA. Also, while the direct transition from βB to βA is possible, it is 3 times less probable than passing through βA2, which acts as an intermediate that rapidly interconverts with βA; see also Fig. S2. This is not the case for the transition from βA′ to βA where the pathway involving any intermediates is 4.5 times less favorable than a direct transition. Finally, we note that the transition between βB′ and βA′ is faster when the transition is direct, rather than via the intermediate βB. These results demonstrate that even in the folded GS10 β-hairpin peptide, the free-energy landscape is complex and involves multiple pathways.
First Passage Times Analysis.
First passage time (FPT) distributions are useful for obtaining a detailed understanding of the dynamics (40). The FPT distribution obtained from the time series of the GS10 peptide is shown in Fig. 2. This plot represents the distribution of the relaxation times to the most populated IS, namely, microstate βA. The distribution is broad, spanning six decades in time from femtoseconds to tens of nanoseconds. The curve shows two “bumps”: one at times of the order of 500 fs and the other one on the 100 ps time scale. From the plot the origin of this behavior is not clear. For this reason, focused versions of the FPT distribution have been calculated as indicated in Methods; i.e., they include the relaxation from only a given subset of microstates.
Fig. 2.

IS FPT distributions to βA focused analysis (see main text). The red, light blue, blue, and gray lines represent the full FPT, focused FPT with Z < 0.2, Z < 0.3, and Z > 0.67, respectively (see text). Dashed lines show different functional fits. The two exponential fits have a characteristic time of 0.6 ps (dashed blue line) and 165 ps (dashed gray line). The fits have been slightly displaced for clarity. The fitting function for the full FPT distribution is f(t) = c0t-αe-t/t0 + c1e-t/t1 + c2e-t/t2, where c0, c1, c2, α, t0, t1, and t2 have a value of 704; 4,035; 88; 0.6; 33; 0.6; and 180, respectively.
A FPT distribution is built considering only the IS that are in the CFEP at values of Z lower than 0.2 (see Fig. 1C), the value at which the first free-energy barrier (to βA2) appears. This distribution is well represented by a power law with an exponential cutoff (light blue lines in Fig. 2; the fit is shown as a dashed line). Such behavior is typical of relaxations within a single well for which the characteristic time to reach the bottom (βA state) is undefined. The exponential cutoff arises from the fact that there is a typical residence time (on the order of 0.1 ps) after which the system jumps to other regions of the landscape. The CFEP, which predicts nearly barrierless transitions to βA, is in agreement with this analysis.
The inclusion of the microstates up to Z < 0.3 (i.e., including βA2) for the calculation of the FPT distribution introduces the first bump. This bump represents the fast exponential decay generated by the partial reorientation of the hydroxyl group of the SER side chain, typical of the microstates found in the βA2 region (blue curves). The time scales of these transitions overlap with the barrierless transitions generating the power-law behavior. However, the two types of transitions are microscopically different because the latter correspond to deformations of the backbone (without changing the β-sheet hydrogen bond pattern), while the former involves the reorientation of the hydroxyl group.
The second bump is also described by an exponential function. It develops when the contributions from the microstates corresponding to values of Z greater than 0.3 are included and it converges once valley 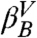 is added. The exponential behavior is even clearer in a distribution that includes only the snapshots beyond the barrier at Z ≈ 0.67 (gray lines), i.e., transitions from the
is added. The exponential behavior is even clearer in a distribution that includes only the snapshots beyond the barrier at Z ≈ 0.67 (gray lines), i.e., transitions from the 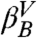 and
and 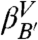 to
to 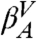 .
.
Finally, the full FPT distribution is fitted by the sum of the two exponentials and the power-law term (red lines in Fig. 2). Thus, each term of the fitting function, which is presented with the fitted parameters in the Fig. 2 caption, has a physical explanation corresponding to specific atomic rearrangements.
Standard Structure-Based Clusterings.
