

Abstract
Mitochondria are known to actively regulate cell death with the final phenotype of demise being determined by the metabolic and energetic status of the cell. Mitochondrial membrane permeabilization (MMP) is a critical event in cell death, as it regulates the degree of mitochondrial dysfunction and the release of intermembrane proteins that function in the activation and assembly of caspases. In addition to the crucial role of proapoptotic members of the Bcl-2 family, the lipid composition of the mitochondrial membranes is increasingly recognized to modulate MMP and hence cell death. The unphysiological accumulation of cholesterol in mitochondrial membranes regulates their physical properties, faciliating or impairing MMP during Bax and death ligand-induced cell death depending on the level of mitochondrial GSH (mGSH), which in turn regulates the oxidation status of cardiolipin. Cholesterol-mediated mGSH depletion stimulates TNF-induced reactive oxygen species and subsequent cardiolipin peroxidation, which destabilizes the lipid bilayer and potentiates Bax-induced membrane permeabilization. These data suggest that the balance of mitochondrial cholesterol to peroxidized cardiolipin regulates mitochondrial membrane properties and permeabilization, emerging as a rheostat in cell death.
Keywords: Cholesterol, cardiolipin, reactive oxygen species, mitochondrial GSH, cell death
1. Introduction
Mitochondria play a central role in various forms of cell death which are characterized by differential biochemical features that include apoptosis (caspase-dependent and independent), necrosis and autophagy. A key event in this role is the breakage of mitochondrial membranes, particularly the outer mitochondrial membrane, which allows the release of proapoptotic factors that engage the apoptosome resulting in apoptotic cell death. Understanding the mechanisms leading to mitochondrial membrane permeabilization (MMP) is thus central to the design of estrategies to control cell death. In pathologies associated with enhanced cell death such as liver diseases (e.g. steatohepatitis) or neurodegeneration (e.g. Alzheimer's disease), the therapeutic goal is to prevent or ameliorate the extent of cell death. In contrast, in cancer the primary interest is to kill tumor cells more efficiently sensitizing them to current cancer therapy. Although a major focus in the field has been paid on the role of Bcl-2 family members in the regulation of MMP and cell death [1, 2], an increasing body of evidence points to a key role of lipids in the regulation of MMP and cell death susceptibility. In particular, mitochondrial cholesterol has emerged as an important modulator of MMP in response to apoptotic stimuli including hypoxia, TNF, amyloid beta peptide (Aβ) or Bax, underlying the importance of this particular bilayer component in disease pathogenesis, including alcoholic and nonalcoholic steatohepatitis, Alzheimer's disease or hepatocellular carcinoma [3, 4]. Although the unphysiological accumulation of cholesterol shields mitochondrial membranes impairing the bilayer permeabilization, as shown in model membranes, this effect can be counterbalanced by the presence of peroxidized cardiolipin, which is in turn regulated by the availability of mitochondrial GSH (mGSH) [3]. Thus, mitochondrial cholesterol plays a paradoxical role in cell death depending on the regulation of mitochondrial GSH (mGSH) transport, mGSH homeostasis and subsequent protection of cardiolipin from peroxidation. In this review, we will briefly summarize the implications of the enrichment of mitochondrial membranes in cholesterol and its impact in mitochondrial membrane properties, mGSH regulation, cardiolipin status, and cell death.
2. Mitochondrial Pathway of Cell Death
Apoptosis is a specific form of cell death which is essential for life as it is required for embryonic development, tissue homeostasis and immune defense. Dysregulation of apoptosis, however, contributes to many pathophysiological states and diseases. The key mediators of apoptotic cell death are cysteine proteases, called caspases, that work in a coordinated cascade to cleave key substrates that dismantle the cell with characteristic biochemical features [5, 6]. Apoptosis can occur through two main pathways, the extrinsic pathway and the intrinsic pathway. The former is initiated upon the binding of an extracellular ligand to transmembrane death receptors of the TNF supefamily, which leads to the assembly of the death-inducing signalling complex (DISC) [7, 8]. The DISC then activates an initiator caspase, which triggers the enzymatic cascade that leads to apoptotic death. The intrinsic pathway is also known as the mitochondrial pathway and is activated by stimuli that lead to the permeabilization of mitochondrial outer membrane (MOM) and the subsequent release of proteins, such as cytochrome c, from the mitochondrial intermembrane space (IMS), which initiate and regulate caspase activation. Cytochrome c normally resides within the cristae of the mitochondrial inner membrane (MIM) and is effectively sequestered by narrow cristae junctions. Within the MIM, cytochrome c participates in the mitochrondrial electron-transport chain, using its haem group as a redox intermediate to shuttle electrons between complex III and complex IV. However, in response to specific apoptotic triggers, such as DNA damage, or metabolic stress, the intrinsic apoptotic pathway is activated and mitochondrial cytochrome c is released into the cytosol [2]. This process is thougth to occur in two phases, first the mobilization of cytochrome c and then its translocation through permeabilized MOM, described below. In addition to cytochrome c, other IMS proteins are mobilized and released into the cytosol. For instance, the release of Smac/Diablo into the cytosol ensures the efficiency of caspase 3 activation in degrading target proteins through inhibition of inhibitor of apoptosis proteins (IAPs) [9, 10]. Furthermore, the mitochondrial protein Omi/HtrA2 promotes cell death in a dual fashion. Besides its IAP activity Omi/HtrA2 also functions as a serine protease, contributing to both caspase-dependent and caspase-independent cell death [11, 12]. Moreover, other specialized mitochondria-resident proteins, such as the apoptosis inducing factor (AIF) [13] and endonuclease G [14], are translocated to the nuclei following their release from mitochondria and promote peripheral chromatin condensation and high molecular weight DNA fragmentation. An important feature of apoptosis is its dependence on energy by sustained ATP supply to support caspase activation [2, 5, 6].
In contrast to apoptotic cell death, necrotic cell death is characterized primarily by the rapid loss of cellular membrane potential, which leads to cytoplasmic swelling, rupture of the plasma membrane, and cytolysis. The extent of mitochondrial dysfunction and the rupture of MIM impairs the ability of mitochondria to generate ATP, needed for the function of homeostatic ion pumps/channels, and the generation of reactive oxygen and nitrogen species (ROS/RNS), which in turn, can regulate caspase activation [15]. The onset of necrotic cell death can also occur in the extrinsic pathway by death receptors, such as TNF or Fas. FADD, a DISC component, has been shown to be involved in TNF-induced necrosis [16, 17]. RIP1 also contributes to death receptor-induced necrosis, as RIP1-deficient T cells are also resistant to death induced by TNF and Fas in the presence of caspase inhibitors [17]. The kinase activity of RIP appears to be required for necrosis induction, although its targets remain to be identified. Necrostatin-1, a small molecule that can inhibit necrosis induced by RIP or by TNF/Fas in the presence of caspase inhibitor [18], may be a valuable tool to disclose signaling pathways involved in death receptor-mediated necrosis. Thus, while mitochondria play a key role in the control of cell death, the phenotype of the dying cell depends on the metabolic and energetic status of the cell that determine the level and extent of caspase activation.
