

Abstract
Macrophage inflammatory protein-3α/C-C chemokine ligand 20 (MIP-3α/CCL20) is an antimicrobial peptide that plays an important role in innate immunity. In addition to direct microbicidal effects, MIP-3α/CCL20 also exhibits cytokine-like functions that are critical during dendritic cell activation. The aim of the present study was to investigate further which signaling pathways are involved in the MIP-3α/CCL20 mRNA expression in response to whole-cell Porphyromonas gingivalis. Primary gingival epithelial cells (GECs) and the immortalized oral keratinocyte cell-line OKF6/TERT-2 were stimulated with whole-cell P. gingivalis. Prior to stimulation, GECs and OKF6/TERT-2 cells were pretreated with specific inhibitors for nuclear-factor-κB (NF-κB), mitogen-activated protein kinase (MAPK), phospholipase C (PLC), and phosphatidylinositol-3-kinase (PI3K). In GECs and OKF6/TERT-2 cells, activation of NF-κB was examined after exposure to P. gingivalis. The gene expression of MIP-3α/CCL20 was significantly induced in response to P. gingivalis (P ≤ 0.05) compared to unstimulated control cells. This induction was specifically blocked when cells were pre-incubated with inhibitors for NF-κB, MAPK, and PLC (P ≤ 0.05), but not for PI3K. These results demonstrate that P. gingivalis induces the MIP-3α/CCL20 mRNA in a NF-κB-, PLC-, and MAPK-dependent manner.
Keywords: C-C chemokine ligand 20, gingival epithelial cells, mitogen activated protein kinase, nuclear factor kappa B, phospholipase C
Introduction
Within the oral cavity, multiple bacterial species interact with epithelia, yet the oral mucosa remains uninflamed in the presence of the commensal microbial flora. Epithelial tissues in general function as the first line of defense protecting the host from the outside microbial environment. To maintain healthy homeostasis, the oral epithelium requires sufficient local immune mechanisms. In addition to a rigid physical barrier, the oral epithelium provides a chemical barrier that is represented by antimicrobial peptides.1,2 Antimicrobial peptides are defined as proteins smaller than 100 amino acids with molecular weights ranging between 3.5–6.5 kDa.1,3 In humans, antimicrobial peptides are represented by a number of different molecules, e.g. human α-defensins, β-defensins, some C-C and C-X-C-chemokines,4 and the cathelicidin LL-37.2,3,5,6
Macrophage inflammatory protein 3α/C-C-chemokine ligand 20 (MIP-3α/CCL20) is a chemokine with regions that are structurally related to hBD-2 and, like hBD-2, it exhibits antimicrobial activity in vitro against Escherichia coli and Staphylococcus aureus.7 Both MIP-3α/CCL20 and hBD-2 are comparable mediators linking innate and adaptive immune responses by interacting with the chemokine receptor 6 (CCR6) and, thereby, activating immature dendritic cells.5,8 In addition, both MIP-3α/CCL20 and hBD-2 are chemo-attractants for immature dendritic cells.8,9
Porphyromonas gingivalis is a Gram-negative, obligate anaerobic bacterium that has been recognized as a major etiological factor in the development and progression of periodontitis.10-12 Proteases (gingipains) synthesized by P. gingivalis are involved in the degradation of the adherens junctions between cells leading to P. gingivalis invading into the epithelium and deeper tissues.13-17 Previously, it has been reported that supernatant from P. gingivalis induced the gene expression of hBD-2 and MIP-3α/CCL20 in gingival epithelial cells (GECs) via protease-activated receptor-2 (PAR-2), and proteases secreted by P. gingivalis were responsible for this up-regulation in GECs.18
The transcription factor NF-κB plays an important key role during cellular responses to inflammatory stimuli and general responses to pathogens in a number of different cell types. In addition to the involvement of PAR-2, it has been shown that induction of the hBD-2 gene expression is mediated by signaling pathways involving NF-κB when gingival epithelial cells were stimulated with P. gingivalis.19 It has been shown that activation of G-protein-coupled receptors, such as PAR-2, may lead to phosphorylation and translocation of NF-κB in endothelial and epithelial cells.20-22 Direct stimulation of PAR-2 using peptide agonists led to NF-κB -mediated up-regulation of the pro-inflammatory cytokines interleukin-6 (IL-6) and interleukin-8 (IL-8).20
It was hypothesized that GECs utilize the transcription factor NF-κB to mediate the gene expression of MIP-3α/CCL20 in response to stimulation with P. gingivalis. Therefore, specific inhibitors for NF-κB were used to block this particular transcriptional pathway in gingival epithelial cells exposed to wild-type P. gingivalis. It was further of interest to elucidate the involvement of phospholipase C (PLC), phosphatidylinositol-3-kinase (PI3K), c-Jun N-terminal kinase-I (JNK I), mitogen-activated protein kinase (MAPK), and nuclear factor for activated T-cells (NFAT) during mediation of the MIP-3α/CCL20 gene expression in GECs.