To compare the present analysis with the standard approach, the 30-ns trajectory of GS10 was clustered using the leader clustering algorithm, including all snapshots without minimization and clustering them based on two all-atom rmsd cutoff values (0.8 Å and 1.0 Å). These cutoff values are small compared to those usually used (12, 41), in accord with the fact that the overall rmsd range is small. A smaller cutoff (e.g., 0.5 Å) cannot be used because the number of clusters increases essentially linearly with the simulation time over the range studied (see Fig. S4), and an astronomical number of rarely visited clusters would result.
The CFEPs corresponding to the two cutoffs are shown in Fig. 3A. These results are very different from that obtained with the IS analysis (see Fig. 1C). They have essentially none of the structure with well-defined barriers found in the latter, which provide a meaningful description of the free-energy surface of the system. In Fig. 3B, the FPT distributions to the most populated microstate of the two rmsd clusterings are shown. These distributions are also very different from that obtained based on the IS (see Fig. 2). The curves are similar to a simple power law with a cutoff, and there is no sign of any dynamical transition involving a barrier.
Fig. 3.

Results of structure-based clustering of room temperature trajectory. (A) rmsd CFEP for the 1.0-Å and 0.8-Å cutoffs are shown as blue and light blue lines, respectively. The CFEP obtained by using the three most relevant degrees of freedom of the system (see text) is shown in gray. (B) FPT distributions to the most populated rmsd cluster. Blue and light blue lines represent the 1.0-Å and 0.8-Å cutoffs realizations, respectively.
One way to recover the correct partition into valleys without introducing the IS is to use the relevant degrees of freedom to define the microstates. Given the structural analysis of the CFEP described above, they are the three dihedral angles χ1, χ2, and ψ. The resulting CFEP (see gray curve in Fig. 3A) is a good approximation to that in Fig. 1C; i.e., it shows the relevant barriers with the correct heights. This result is a confirmation of the IS methodology, which does not require prior knowledge of the “relevant degrees of freedom.” An alternative way of selecting these coordinates could be based on principal components.
Application of the IS Framework to the PDZ2 Domain.
To illustrate the utility of the IS approach for describing the free-energy surface of a protein, we have applied it to a 1.6 ns MD trajectory (for a total of 8 × 104 snapshots) of the PDZ2 domain in its native state [Protein Data Base (PDB) ID code 3PDZ; see SI Text for the simulation details]. This short trajectory is not long enough to provide equilibrium sampling of the native state free-energy surface, but it suffices for the present purpose.
Since the number of degrees of freedom is very large (96 residues for a total of 1,422 atoms) and the sampling is limited, we use a reduced set of atoms; i.e., the backbone heavy atoms and Cβ (BB-CB). As for the GS10 peptide, the IS microstates are defined by the application of the leader clustering algorithm to the minimized trajectory with a small cutoff (0.2 Å). A total of 3,029 IS are found. Fig. 4A shows a sample window of the IS visited as a function of time and Fig. 4B presents the CFEP of the PDZ2 domain obtained with the IS. The profile shows a complex multiminimum free-energy surface; four major ones are labeled in the figure. The two most populated microstates belonging to the α and γ valleys are structurally compared (BB-CB rmsd difference of 0.72 Å; see Fig. S5). The residues primarily involved in the conformational change are in the loop regions. Fig. 4B also shows the structureless CFEP obtained from the trajectory without minimization with a BB-CB rmsd cutoff of 0.6 Å.
Fig. 4.

(A) Time series of IS; (B) CFEP of the PDZ2 domain. The IS and unminimized based profiles are shown in red and blue, respectively. The IS CFEP indicates that the protein visited four major valleys (labeled α, β, γ, and δ) during the simulation.