3. Mitochondrial Membrane Permeabilization and Cytochrome C Release
The mechanisms of MMP and cytochrome c release from IMS have been an intense area of research, whose understanding may be important to control cell death. Indeed, this is a critical issue because, in addition to cytochrome c, other proteins are also released from the intermembrane space into the cytosol, where they engage in a strategic battle to promote or counteract caspase activation and hence cell death, as described above. The intricacy of this pathway highlights the central role of mitochondria in regulating cell death, regardless of the phenotype of death (caspase-dependent apoptosis, caspase-independent apoptosis, or necrosis). Since these proapoptotic proteins are normally confined in the IMS, the events that culminate in the rupture of the physical barrier (MOM), which limits their release into the cytosol, constitute a point-of-no-return in cell death [2]. While the underlying mechanisms are not completely understood, there has been evidence for two possible mechanisms leading to the breakage of MOM: the mitochondrial permeability transition (MPT), and the permeabilization of MOM without disruption of the inner membrane, which is regulated by Bcl-2 family. The usually impermeable MIM prevents unrestrained influx of low-molecular-weight solutes into the mitochondria. The MPT features mitochondrial swelling, uncoupling and MIM permeabilization to small solutes, which results in a colloidal osmotic pressure that leads primarily to massive swelling of the mitochondrial matrix sensitive to cyclosporine A [2, 19, 20]. As a consequence of MPT the MOM ruptures and cytochrome c and other IMS proteins are released into the cytosol. MTP is most likely assembled at the contact sites of the MOM and the MIM and the actual components of the MTP remain ill defined. Although MPT has a definitive role in regulating MMP, mitochondrial dysfunction and cell death, this mechanism seems to play a minor role in apoptotic cell death as opposed to necrosis. For instance, deficiency of VDAC isoforms or cyclophilin D, two essential MPT components, does not prevent apoptosis induced by Bcl-2 family members, but impairs Ca2+-mediated MPT and ischemia-reperfusion necrosis [21-23]. Alternatively to MPT, Bcl-2 family members control MOM permeabilization. Bcl2-family death agonists induce MOM permeabilization, thereby promoting cytochrome c release, whereas Bcl2-family death antagonists prevent it. Thus, Bcl2-family proteins control mitochondrial integrity, regulate cytochrome c release and intrinsic apoptosis [1]. Under non-apoptotic conditions, Bax is inactive and present in the cytosol as a monomer. Following an apoptotic stimulus, Bax is activated and translocates to the mitochondria, where it undergoes a conformational change and inserts into the MOM. Bax oligomerization is associated with the formation of openings in the MOM to allow the release of cytochrome c and other IMS proteins into the cytosol, and hence Bax oligomerization is considered a critical regulatory point in cell death [1, 2]. In contrast to Bax, Bak resides on the MOM rather than in the cytosol but, like Bax, it undergoes a conformational change in response to apoptotic stimuli. This change allows Bak to oligomerize, contributing to MOM permeabilization. One potential mechanism involved in the permeabilizing activities of Bax/Bak oligomers is the formation of pores in MOM. However, attempts to visualize these oligomers have shown that large clusters of Bax are localized adjacent, but not on, the MOM [1]. An alternative model suggests that the insertion of activated, oligomerized Bax and/or Bak into the MOM creates a positive curvature stress on the membrane, leading to supramolecular pores that include lipids in the MOM [24-26]. Clearly, understanding the mechanisms underlying MOM permeabilization may provide novel strategies to regulate cytochrome c release and hence apoptosis. The relative prevalence of these pathways in the regulation of cell death is not definitively established. One important feature of mitochondrial membrane permeabilization is the loss of function resulting in the inability of mitochondria to synthesize ATP through the oxidative phosphorylation. However, while the final outcome of mitochondrial dysfunction is cell death, as mentioned above, the phenotype of death, apoptosis and/or necrosis, is dependent on the level of cellular ATP as ATP is required for the efficient assembly of the apoptosome and/or the extent of ROS generation [1, 2].
In addition to understanding the mechanisms leading to MOM permeabilization to design strategies to control cell death, another level of control centers on the mechanisms underlying the mobilization of cytochrome c from IMS. It has been proposed that during mobilization cytochrome c detaches from the MIM and dissociates from the membrane phospholipid cardiolipin [27]. Indeed, it has been estimated that a relatively low proportion of cytochrome c in the mitochondria seems to be associated with cardiolipin, involving two major mechanisms [27]. At physiological pH, cytochrome c has 8 positive net charges, establishing an electrostatic bond with the anionic cardiolipin. In addition, cytochrome c has a hydrophobic channel through which one acyl chain of cardiolipin inserts. The other chains of cardiolipin remain in the membrane, thereby anchoring cytochrome c to the MIM [28]. One mechanism involved in cytochrome c detachment from MIM involves cardiolipin oxidation because oxidized cardiolipin has a lower affinity for cytochrome c than the unoxidized form [29, 30]. Cardiolipin can be oxidized by ROS, which are controlled by mitochondrial antioxidants such as GSH [30, 31] or by the cardiolipin–cytochrome c complex [32]. Detachment of cytochrome c from cardiolipin might also be triggered by increased cytosolic Ca2+, which weakens the electrostatic interaction between cytochrome c and cardiolipin. In addition, as described below, oxidized cardiolipin modulates the biophysical properties of MOM to allow oligomerized Bax to insert and permeabilized MOM [30].
In spite that only a small fraction of cytochrome c is bound to cardiolipin (about 15%), the oxidation of this lipid is thought to impact the mobilization of cytochrome c from MIM, although the relevance of this step in the release of cytochrome c to the cytosol is controversial. For instance, in isolated mitochondria, exposure to 50–80 mM K+, a concentration of intracellular K+ that is within physiological limits, causes the release of cytochrome c from the IMM [33]. In addition, live-cell imaging has revealed no differences in the kinetics of the release of cytochrome c compared with other IMS proteins in cells undergoing apoptosis [34]. These results imply that cytochrome c mobilization might not be required as an additional step in the release of cytochrome c from the mitochondria. In addition, mobilization of cytochrome c might also involve its removal from narrow cristae junctions, although this specific issue is unsettled. In this regard, it has been suggested that the remodelling of cristae is required in the release of this interior pool of cytochrome c [35], and recent studies correlated the disassembly of Opa1 oligomers, a structural determinant of cristae morphology, with the remodelling of cristae [36]. However, recent data using three-dimensional electron microscope tomograpy, have disclosed that cristae remodelling is not required for efficient release of cytochrome c [37]. Moreover, BH3-only proteins Bid and Bim induced full cytochrome c release but only a subtle alteration in cristae junctions, which involved the disassembly of Opa1 complexes [38].