Materials and Methods
Reagents
Specific inhibitors for NF-κB (inhibitor name: MG132 and JSH 23, respectively), NFAT (inhibitor name: VIVIT peptide), JNK I PI3K (inhibitor name: LY294002), PLC (inhibitor name: U73122 and its corresponding control U73343), and p38/MAPK (inhibitor name: ML 3163) were purchased from Calbiochem (San Diego, CA, USA). Interleukin-1β was obtained from Sigma (St Louis, MO, USA).
Human gingival epithelial cell cultures
The immortalized human oral keratinocyte cell line OKF6/TERT-2 was kindly provided by Dr J. Rheinwald (Harvard University). Cells were cultured in accordance with described protocols.23 Briefly, cell culture was performed using keratinocyte serum-free medium (kersfm; GIBCO/Invitrogen, Carlsbad, CA, USA) consisting of 25 μg/ml bovine pituitary extract, 0.2 ng/ml epidermal growth factor, penicillin/streptomycin (1%) and 0.4 mM CaCl2. For passaging the media, Dulbecco’s modified Eagle medium/F-12 medium including 5%/ml hydrocortisone, 10 ng/ml of epidermal growth factor, 0.4 mM CaCl2, and 10% heat inactivated fetal bovine serum (heat inactivation for 30 min at 55° C) was applied (GIBCO/Invitrogen). For both experimental and control groups, the cells were grown in keratinocyte serum-free medium (ker-sfm).
For obtaining primary gingival epithelial cells (GECs), gingival biopsies were collected from healthy patients who underwent third-molar extraction in the Department of Oral Surgery, School of Dentistry, University of Washington in accordance with a University of Washington Institutional Review Board approved study (IRB, UW Human Subjects Number: 98-7483). For culture of primary GECs, the tissue was prepared as described previously by our group.19 Epithelial cells were cultured in keratinocyte growth medium (KGM) with 0.15 mM Ca2+ using the supplements from the KGM-bullet kit (Cambrex, Walkersville, MD, USA). OKF6/TERT-2 and primary GECs were cultured at 37° C in a humidified atmosphere (5% CO2). For stimulation experiments, 200,000 cells were plated using a 6-well cell culture dish.
GECs transfection with siRNA
Previously, it has been reported that gene expression of MIP-3α/CCL20 was increased in GECs when exposed to cell-free supernatant of P. gingivalis, and that this increase was mediated via protease-activated receptor-2 (PAR-2), but not PAR-1.18 To characterize the MIP-3α/CCL20 mRNA expression further, GECs were stimulated with whole-cell P. gingivalis to confirm that the mRNA expression of MIP-3α/CCL20 is mediated via PAR-2. For gene silencing, HP-guaranteed-siRNA® tagged with Alexa Fluor 488 (Qiagen, Valencia, CA, USA) was used to target the human PAR-2 gene in primary GECs. siRNA-sequences were published previously.18 The fast forward transfection protocol was performed according to the manufacturer’s instructions. Scrambled non-silencing RNA served as a negative control and was transfected using the same concentration as for PAR-2 siRNA. GECs treated with transfection agent alone served as an additional control for all experiments. Transfection efficiency was monitored using a fluorescence microscope (Eclipse TS100; Nikon, Melville, NY, USA) and confirmed by real-time PCR. siRNA (25 nM) specific for PAR-2 was introduced to GECs, and stimulation experiments were performed 48 h after transfection.18
For inhibition experiments, both OKF6/TERT-2 and GECs were pretreated with specific inhibitors for signaling pathways 1 h prior to stimulation with P. gingivalis. Concentrations of inhibitors were as follows: MG132, 10 μM;19 JSH 23, 5 μM, 10 μM, and 30 μM; VIVIT peptide, 1 μM, 2 μM, and 4 μM; JNK I, 1μM, 4 μM;19 LY294002, 20 μM, 40 μM, and 80 μM; U73122 and U73343, 400 nM each; ML 3163, 1 μM, 3 μM, and 9 μM.