Conclusions
An understanding of how a protein functions at an atomic level of detail requires a knowledge of the free-energy surface. It is now recognized that equilibrium room temperature molecular dynamics simulations, combined with networks-based approaches, can be used for that purpose. In this type of analysis, the configurations visited during the trajectory have to be discretized to obtain statistically meaningful results. The standard approach, which has been used with considerable success, is to cluster the configurations in terms of an rmsd cutoff, whose definition is often arbitrary. In the present paper we show that an alternative approach based on the use of IS provides a natural, physically meaningful discretization. For two examples, a 10-residue peptide in a β-hairpin geometry and a PDZ2 domain, it is demonstrated that the use of the IS makes possible a detailed description of the free-energy surface, which is not obtainable when the rmsd criterion is applied directly to the trajectory. Importantly, the approach based on the IS does not require any a priori knowledge of the relevant degrees of freedom.
A difference in the application of the IS analysis to peptides and proteins, as compared with the original work of Stillinger and Weber 23, is that we introduce an rmsd criterion for clustering the IS, rather than using the energies per se. A reason for doing so is that the precise minimization required for the latter approach is very difficult to achieve for peptides and proteins. Moreover, the rmsd criterion, which permits the use of much smaller cutoff values than can be applied to the unminimized trajectory, is a robust way of focusing on the aspects of the structural changes of primary interest, e.g., the backbone plus Cβ atoms of the PDZ2 domain.
For the β-hairpin peptide, the one-dimensional CFEP obtained using the IS as microstates show the presence of several valleys separated by energy barriers as high as 3 kcal/mol; by contrast, the free-energy profile obtained from the standard room temperature analysis is a single structureless valley. Correspondingly, the distribution of the relaxation times to the most populated microstate is a multiexponential function. Analysis of the conformational changes by a simplified (coarse-grained) version of the conformation space network indicated the presence of multiple pathways. Thus, the present approach applied to the, perhaps surprisingly, complex dynamics of this simple system makes possible the determination of the relation among the different relaxation times, the barriers along the free-energy profile, and the peptide conformational changes.
The major conclusion of this paper is that energy minimization of the configurations visited along an equilibrium trajectory is an effective discretization method that does not require introduction of a knowledge of the essential degrees of freedom to obtain high-resolution representations of the underlying free-energy surface. This is of particular importance when the conformational changes are small, as they often are in the native state, in contrast to the much larger structural changes involved in protein folding. Moreover, there are an increasing number of experimental data for systems where small conformational changes in the native state are essential to function. They include the catalytic cycle of dihydrofolate reductase (3), where the rmsd between the five active states is in the range 0.5 to 2.0 Å, and the structural transition occurring in PDZ2 domains on ligand binding, where the rmsd is on the order of 2.0 Å (42). The present approach should then be of widespread interest for understanding the free-energy surface and its role in protein function. An experimental test of the results for the GS10 peptide should be possible in the near future by the use of time-resolved multidimensional infrared spectroscopy (43–45).
Methods
In this section, we outline briefly the method used, including details about aspects that are previously undescribed; more standard methodologies are described in SI Text.
Simulation Setup.
The system investigated is a 10-residue peptide (called GS10) with the sequence AAAGSAAA and N-acetylated (ACE) and C-amidated (CBX) blocking groups. The GS motif ensures increased stability of the β-hairpin structure because the G and S residues favor a turn (37). MD simulations, using the Langevin algorithm with a friction coefficient equal to 50 ps-1, were calculated with the CHARMM program (46, 47), the polar hydrogen energy function (PARAM19) was used, and the effective solvation free energy was approximated with the SASA implicit solvation model (48). SHAKE was employed so that an integration step of 2 fs could be used.
A simulation of 30 ns at 300 K was performed and snapshots were saved every time step for a total of 1.5 × 107 conformations. The trajectory was started from a minimized β-hairpin structure. The trajectory is long enough to provide approximate equilibrium sampling of the conformation space accessible to the system. During the simulation the all-atom rmsd remains less than 3.5 Å, indicating that no unfolding events occurred. Thus, the present study focuses on the atomic rearrangements of the β-hairpin structure, rather than on folding/unfolding processes. For details, see SI Text.
Inherent Structures.