4. Cholesterol in Mitochondrial Membrane Properties, MMP and Cell Death
Cholesterol is a critical component of membranes with key structural and functional roles [39]. Compared to plasma membrane mitochondria are much less enriched in cholesterol with estimates of about 3-5% of the total cholesterol cell load. However, this restricted pool of cholesterol in mitochondria plays a fundamental physiological role such as in the synthesis of bile acids in hepatocytes or steroidogenic hormones in specialized tissues [3]. Given the role of cholesterol in the regulation of membrane dynamics and physical properties, the synthesis and supply of membrane cholesterol is highly regulated under physiological conditions. Cholesterol is synthesized in the ER and delivered to other organelles by a combination of vesicular and non-vesicular transport processes [39, 40]. Since cholesterol is very insoluble in water, it must be shuttled by carriers. The mechanisms of non-vesicular cholesterol transport are poorly understood, although there is increasing evidence that this is the major route for cholesterol movement between organelles. In the case of cholesterol trafficking to mitochondria, the best characterized example of non-vesicular mechanism involves the steroidogenic acute regulatory protein (StAR), which is the prototype for the StAR-related lipid transfer (START) family of transport proteins. StAR plays an essential role in the delivery of cholesterol to mitochondria, where it is used in steroid hormone synthesis in steroidogenic tissues or in the acidic pathway of bile acid synthesis in hepatocytes [39, 41]. Cholesterol is known to regulate membrane organization. In particular cholesterol modulates the coexistence within membranes of lipid-disordered and lipid-ordered phases, which regulates membrane permeability and function of resident proteins. In this regard, it has been shown that cholesterol loading in mitochondria results in increased membrane order parameter which impacts negatively in specific membrane carriers, such as the GSH transport system without effect in others, including the S-adenosyl-L-methionine transport system [42]. Since GSH is synthesized de novo exclusively in the cytosol but not in mitochondria, the mitochondrial source of GSH depends on its transport from cytosol by the 2-oxoglutarate and dicarboxylates carriers, which have been previously characterized [43, 44]. Functional expression studies in Xenopus laevis oocytes provided clear evidence that the 2-oxoglutarate carrier is highly sensitive to mitochondrial membrane fluidity loss induced by cholesterol loading [44]. Coupled with previous findings showing low and high affinity components for the mitochondrial transport of GSH into rat liver mitochondria, the 2-oxoglutarate carrier accounts for the low affinity transport site with the dicarboxylate playing a minor role. Thus, one of the functional consequences of mitochondrial cholesterol enrichment is the impairment of mitochondrial GSH (mGSH) transport which results in its depletion in the mitochondrial matrix, promoting the stimulation of mitochondrial ROS induced by death ligands (e.g. TNF) (Figure 1). This paradigm would favor the peroxidation of cardiolipin by enhanced ROS, which destabilizes the lipid bilayer potentiating MOM and subsequent release of cytochrome c. Since GD3 is a key component of raft like domains and shown to promote MOM [15], it is conceivable that the enhanced mitochondrial cholesterol loading may result also in the enrichment of GD3 in specific domains of mitochondrial membranes possibly associated with peroxidized cardiolipin, although this remains to be established.
Figure 1. Cholesterol regulates the cardiolipin/peroxidized cardiolipin status.
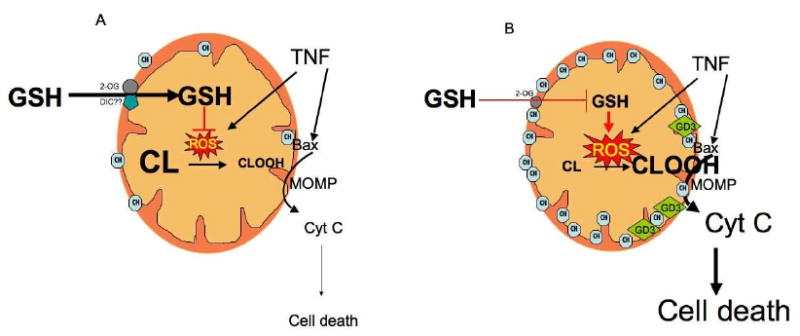
A, under physiological settings the pool of mitochonrial cholesterol is low allowing unimpaired transport of GSH into the mitochondrial matrix by the 2-oxoglutarate (2-OG) carrier, with a minor role for the dicarboxylate carrier (DIC), at least in rat liver mitochondria. In this scenario, the generation of ROS by cell death stimuli such as TNF, hypoxia or Aβ, is controlled by GSH availability, ensuring the presence of intact cardiolipin in mitochondrial membranes. B, when the trafficking of cholesterol is stimulated as seen in different pathologies (e.g. ASH/NASH or Alzheimer's disease), the mitochondrial transport of GSH via 2-OG is impaired resulting in GSH depletion which stimulates the generation of ROS in response to TNF causing cardiolipin peroxidation (CLOOH), which destabilizes the lipid bilayer and contributes to MOM permeabilization (MOMP) mediated by Bax.
4.1 Mitochondrial cholesterol in liver diseases and neurodegeneration
One of the most common forms of liver disease worldwide is steatohepatitis, which encompasses both alcoholic (ASH) and nonalcoholic steatohepatitis (NASH). While the pathogenesis of these diseases are obviously different, they exhibit indistinguisable histological features and commom underlying mechanisms of disease progression that begin with hepatic steatosis. Although fat infiltration in these diseases is heterogenous consisting primarily of triglyceride, free fatty acids and cholesterol, recent studies have pointed to an emerging role for cholesterol, particularly in mitochondria, in the progression from steatosis to steatohepatitis as it sensitizes hepatocytes to inflammatory cytokines (e.g. TNF/Fas ligand) [45]. Moreover, massive triglyceride accumulation in the liver of transgenic mice overexpressing DGAT2 did not impair insulin sensitivity, a hallmark feature of NASH [46]. In addition, unchanged hepatic free fatty acids levels were reported in patients with NASH compared to healthy controls, while the degree of free cholesterol accumulation correlated with disease progression [47, 48]. The underlying mechanism for the sensitization of mitochondrial cholesterol-loaded hepatocytes to TNF/Fas and hence steatohepatitis involved the selective mGSH depletion, an outcome also observed in models of ASH [43, 45]. Two pieces of evidence supported this contention. First, the replenishment of mGSH by permeable precursors, such as GSH ethyl ester, protected mitochondrial cholesterol-loaded hepatocytes induced by alcohol feeding or genetic models against TNF/Fas-mediated cell death. Second, control hepatocytes selectively depleted of mGSH reproduced the sensitization to TNF/Fas observed in the nutritional and genetic models of hepatic steatosis characterized by cholesterol accumulation. In this paradigm, we have observed that mGSH depletion sensitizes to TNF/Fas by enhancing the mitochondrial generation of ROS via acidic sphingomyelinase-induced ceramide generation, without inactivation of NF-κB-dependent survival pathway [30]. Moreover, mGSH depletion potentiated TNF-induced cardiolipin peroxidation, which facilitated the permeabilizing activity of oligomerized BAX in the MOM (see below).