IS are potential energy minima on the free-energy surface and are obtained by minimizing each snapshot of the trajectory (23). All configurations that under this mapping end up in the same minimum determine the basin (of attraction) of the IS. The minimization in the present study was performed by the steepest descent method followed by application of the adopted basis Newton-Raphson algorithm (46). The former is used to quench the system to the closest potential energy minimum, while the latter is required for approximate convergence of the minimization. Interestingly, structurally different IS (i.e., conformations belonging to different regions of the energy surface) were found to have very similar energies in some cases; e.g., snapshots characterized by energy differences within 10-4 kcal/mol have an average all-atom rmsd of 1.84 Å, which is a value close to the overall average rmsd pairwise difference of 1.92 Å. The low-lying minima (such as βA, βB, etc.) are both energetically and structurally different, whereas the higher-energy ones can have similar energies but different structures. Since this value of the energy difference threshold is near the limiting value that can be achieved with the present potential because of the nature of the solvation model (see Fig. S6), we defined the set of IS as the clusters obtained by the application of the leader clustering algorithm (49, 50) to the time series of the minimized snapshots with a very small cutoff of 0.15 Å. This procedure is robust; i.e., it is not sensitive to the value of the cutoff, provided that it is small enough; e.g., clustering with values of the cutoff as small as 0.05 give quantitatively similar results for the free-energy profile (see Fig. S7). To verify the rmsd analysis, we have used the same energy function, but without the solvation correction, which made possible the use of converged minimization to determine the IS. Identical results for the CFEP are obtained with a 10-5 kcal/mol energy cutoff and with rmsd cutoffs of 0.15 and 0.015 Å (see Fig. S8).
Conformation Space Networks.
The IS provide a natural discretization of the trajectory snapshots into a set of microstates. Each microstate (IS) represents a node of the CSN (11, 14), and a link is present if a direct transition between two microstates has been observed during the time series in a time step of a given size St (36). For details, see SI Text.
Mincut-Based Free-Energy Profile.
In a mincut-based free-energy profile, which shows the free energy as a function of the cumulative partition function (34, 38), microstates are ranked according to their kinetic proximity with respect to a reference microstate (R). In this work, the mean first passage time (m) version of the algorithm was employed (38); details are given in SI Text.
Coarse CSN.
The essential element of the CCSN is to keep only a single microstate near the bottom of each valley (e.g., the lowest energy microstate) and to determine the transitions between this selected set of states. In this way one excludes, in particular, the many IS in the neighborhood of the top of the barrier (see Fig. S9) where multiple crossings occur. Thus, introduction of the CCSN is a simple procedure for determining the meaningful transitions between pairs of valleys. If there are intermediates that play a role, they will be missed in the present CCSN, if they are not explicitly included (e.g., microstate βA2; see Results). Alternatively, the iterative mincut method for determining SEKN could be used (12, 51). A test for the correct description of the intervalley transitions by the CCSN is provided by the agreement between the first passage time distributions from a Markov model of the CCSN and the MD simulation; see Fig. S10.
First Passage Time Distributions.
The distribution of FPTs to a given target microstate (R) is calculated as the probability distribution of the time intervals to reach R for the first time along the time series. Hence, for every time t the time difference T = (tR - t) contributes to the distribution, where tR is the first occurrence of microstate R along the time series with respect to time t. Given this procedure and the time series, the FPT depends on the definition of R only. FPT distributions are relevant for probing the dynamical behavior of a system. For example, exponential FPT distributions indicate the presence of a single rate-limiting barrier to reach R, whereas power-law distributions indicate barrierless relaxations (or multiple pathways with a range of barriers). To investigate the contribution to the FPT distribution of a given set of microstates with a desired property (e.g., a subset of snapshots with specific values of the cumulative partition function Z) only the snapshots fulfilling the property are included in the calculation; this is referred as a focused FPT analysis.
Supplementary Material
Acknowledgments.
We thank Dr. S. Muff and Dr. M. Seeber, who wrote the code for the CFEP analysis in the WORDOM program (50), based on refs. 34 and 38). The amino acid composition of the GS10 peptide was inspired by the work of Guarnera et al. on simplified sequences (52). F.R. was supported by the European Molecular Biology Organization (EMBO) and the CNRS.