Neurodegeneration reflects a number of hereditary or sporadic diseases characterized by progressive atrophy and neuronal loss, which include major diseases such as Alzheimer's disease (AD), Parkinson's disease (PD), and Huntington's disease (HD). AD is characterized by progressive memory loss and cognitive impairment whose incidence is associated with aging. Despite intense research, the molecular pathogenesis of AD is still incompletely unknown. One of the main features of AD involves the deposition in the brain of senile plaques, consisting predominantly of the Aβ peptide of 40-42 amino acid, which is thought to play a key role in AD [4]. Indeed, using longitudinal in vivo multiphoton microscopy to sequentially image young APPswe/PS1d9xYFP (B6C3-YFP) transgenic mice, it was observed that Aβ plaques formed very quickly, within 24 h. The appearance of plaques is followed by the activation and recruitment of microglia to this specific sites. Progressive neuritic changes ensue, leading to increasingly dysmorphic neurites which appears within days to weeks [49]. Thus, these findings critically establish that Aβ plaque formation precedes disease progression [49]. In addition to aging, epidemiological observations identified hypercolesterolemia as a major risk factor for sporadic AD [4, 50, 51]. In this regard, the role of statins in AD, however, has been controversial, although the epidemiological evidence linking cholesterol to AD has been supported by in vivo and in vitro data. Indeed, since the amiloidogenic processing of the amyloid precursor protein by the presenilin1/γ-secretase is thought to occur in specific microdomains of the plasma membrane, cholesterol enrichment favors the generation of Aβ peptide and hence neurite degeneration [52]. Consistent with the mitochondrial targeting of Aβ as a major mechanism involved in Aβ-induced neurotoxicity, the specific role of mitochondrial cholesterol in this context has been recently evaluated. Using genetic mouse models of cholesterol accumulation, it was observed mitochondrial cholesterol enrichment in transgenic SREBP-2 and NPC1 knockout mice which determined enhanced susceptibility to Aβ-mediated neuroinflammation and neurotoxicity due to mGSH depletion [53]. Moreover, APP/PS1 transgenic mice exhibited enhanced mitochondrial cholesterol loading and subsequent mGSH depletion. Isolated mitochondria from these mice exhibited increased susceptibility to Aβ-mediated cytochrome c and Smac/Diablo release, indicative of MMP, and caspase activation compared to wild type mice. Of relevance, the onset of AD symptons in vivo were prevented by mGSH recovery with GSH ethyl ester, thus pointing that strategies aimed to boost this particular pool of GSH may be of potential relevance in AD.
4.2 Mitochondrial cholesterol and cancer therapy
Cancer cells exhibit critical metabolic transformations regarding the cholesterol homeostasis best illustrated by the ability of solid tumors to synthesize cholesterol under hypoxia, which contrasts with the physiological requirement for oxygen in the transformation of lanosterol into cholesterol via 9 redox reactions [3, 39]. Moreover, recent observations indicated that hypoxia stimulates the degradation of HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis [54]. In addition to this continued cholesterol synthesis in growing tumor cells, we have recently observed the stimulation of cholesterol trafficking to mitochondria from hepatocellular carcinoma (HCC) cells due to the overexpression of StAR [55]. Previous studies have shown that cholesterol enrichment of rat liver mitochondria impairs the ability of atractyloside to induce MPT [56], which suggest that mitochondrial cholesterol loading may actually account for the recognized mitochondrial dysfunction occurring in cancer cells (Figure 2). In addition to this putative effect of cholesterol in the regulation of mitochondrial function of cancer cells, we focused on the role of mitochondrial cholesterol in chemotherapy sensitivity and cell death regulation. Indeed, HCC mitochondria exhibit increased membrane order and resistance to Bax-mediated MMP, which are reversed either by cholesterol extraction with methylcyclodextrin or membrane fluidification by A2C [55]. Observations in HeLa cells treated with U18666A, which results in mitochondrial cholesterol loading, indicated a delayed release of Smac/Diablo and cytochrome c, as well as in BAX oligomerization and partial protection against stress-induced apoptosis [57]. Consistent with our findings in HCC, data from liposomes entrapping fluorescent dextrans indicated that the presence of cholesterol in the bilayer impaired Bax-mediated permeabilization due to the combination of reduced membrane dynamics and decreased Bax integration into the bilayer [55]. Moreover, the silencing of StAR downregulated mitochondrial cholesterol levels and sensitized HCC cells to chemotherapy, similar to the findings observed with statins or squalene synthase inhibition, which resulted in downregulation of cholesterol loading [55]. Thus these findings clearly identify the mitochondrial cholesterol pool as a novel mechanism whereby cancer cells, particularly HCC, resist chemotherapy and evade Bax-mediated MMP and subsequent apoptosis.
Figure 2. Paradoxical role of mitochondrial cholesterol in cell death.
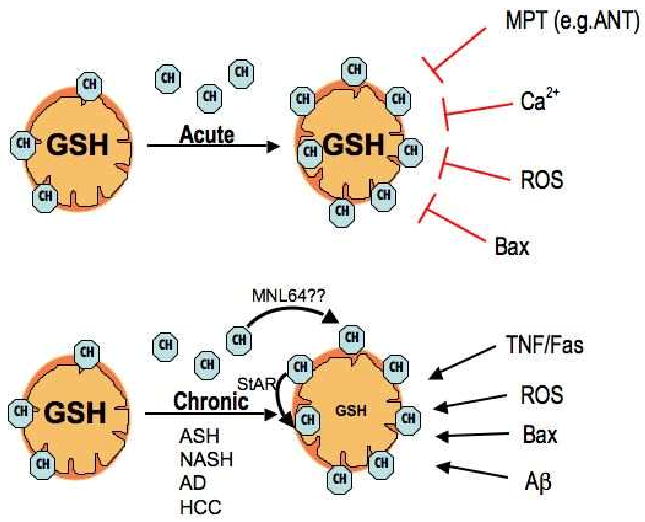
Since cholesterol regulates membrane dynamics and physical properties, the in situ enrichment in cholesterol impairs mitochondrial membrane permeabilization via MPT induced in response to atractyloside, which acts via adenine nucleotide translocator (ANT), Ca2+ or ROS, as well as the permeabilization of the outer membrane by active Bax. However, in pathological conditions associated with mitochondrial cholesterol trafficking and accumulation in both the inner and outer membranes via StAR expression potentiates mitochondrial membrane permeabilization in response to TNF/Fas, ROS, Bax or Aβ due to the mitochondrial GSH depletion. A role for the StAR family member MLN64 in the delivery of cholesterol to mitochondria remains to be fully established.