Footnotes
The authors declare no conflict of interest.
*This Direct Submission article had a prearranged editor.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.0915087107/-/DCSupplemental.
References
- 1.Schuler B, Eaton WA. Protein folding studied by single-molecule FRET. Curr Opin Struct Biol. 2008;18:16–26. doi: 10.1016/j.sbi.2007.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Colletier JP, et al. Shoot-and-Trap: Use of specific x-ray damage to study structural protein dynamics by temperature-controlled cryo-crystallography. Proc Natl Acad Sci USA. 2008;105:11742–11747. doi: 10.1073/pnas.0804828105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boehr DD, McElheny D, Dyson HJ, Wright PE. The dynamic energy landscape of dihydrofolate reductase catalysis. Science. 2006;313:1638–1642. doi: 10.1126/science.1130258. [DOI] [PubMed] [Google Scholar]
- 4.Goldstein M. Viscous liquids and glass transition: A potential energy barrier picture. J Chem Phys. 1969;51:3728–3739. [Google Scholar]
- 5.Frauenfelder H, Sligar S, Wolynes P. The energy landscapes and motions of proteins. Science. 1991;254:1598–1603. doi: 10.1126/science.1749933. [DOI] [PubMed] [Google Scholar]
- 6.Elber R, Karplus M. Multiple conformational states of proteins: A molecular dynamics analysis of myoglobin. Science. 1987;235:318–321. doi: 10.1126/science.3798113. [DOI] [PubMed] [Google Scholar]
- 7.Zagrovic B, Snow CD, Shirts MR, Pande VS. Simulation of folding of a small alpha-helical protein in atomistic detail using worldwide-distributed computing. J Mol Biol. 2002;323:927–937. doi: 10.1016/s0022-2836(02)00997-x. [DOI] [PubMed] [Google Scholar]
- 8.Karplus M, Kuriyan J. Molecular dynamics and protein function. Proc Natl Acad Sci USA. 2005;102:6679–6685. doi: 10.1073/pnas.0408930102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dror RO, et al. Identification of two distinct inactive conformations of the beta2-adrenergic receptor reconciles structural and biochemical observations. Proc Natl Acad Sci USA. 2009;106:4689–4694. doi: 10.1073/pnas.0811065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Klepeis J, Lindorff-Larsen K, Dror R, Shaw D. Long-timescale molecular dynamics simulations of protein structure and function. Cur Opin Struc Biol. 2009;19:120–127. doi: 10.1016/j.sbi.2009.03.004. [DOI] [PubMed] [Google Scholar]
- 11.Rao F, Caflisch A. The protein folding network. J Mol Biol. 2004;342:299–306. doi: 10.1016/j.jmb.2004.06.063. [DOI] [PubMed] [Google Scholar]
- 12.Krivov S, Karplus M. Hidden complexity of free energy surfaces for peptide (protein) folding. Proc Natl Acad Sci USA. 2004;101:14766–14770. doi: 10.1073/pnas.0406234101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Caflisch A. Network and graph analyses of folding free energy surfaces. Curr Opin Struct Biol. 2006;16:71–78. doi: 10.1016/j.sbi.2006.01.002. [DOI] [PubMed] [Google Scholar]
- 14.Gfeller D, De Los Rios P, Caflisch A, Rao F. Complex network analysis of free-energy landscapes. Proc Natl Acad Sci USA. 2007;104:1817–1822. doi: 10.1073/pnas.0608099104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Settanni G, Fersht AR. High temperature unfolding simulations of the TRPZ1 peptide. Biophys J. 2008;94:4444–4453. doi: 10.1529/biophysj.107.122606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang S, Roux B. Src kinase conformational activation: Thermodynamics, pathways, and mechanisms. PLOS Comput Biol. 2008;4(3):e1000047. doi: 10.1371/journal.pcbi.1000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Prada-Gracia D, Gomez-Gardenes J, Echenique P, Falo F. Exploring the free energy landscape: From dynamics to networks and