5. Role of Cardiolipin/Peroxidized Cardiolipin in Cell Death
Cardiolipin is a mitochondria-specific phospholipid, which plays an essential role in the organization of mitochondrial electron transport complexes involved in the generation of energy [27]. Indeed, cardiolipin dynamics regulate mitochondrial structural reorganization in prokaryotes and eukaryotes at different levels, including oxidative phosphorylation complexes, cristae remodelling and energy transducing systems, as reviewed recently [58]. Accumulating evidence suggests that this unique lipid also has an active role in several of the mitochondria-dependent steps of apoptosis, namely, in the regulation of MOM, mobilization of cytochrome, and more recently, in the anchoring of caspase-8 to mitochondria during death receptor-induced apoptosis [59]. Mature cardiolipin is synthesized by the sequential participation of phospholipase A, which removes one saturated acyl chain to generate monolysocardiolipin, and by tafazzin, a mitochondrial enzyme that catalyzes the addition of an unsaturated chain to monolysocardiolipin [60]. It provides a docking site for the interaction and activation of proapoptotic Bcl-2 proteins (tBid) in intact mitochondria [61], while studies in liposomes and outer mitochondrial membrane vesicles demonstrated the requirement for intact cardiolipin in the bilayer for efficient poration by active Bax [26]. Thus, these data indicate that cardiolipin cooperates with Bax to form large-scale openings independently of the formation of large (>100kDa) Bax aggregates. Besides the role of cardiolipin in Bax-mediated MOM permeabilization, cardiolipin has an important function in the attachment of cytochrome c to MIM and in the modulation of its release from IMS. An additional role involves the generation of a peroxidase complex brought about between cytochrome c-cardiolipin interaction which contributes to the oxidation of cardiolipin into peroxidized cardiolipin [32]. Although the generation of peroxidized cardiolipin from intact cardiolipin contributes to the detachment of cytochrome c from the inner membrane, the exact mechanism whereby peroxidized cardiolipin facilitated MOM permeabilization and cytochrome c release was unclear. In this regard, we examined this critical issue using primary hepatocytes which have been selectively depelted of mGSH. First, we observed that mGSH controls the formation of cardiolipin peroxidation during TNF-mediated hepatocellular cell death by regulating ROS generation [30] (Figure 1). Hepatocytes selectively depleted of mGSH pharmacologically potentiated cardiolipin peroxidation induced by TNF by a mechanism dependent on acidic sphingomyelinase (ASMase), as ASMase defiency impaired TNF-induced peroxidized cardiolipin despite mGSH depletion. Intriguingly, this outocome was observed despite the maintenance of glutathione peroxidase and thioredoxin-III functional activities, indicating the critical role of mGSH in the scavenging of mitochondrial peroxides generation. In addition, using liposomes mimicking the composition of mitochondrial outer membrane entrapping fluorescent dextrans, we observed that the incorporation of peroxidized cardiolipin in the bilayer enhanced the pore forming activity of active Bax by restructuring the lipid bilayer through a mechanism promoting the lamellar-to-inverted hexagonal lipid phase transition [30]. Thus, peroxidized cardiolipin seems to play a dual role in mitochondrial apoptosis, facilitating the mobilization of cytochrome c from the inner membrane available for release while promoting the ability of Bax to permeabilize MOM.
Another novel function of cardiolipin in cell death has been the recently described effect in regulating the activation of apycal caspase-8 in type II cells [59]. Following an external stimuli, caspase-8 translocated to mitochondria to bind to sites enriched in cardiolipin resulting in its proteolytic activation. Interestingly, recent evidence indicated that these activating platforms enriched in cardiolipin corresponds to sites also enriched in ganglioside GD3 and cholesterol, suggesting the presence of raft-like domains, which have been described in cells undergoing apoptosis [62, 63]. Therefore, cardiolipin is an essential constituent of specific microdomains, localized at contact sites between the inner and outer mitochondrial membranes, from which it orchestrates apoptosis by integrating signals from a variety of death-inducing proteins to bring in close proximity caspase-8 and Bid to generate tBid where it is needed. In line with this emerging view, the oxidation of cardiolipin would imply the generation of its peroxidized form in sites close to MOM, facilitating the permeabilizing activity of oligomerized Bax. A critical aspect derived from the above considerations implies the redistribution of cardiolipin from the inner to the outer membrane to comply with the various steps of mitochondrial apoptosis it regulates (e.g. MOM permeabilization, cytochrome c mobilization). The underlying mechanism involved in the intramitochondrial trafficking of cardiolipin during apoptosis is not yet fully understood, although there is evidence indicating a multifactorial nature in this process. Mitochondrial creatine kinase has been shown to regulate the density of contact sites within mitochondrial membranes where cardiolipin has been shown to reside [64]. In addition, Bid and mostly tBid exhibit lipid transfer activity contributing to the transbilayer movement of cardiolipin to the outer membrane [65, 66]. Moreover, phospholipid scramblases (PLS) induce non-specific bi-directional movement of phospholipids across the membrane. In particularly PLS3, a mitochondrial located PLS, has been shown to translocate cardiolipin from the MIM to the MOM during apoptosis induced by UV-irradiation [67, 68]. Nevertheless, regardless of the exact mechanism(s) involved, the translocation of cardiolipin between mitochondrial membranes has profound consequences in the mitochondrial apoptotic pathway, regulating the release of cytochrome c, mitochondrial bioenergetics and membrane perturbing effects such as the formation of cardiolipin-enriched microdomains, cristae remodelling and the accumulation of lyso-phospholipids. Further studies will be needed to establish if cholesterol, sphingolipids (e.g. GD3) and (peroxidized) cardiolipin are part of specific signaling domains which regulate the critcal steps of mitochondrial pathways of cell death.
Thus, consistent with the fundamental role of cardiolipin in mitochondrial structural organization, it is conceivable that the oxidation of cardiolipin contributes to the loss of mitochondrial function and the regulation of the different steps of mitochondrial apoptosis, such as MOMP, cytochrome c mobilization and release. However, this scenario may be specific for Bax mediated but not for oxidant induced MOMP and cell death. In the case of Bax activation brought about by TNF, cardiolipin peroxidation stimulated the release of cytochrome c and Smac/Diablo in mouse hepatocytes following TNF exposure [30]. Intriguingly, recent findings in oxidant-mediated release of mitochondrial apoptogenic proteins indicated a correlation between the mitochondrial release of cytochrome c and the content of Gpx4 in mitochondria from mice overexpressing or lacking Gpx4 [69]. Although, the level of ROS generation was not reported and the content of peroxidized cardiolipin was indirectly assessed by 10-N-nonyl acridine orange, the release of Smac/Diablo and Omi/HtrA2 was unaffected by the extent of Gpx4 content, indicating that the effect of peroxidized cardiolipin is specific for cytochrome c. Moreover, these findings imply that MOMP is independent of Gpx4 and presumably from ROS and peroxidized cardiolipin [69]. Hence while peroxidized cardiolipin may modulate cytochrome c mobilization, its effect on MOMP via destabilization of the bilayer may be restricted to Bax-mediated MOMP.
6. Conclusions
As an integral component, cholesterol plays an important role in the structure, physical properties and function of biological membranes. In particular, the mitohondrial cholesterol pool, which represents a minor proportion of the total cholesterol load, is emerging as an important factor in the regulation of mitochondrial membrane permeabilization and cell death. The enrichment of cholesterol in mitochondrial membranes determines the progression of liver and neurodegenerative diseases, and the resistance of cancer cells to current cancer therapy. Much of this effect of cholesterol is due to the changes in physical properties of mitochondrial membranes, especially in the appearance of lipid-ordered phases and subsequent loss of membrane fluidity that impairs mitochondrial function and specific membrane carriers. In particular, cholesterol loading in mitochondrial membranes selectively affects the transport of GSH into the matrix resulting in its depletion, which has a major impact on the stimulation of oxidant species derived from mitochondrial electron transport chain. As a consequence, cardiolipin becomes peroxidized, facilitating the redistribution of free cytochrome c to the intermembrane space and the permeabilization of mitochondrial outer membrane by Bax. The novelty of the findings described thus far centers on the dual role of cholesterol in cell death regulation. While in certain pathological conditions such as steatohepatitis or neurodegeneration the accumulation of cholesterol in mitochondrial membranes promotes apoptotic cell death, cholesterol loading in mitochondria of cancer cells contributes to resistance to cancer therapy. Much of this paradoxical behavior of cholesterol resides on the regulation of mitochondrial GSH stores, which it is known to play a fundamental role in oxidative stress, cardiolipin maintenance and cell death susceptibility. Accounting for its proapoptotic function, mitochondrial cholesterol enrichment shown in liver diseases or neurodegeneration leads to mitochondrial GSH depletion. In contrast, cancer cells manage to maintain unrestricted transport of GSH in mitochondria despite increased trafficking of cholesterol resulting in decreased membrane fluidity. The mechanisms, however, responsible for the unexpected maintenance of mitochondrial GSH in cancer cells in the face of increased cholesterol loading is currently under investigation. Thus, targeting mitochondrial cholesterol either by restraining its trafficking to or within mitochondrial membranes via StAR family members or by decreasing its availability (e.g. ezetimide or statins) may be of relevance for a number of pathologies such as steatohepatitis, neurodegenerative diseases or cancer. Alternatively, modulating the content of mitochondrial GSH either increasing its levels by permeable GSH prodrugs or selectively depleting pharmacologically may be of potential benefit in these pathologies.
Acknowledgments
The work was supported by grants: SAF2006-06780, SAF2008-02199, SAF2008-04974 and SAF2009-11417, BFU2005-06095 (Plan Nacional de I+D), PI070193 and PI060395 (Instituto de Salud Carlos III), P50-AA-11999 (Research Center for Liver and Pancreatic Diseases, US National Institute on Alcohol Abuse and Alcoholism) and by CIBEREHD from the Intituto Carlos III.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nature Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 2.Kroemer G, Galluzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev. 2007;87:99–163. doi: 10.1152/physrev.00013.2006. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Ruiz C, Mari M, Colell A, Morales A, Caballero F, Montero J, Terrones O, Basañez G, Fernández-Checa JC. Mitochondrial cholesterol in health and disease. Histol Histopathol. 2009;24:117–132. doi: 10.14670/HH-24.117. [DOI] [PubMed] [Google Scholar]
- 4.Colell A, Fernández A, Fernández-Checa JC. Mitochondria, cholesterol and amyloid beta peptide: a dangerous trio in Alzheimer disease. J Bioenerg Biomemb. 2009;41:417–23. doi: 10.1007/s10863-009-9242-6. [DOI] [PubMed] [Google Scholar]
- 5.Pop C, Salvesen GS. Human caspases: activation, specificity, and regulation. J Biol Chem. 2009;284:21777–81. doi: 10.1074/jbc.R800084200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Denault JB, Salvesen GS. Apoptotic caspase activation and activity. Methods Mol Biol. 2008;414:191–220. doi: 10.1007/978-1-59745-339-4_15. [DOI] [PubMed] [Google Scholar]
- 7.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Meter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schutze S, Tchikov V, Schneider-Brachert W. Regulation of TNFR1 and CD95 signaling by receptor compartimentalization. Nat Rev Mol Cell Biol. 2008;9:655–662. doi: 10.1038/nrm2430. [DOI] [PubMed] [Google Scholar]
- 9.Du C, Fang M, Li Y, Wang X. Smac, a mitochondrial protein that promotes cytochrome c-dependent caspase activation by eliminating IAP inhibition. Cell. 2000;102:33–42. doi: 10.1016/s0092-8674(00)00008-8. [DOI] [PubMed] [Google Scholar]
- 10.Verhagen AM, Ekert PG, Pakusch M, Silke J, Connolly LM, Reid GE, Moritz RL, Simpson RJ, Vaux DL. Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to and antagonizing IAP proteins. Cell. 2000;102:43–53. doi: 10.1016/s0092-8674(00)00009-x. [DOI] [PubMed] [Google Scholar]
- 11.Hedge R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, DuBois G, Lazebnik Y, Zervos AS, Fernandes-Alnemri T, Alnemri E. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts IAP-caspase interaction. J Biol Chem. 2002;277:432–438. doi: 10.1074/jbc.M109721200. [DOI] [PubMed] [Google Scholar]
- 12.Van Loo G, van Gurp M, Depuydt B, Srinivasula SM, Rodriguez I, Alnemri ES, Gevaert K, Vandekerckhove J, Declercq W, Vandenabeele P. The serine protease Omi/HtrA2 is released from mitochondria during apoptosis. Omi interacts with caspase-inhibitor XIAP and induces enhanced caspase activity. Cell Death Differ. 2002;9:20–26. doi: 10.1038/sj.cdd.4400970. [DOI] [PubMed] [Google Scholar]
- 13.Susi SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G. Molecular characterization of mitochondrial apoptosis-inducing factor. Nature. 1999;397:441–446. doi: 10.1038/17135. [DOI] [PubMed] [Google Scholar]
- 14.Li LY, Luo X, Wang X. Endonuclease G is an apoptotic Dnase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- 15.Mari M, Colell A, Morales A, Von Montfort C, Garcia-Ruiz C, Fernandez-Checa JC. Redox control of liver function in health and disease. Antioxid Redox Signal. 2009 Oct 5; doi: 10.1089/ars.2009.2634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kawahara A, Ohsawa Y, Matsumura H, Uchiyama Y, Nagata S. Caspase-independent cell killing by Fas-associated protein with death domain. J Cell Biol. 1998;143:1353–1360. doi: 10.1083/jcb.143.5.1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holler N, Zaru R, Micheau O, Thome M, Attinger A, Valitutti S, Bodmer JL, Schneider P, Seed B, Tschopp J. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 18.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, Cuny GD, Mitchison TJ, Moskowitz MA, Yuan J. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 19.Lemasters JJ. V. Necrapoptosis and the mitochondrial permeability transition: Shared pathways to necrosis and apoptosis. Am J Physiol. 1999;276:G1–G6. doi: 10.1152/ajpgi.1999.276.1.G1. [DOI] [PubMed] [Google Scholar]