back. PLOS Comput Biol. 2009;5:e1000415. doi: 10.1371/journal.pcbi.1000415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li CB, Yang H, Komatsuzaki T. Multiscale complex network of protein conformational fluctuations in single-molecule time series. Proc Natl Acad Sci USA. 2008;105:536–541. doi: 10.1073/pnas.0707378105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ihalainen JA, et al. Folding and unfolding of a photoswitchable peptide from picoseconds to microseconds. Proc Natl Acad Sci USA. 2007;104:5383–5388. doi: 10.1073/pnas.0607748104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ihalainen JA, et al. Alpha-helix folding in the presence of structural constraints. Proc Natl Acad Sci USA. 2008;105:9588–9593. doi: 10.1073/pnas.0712099105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowman GR, Beauchamp KA, Boxer G, Pande VS. Progress and challenges in the automated construction of Markov state models for full protein systems. J Chem Phys. 2009;131:124101. doi: 10.1063/1.3216567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Noé F, et al. Constructing the equilibrium ensemble of folding pathways from short off-equilibrium simulations. Proc Nat Acad Sci USA. 2009;106:19011–19016. doi: 10.1073/pnas.0905466106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stillinger FH, Weber TA. Hidden structure in liquids. Phys Rev A. 1982;25:978–989. [Google Scholar]
- 24.Stillinger F, Weber T. Dynamics of structural transitions in liquids. Phys Rev A. 1983;28:2408–2416. [Google Scholar]
- 25.Denny R, Reichman D, Bouchaud J. Trap models and slow dynamics in supercooled liquids. Phys Rev Lett. 2003;90(2):025503. doi: 10.1103/PhysRevLett.90.025503. [DOI] [PubMed] [Google Scholar]
- 26.Berthier L, Garrahan J. Nontopographic description of inherent structure dynamics in glassformers. J Chem Phys. 2003;119(8):4367–4371. [Google Scholar]
- 27.Caves LS, Evanseck JD, Karplus M. Locally accessible conformations of proteins: Multiple molecular dynamics simulations of crambin. Protein Sci. 1998;7:649–66. doi: 10.1002/pro.5560070314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baumketner A, Shea JE, Hiwatari Y. Glass transition in an off-lattice protein model studied by molecular dynamics simulations. Phys Rev E. 2003;67:011912. doi: 10.1103/PhysRevE.67.011912. [DOI] [PubMed] [Google Scholar]
- 29.Nakagawa N, Peyrard M. the inherent structure landscape of a protein. Proc Nat Acad Sci USA. 2006;103(14):5279–5284. doi: 10.1073/pnas.0600102103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim J, Keyes T. Inherent structure analysis of protein folding. J Phys Chem B. 2007;111:2647–2657. doi: 10.1021/jp0665776. [DOI] [PubMed] [Google Scholar]
- 31.Kim J, Keyes T, Straub J. Relationship between protein folding thermodynamics and the energy landscape. Phys Rev E . 2009;79(3):030902. doi: 10.1103/PhysRevE.79.030902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krivov SV, Chekmarev SF, Karplus M. Potential energy surfaces and conformational transitions in biomolecules: A successive confinement approach applied to a solvated tetrapeptide. Phys Rev Lett. 2002;88:038101. doi: 10.1103/PhysRevLett.88.038101. [DOI] [PubMed] [Google Scholar]
- 33.Rao F, Caflisch A. Replica exchange molecular dynamics simulations of reversible folding. J Chem Phys. 2003;119:4035–4042. doi: 10.1063/1.1809588. [DOI] [PubMed] [Google Scholar]
- 34.Krivov SV, Karplus M. One-dimensional free-energy profiles of complex systems: Progress variables that preserve the barriers. J Phys Chem B. 2006;110:12689–12698. doi: 10.1021/jp060039b. [DOI] [PubMed] [Google Scholar]