- 20.Halestrap AP, Connern CP, Griffiths EJ, Kerr PM. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol Cell Biochem. 1997;174:167–172. [PubMed] [Google Scholar]
- 21.Cheng EH, Sheiko TV, Fisher JK. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 22.Baines CP, Kaiser RA, Sheiko T. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol. 2007;9:550–555. doi: 10.1038/ncb1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakagawa T, Shimizu S, Watanabe T. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature. 2005;434:652–658. doi: 10.1038/nature03317. [DOI] [PubMed] [Google Scholar]
- 24.Basañez G, Sharpe JC, Gallanis J, Brandt TB, Hardwick JM, Zimmerberg J. Bax-type apoptotic proteins porate pure lipid bilayers through a mechanism sensitive to intrinsic monolayer curvature. J Biol Chem. 2002;277:49360–49365. doi: 10.1074/jbc.M206069200. [DOI] [PubMed] [Google Scholar]
- 25.Basañez G, Hardwick JM. Unravelling the Bcl-2 apoptosis code with a simple model system. PLoS Biology. 2008;6:e154. doi: 10.1371/journal.pbio.0060154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kuwana T, Mackey MR, Perkins G, Ellisman MH, Latterich M, Schneider R, Green DR, Newmeyer DD. Bid, Bax and lipids cooperate to form supramolecular openings in the outer mitochondrial membrane. Cell. 2002;111:331–342. doi: 10.1016/s0092-8674(02)01036-x. [DOI] [PubMed] [Google Scholar]
- 27.Schug ZT, Gottlieb E. Cardiolipin acts as a mitochondrial signalling platform to launch apoptosis. Biochim Biophys Acta. 2009;1788:2022–2031. doi: 10.1016/j.bbamem.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 28.Kalanxhi E, Wallace CJ. Cytochrome c impaled: investigation of the extended lipid anchorage of a soluble protein to mitochondrial membrane models. Biochem J. 2007;407:179–187. doi: 10.1042/BJ20070459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kriska T, Korytowski W, Girotti AW. Role of mitochondrial cardiolipin peroxidation in apoptotic photokilling of 5-aminolevulinate-treated tumor cells. Arch Biochem Biophys. 2005;433:435–446. doi: 10.1016/j.abb.2004.09.025. [DOI] [PubMed] [Google Scholar]
- 30.Marí M, Colell A, Morales A, Caballero F, Moles A, Fernández A, Terrones O, Basañez G, Antonsson B, García-Ruiz C, Fernández-Checa JC. Mechanism of mitochondrial glutathione-dependent hepatocellular susceptibility to TNF despite NF-kappaB activation. Gastroenterology. 2008;134:1507–20. doi: 10.1053/j.gastro.2008.01.073. [DOI] [PubMed] [Google Scholar]
- 31.Marí M, Morales A, Colell A, García-Ruiz C, Fernández-Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxid Redox Signal. 2009;11:2685–700. doi: 10.1089/ars.2009.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kagan VE, Tyurin VA, Jiang J, Tyurina YY, Ritov VB, Amoscato AA, Osipov AN, Belikova NA, Kapralov AA, Kini V, Vlasova IL, Zhao Q, Zou M, Di P, Svistunenko DA, Kurnikov IV, Borisenko GG. Cytochrome c acts as a cardiolipin oxygenase required for release of 42. proapoptotic factors. Nat Chem Biol. 2005;1:223–232. doi: 10.1038/nchembio727. [DOI] [PubMed] [Google Scholar]
- 33.Uren RT, Dewson G, Bonson C, Lithgow T, Newmeyer DD, Kluck RM. Mitochondrial release of pro-apoptotic proteins: electrostatic interactions can hold cytochrome c but not Smac/DIABLO to mitochondrial membranes. J Biol Chem. 2005;280:2266–2274. doi: 10.1074/jbc.M411106200. [DOI] [PubMed] [Google Scholar]
- 34.Muñoz-Pinedo C, Guio-Carrion A, Goldstein JC, Fitzgerald P, Newmeyer DD, Green DR. Different mitochondrial intermembrane space proteins are released during apoptosis in a manner that is coordinately initiated but can vary in duration. Proc Natl Acad Sci USA. 2006;103:11573–11578. doi: 10.1073/pnas.0603007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scorrano L, Ashiya M, Buttle K, Weiler S, Oakes SA, Manella CA, Korsmeyer SJ. A distinct pathway remodels mitochondrial cristae and mobilizes cytochrome c during apoptosis. Dev Cell. 2002;2:55–67. doi: 10.1016/s1534-5807(01)00116-2. [DOI] [PubMed] [Google Scholar]
- 36.Frezza C, Cipolat S, Martins de Brito O, Micaroni M, Beznoussenko GV, Rudka T, Bartoli D, Polishuck RS, Danial NN, De Strooper B, Scorrano L. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006;126:177–189. doi: 10.1016/j.cell.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 37.Sun MG, Williams J, Muñoz-Pinedo C, Perkins GA, Brown JM, Ellisman MH, Green DR, Frey TG. Correlated three-dimensional light and electron microscopy reveals transformation of mitochondria during apoptosis. Nat Cell Biol. 2007;9:1057–1072. doi: 10.1038/ncb1630. [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi R, Lartigue L, Perkins G, Scott RT, Dixit A, Kushnarena Y, Kuwana T, Ellisman ME, Newmeyer DD. OPA1-mediated cristae opening is Bax/Bak and BH3 dependent, required for apoptosis, and independent of Bak oligomerization. Mol Cell. 2008;31:557–569. doi: 10.1016/j.molcel.2008.07.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol. 2008;9:125–138. doi: 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- 40.Maxfield FR, Tabas I. Role of cholesterol and lipid organization in disease. Nature. 2005;438:612–621. doi: 10.1038/nature04399. [DOI] [PubMed] [Google Scholar]
- 41.Pandak WM, Ren S, Marquez D, Hall E, Redford K, Mallonee D, Bohdan P, Heuman D, Gil G, Hylemon P. Transport of cholesterol into mitochondria is rate-limiting for bile acid synthesis via the alternative pathway in primary rat hepatocytes. J Biol Chem. 2002;277:48158–48164. doi: 10.1074/jbc.M205244200. [DOI] [PubMed] [Google Scholar]
- 42.Fernández A, Colell A, Caballero F, Matías N, García-Ruiz C, Fernández-Checa JC. Mitochondrial S-adenosyl-l-methionine transport is insensitive to alcohol-mediated changes in membrane dynamics. Alcohol Clin Exp Res. 2009;33:1169–80. doi: 10.1111/j.1530-0277.2009.00940.x. [DOI] [PubMed] [Google Scholar]
- 43.Fernández-Checa JC, Kaplowitz N. Hepatic mitochondrial glutathione: transport and role in disease and toxicity. Toxicol Appl Pharmacol. 2005;204:263–273. doi: 10.1016/j.taap.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 44.Coll O, Colell A, García-Ruiz C, Kaplowitz N, Fernández-Checa JC. Sensitivity of the 2-oxoglutarate carrier to alcohol intake contributes to mitochondrial glutathione depletion. Hepatology. 2003;38:692–702. doi: 10.1053/jhep.2003.50351. [DOI] [PubMed] [Google Scholar]
- 45.Marí M, Caballero F, Colell A, Morales A, Caballeria J, Fernandez A, Enrich C, Fernandez-Checa JC, García-Ruiz C. Mitochondrial free cholesterol loading sensitizes to TNF- and Fas-mediated steatohepatitis. Cell Metab. 2006;4:185–98. doi: 10.1016/j.cmet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 46.Monetti M, Levin M, Watt MJ, Sajan MP, Marmor S, Hubbard BK, Stevens RD, Bain JR, Newgard CB, Farese RV, Hevener AL, Farese RV. Dissociation of hepatic steatosis and insulin resistance in mice overexpressing DGAT in the liver. Cell Metab. 2007;6:69–78. doi: 10.1016/j.cmet.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 47.Puri P, Baillie RA, Wiest MM, Mirshahi F, Choudhury J, Cheung O, Sargeant C, Contos MJ, Sanyal AJ. A lipidomic analysis of nonalcoholic fatty liver disease. Hepatology. 2007;46:1081–90. doi: 10.1002/hep.21763. [DOI] [PubMed] [Google Scholar]
- 48.Caballero F, Fernández A, De Lacy AM, Fernández-Checa JC, Caballería J, García-Ruiz C. Enhanced free cholesterol, SREBP-2 and StAR expression in human NASH. J Hepatol. 2009;50:789–96. doi: 10.1016/j.jhep.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 49.Meyer-Luehmann M, Spires-Jones TL, Prada C, Garcia-Alloza M, de Calignon A, Rozkalne A, Koenigsknecht-Talboo J, Holtzman DM, Bacskai BJ, Hyman BT. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer's disease. Nature. 2008;4541:720–724. doi: 10.1038/nature06616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Notkola IL, Sulkava R, Pekkanen J, Erkinjuntti T, Ehnholm C, Kivinen P. Serum total cholesterol, apolipoprotein E 4 allele, and Alzheimer's disease. Neuroepidemiology. 1998;17:14–20. doi: 10.1159/000026149. [DOI] [PubMed] [Google Scholar]
- 51.Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol. 2000;57:1439–1443. doi: 10.1001/archneur.57.10.1439. [DOI] [PubMed] [Google Scholar]
- 52.Uemura K, Lill CM, Li X, Peters JA, Ivanov A, Fan Z, DeStrooper B, Bacskai BJ, Hyman BT, Berezovska O. Allosteric modulation of PS1/gamma-secretase conformation correlates with amyloid beta(42/40) ratio. PLoS ONE. 2009;8:7893. doi: 10.1371/journal.pone.0007893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fernández A, Llacuna L, Fernández-Checa JC, Colell A. Mitochondrial cholesterol loading exacerbates amyloid beta peptide-induced inflammation and neurotoxicity. J Neurosci. 2009;29:6394–405. doi: 10.1523/JNEUROSCI.4909-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nguyen AD, McDonald JG, Bruick RK, DeBose-Boyd RA. Hypoxia stimulates degradation of 3-hydroxy-3-methylglutaryl Coenzyme A reductase through accumulation of lanosterol and hypoxia-inducible factor (HIF)-mediated induction of Insigs. J Biol Chem. 2007;282:27436–27446. doi: 10.1074/jbc.M704976200. [DOI] [PubMed] [Google Scholar]
- 55.Montero J, Morales A, Llacuna L, Lluis JM, Terrones O, Basañez G, Antonsson B, Prieto J, García-Ruiz C, Colell A, Fernández-Checa JC. Mitochondrial cholesterol contributes to chemotherapy resistance in hepatocellular carcinoma. Cancer Res. 2008;68:5246–56. doi: 10.1158/0008-5472.CAN-07-6161. [DOI] [PubMed] [Google Scholar]
- 56.Colell A, García-Ruiz C, Lluis JM, Coll O, Mari M, Fernández-Checa JC. Cholesterol impairs the adenine nucleotide translocator-mediated mitochondrial permeability transition through altered membrane fluidity. J Biol Chem. 2003;278:33928–35. doi: 10.1074/jbc.M210943200. [DOI] [PubMed] [Google Scholar]
- 57.Lucken-Ardjomande S, Montessuit S, Martinou JC. Bax activation and stress-induced apoptosis delayed by the accumulation of cholesterol in mitochondrial membranes. Cell Death Differ. 2008;15:484–493. doi: 10.1038/sj.cdd.4402280. [DOI] [PubMed] [Google Scholar]
- 58.Mileykovskaya E, Dowhan W. Cardiolipin membrane domains in prokaryotes and eukaryotes. Biochim Biophys Acta-Biomembranes. 2009;1788:2084–2091. doi: 10.1016/j.bbamem.2009.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gonzalvez F, Schug ZT, Houtkooper RH, MacKenzie ED, Brooks DG, Wanders RJ, Petit PX, Vaz FM, Gottlieb E. Cardiolipin provides an essential activating platform for caspase-8 on mitochondria. J Cell Biol. 2008;183:681–696. doi: 10.1083/jcb.200803129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Xu Y, Malhotra A, Ren M, Schlame M. The enzymatic function of tafazzin. J Biol Chem. 2006;281:39217–39224. doi: 10.1074/jbc.M606100200. [DOI] [PubMed] [Google Scholar]
- 61.Lutter M, Fang M, Luo X, Nishijima M, Xie X, Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. 2000;22:754–761. doi: 10.1038/35036395. [DOI] [PubMed] [Google Scholar]
- 62.Garofalo T, Giammarioli AM, Misasi R, Tinari A, Manganelli V, Gambardella L, Pavan A, Malorni W, Sorice M. Lipid microdomains contribute to apoptosis-associated modifications of mitochondria in T cells. Cell Death Differ. 2005;12:1378–1389. doi: 10.1038/sj.cdd.4401672. [DOI] [PubMed] [Google Scholar]
- 63.Sorice M, Manganelli V, Matarrese P, Tinari A, Misasi R, Malorni W, Garofalo T. Cardiolipin-enriched raft-like microdomains are essential activating platforms for apoptotic signals on mitochondria. FEBS Lett. 2009;583:2447–2450. doi: 10.1016/j.febslet.2009.07.018. [DOI] [PubMed] [Google Scholar]
- 64.Speer O, Back N, Buerklen T, Brdiczka D, Koretsky A, Wallimann T, Eriksson O. Octameric mitochondrial creatine kinase induces and stabilizes contact sites between the inner and outer membrane. Biochem J. 2005;385:445–450. doi: 10.1042/BJ20040386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sorice M, Circella A, Cristea IM, Garofalo T, Renzo LD, Alessandri C, Valesini G, Esposti MD. Cardiolipin and its metabolites move from mitochondria to other cellular membranes during death receptor-mediated apoptosis. Cell Death Differ. 2004;11:1133–1145. doi: 10.1038/sj.cdd.4401457. [DOI] [PubMed] [Google Scholar]
- 66.Esposti MD, Erler JT, Hickman JA, Dive C. Bid, a widely expressed proapoptotic protein of the Bcl-2 family, displays lipid transfer activity. Mol Cell Biol. 2001;21:7268–7276. doi: 10.1128/MCB.21.21.7268-7276.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu J, Chen J, Dai Q, Lee RM. Phospholipid scramblase 3 is the mitochondrial target of protein kinase C {delta}-induced apoptosis. Cancer Res. 2003;63:1153–1156. [PubMed] [Google Scholar]
- 68.Liu J, Dai Q, Chen J, Durrant D, Freeman A, Liu T, Grossman D, Lee RM. Phospholipid scramblase 3 controls mitochondrial structure, function, and apoptotic response. Mol Cancer Res. 2003;1:892–902. [PubMed] [Google Scholar]
- 69.Liang H, Ran Q, Jang YC, Holstein D, Lechleiter J, McDonald-Marsh T, Musatov A, Song W, Van Remmen H, Richardson A. Glutathione peroxidase 4 differentially regulates the release of apoptogenic proteins from mitochondria. Free Radic Biol Med. 2009;47:312–320. doi: 10.1016/j.freeradbiomed.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]