- 35.Debenedetti PG, Stillinger FH. Supercooled liquids and the glass transition. Nature. 2001;410:259–267. doi: 10.1038/35065704. [DOI] [PubMed] [Google Scholar]
- 36.Gfeller D, de Lachapelle DM, De Los Rios P, Caldarelli G, Rao F. Uncovering the topology of configuration space networks. Phys Rev E. 2007;76:026113. doi: 10.1103/PhysRevE.76.026113. [DOI] [PubMed] [Google Scholar]
- 37.Ferrara P, Caflisch A. Folding simulations of a three-stranded antiparallel beta-sheet peptide. Proc Natl Acad Sci USA. 2000;97:10780–10785. doi: 10.1073/pnas.190324897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Krivov SV, Muff S, Caflisch A, Karplus M. One-dimensional barrier-preserving free-energy projections of a beta-sheet miniprotein: New insights into the folding process. J Phys Chem B. 2008;112:8701–8714. doi: 10.1021/jp711864r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Muff S, Caflisch A. Identification of the protein folding transition state from molecular dynamics trajectories. J Chem Phys. 2009;130:125104–125111. doi: 10.1063/1.3099705. [DOI] [PubMed] [Google Scholar]
- 40.Chekmarev SF, Krivov SV, Karplus M. Folding time distributions as an approach to protein folding kinetics. J Phys Chem B. 2005;109:5312–5330. doi: 10.1021/jp047012h. [DOI] [PubMed] [Google Scholar]
- 41.Rao F, Settanni G, Guarnera E, Caflisch A. Estimation of protein folding probability from equilibrium simulations. J Chem Phys. 2005;122:184901–184905. doi: 10.1063/1.1893753. [DOI] [PubMed] [Google Scholar]
- 42.Kong Y, Karplus M. Signaling pathways of PDZ2 domain: A molecular dynamics interaction correlation analysis. Proteins: Struct Funct Bioinformatics. 2008;74:145–54. doi: 10.1002/prot.22139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hamm P, Lim M, Hochstrasser RM. Structure of the amide I band of peptides measured by femtosecond nonlinear-infrared spectroscopy. J Phys Chem B. 1998;102:6123–6138. [Google Scholar]
- 44.Kolano C, et al. Watching hydrogen-bond dynamics in a beta-turn by transient two-dimensional infrared spectroscopy. Nature. 2006;444:469–472. doi: 10.1038/nature05352. [DOI] [PubMed] [Google Scholar]
- 45.Bagchi S, Falvo C, Mukamel S, Hochstrasser RM. 2D-IR experiments and simulations of the coupling between amide-I and ionizable side chains in proteins: Application to the Villin headpiece. J Phys Chem B. 2009;113:11260–11273. doi: 10.1021/jp900245s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brooks BR, et al. Charmm: A program for macromolecular energy, minimization, and dynamics calculations. J Comput Chem. 1983;4:187–217. [Google Scholar]
- 47.Brooks BR, et al. CHARMM: The biomolecular simulation program. J Comput Chem. 2009;30:1545–1614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ferrara P, Apostolakis J, Caflisch A. Evaluation of a fast implicit solvent model for molecular dynamics simulations. Proteins: Struct Funct Genet. 2002;46:24–33. doi: 10.1002/prot.10001. [DOI] [PubMed] [Google Scholar]
- 49.Hartigan J. Clustering Algorithms. New York: Wiley; 1975. [Google Scholar]
- 50.Seeber M, Cecchini M, Rao F, Settanni G, Caflisch A. Wordom: A program for efficient analysis of molecular dynamics simulations. Bioinformatics. 2007;23:2625–2627. doi: 10.1093/bioinformatics/btm378. [DOI] [PubMed] [Google Scholar]
- 51.Allen LR, Krivov SV, Paci E. Analysis of the free-energy surface of proteins from reversible folding simulations. PLOS Comput Biol. 2009;5:e1000428. doi: 10.1371/journal.pcbi.1000428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guarnera E, Pellarin R, Caflisch A. How does a simplified-sequence protein fold? Biophys J. 2009;97(6):1737–1746. doi: 10.1016/j.bpj.2009.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.