P. gingivalis culture condition and treatment
Wild-type P. gingivalis strain 33277 was cultured to the late logarithmic growth phase as described previously.19 Bacterial numbers were estimated by absorbance measurement using TECAN, GENios Multidetection Reader (v.4.51; Phoenix, Hayward, CA, USA). Subsequently, aliquots of the bacteria were used for pre-incubation (10 min) with 1 mmol/l of the serine and cysteine protease inhibitor tosyl-L-lysine chloromethyl ketone (TLCK; Sigma), which inhibits the gingipains.24,25 The protease inhibitor was diluted in endotoxin-free water (HyPure™; HyClone, Logan, UT, USA). The GECs were grown to 80% confluence and stimulated with either P. gingivalis or TLCK-pre-incubated P. gingivalis using an amount equivalent to a ‘multiplicity of infection of 50:1’ (MOI50:1) for 16 h. Blank medium served as a negative control for the stimulation experiments. Each experiment was performed in triplicate, and the immortalized cell line OKF6/TERT-2 as well as primary gingival epithelial cells from one to three different donors were tested.
Assay for NF-aB activity
After stimulation of OKF6/TERT-2 and primary GECs with whole-cell P. gingivalis, cells were harvested using RIPA-buffer (Sigma-Aldrich, St Louis, MO, USA) at 4°C with gentle agitation to obtain whole cell extracts. Protein concentration was determined by BCA (bichinoninic acid)/BSA (bovine serum albumin) protein assay (Pierce, Rockford, IL, USA). For concentration determination, 10 μl of the sample as well as dilution series of BSA (stock solution 2 mg/ml) were prepared and analyzed by absorbance measurement (560 nm, TECAN) using 80 μl of BCA reagent per well (96-well plate; developed over 30 min at 37°C). NF-κB activity was measured using the TransAM™ NF-κB p65 assay (Active Motif, Carlsbad, CA, USA) specific for the p65 (RelA) subunit of NF-κB, according to the manufacturer’s instructions. The specificity of binding was examined using an oligonucleotide containing a wild-type or mutated NF-κB consensus binding sequence (provided with the kit). Analysis for NF-κB/p65 activation was performed by photometric measurements at OD 450 nm. Values were normalized to the assay control according to the manufacturer’s instruction.
Conditions for RT-PCR and real-time PCR
After stimulation of OKF6/TERT-2 and primary GECs, total RNA was extracted using the RNeasy Mini Kit (Qiagen). The reverse transcription reaction was performed using 500 ng of total RNA. The reaction mix contained 1 × reverse transcriptase (RT) buffer, 250 nM oligo-(dT)-primer, 10 mM dNTP mix, 50 U of RT, and 13 U of RNase inhibitor (Ambion, Austin, TX, USA) and was carried out following standard protocols as previously described.26 Controls without RT enzyme were included with every experiment.
Quantitative analysis of the cDNA was performed using the MyiQ®iCycler (Bio-Rad, Hercules, CA, USA) and iQ® SYBR Green Supermix® (Bio-Rad) according to the manufacturer’s instructions. PCR reactions were carried out in 96-well plates in a total volume of 25 μl, including 1 μl of cDNA and 250 nM primers. At the end of every real-time PCR, melting curve analysis was performed to confirm the amplified product was specific. Standard curve analysis was conducted confirming a linear dependency (efficiency) between the cDNA concentration and the threshold cycle (Ct) calculated by the iQ5® software (Bio-Rad). All reactions were carried out in duplicate, and average Ct-values were calculated. Sample values were normalized to the expression of the house-keeping gene ribosomal phosphoprotein (RPO) and relative expression was calculated using the mathematical model proposed by Pfaffl.26,27
Previously, IL-8 expression has been implicated as p38/MAPK dependent.28,29 In addition, it was shown that the PAR-2-mediated gene expression of IL-8 was up-regulated via the NF-κB pathway.20 In addition to MIP-3α/CCL20, the gene expression of IL-8 was tested in GECs. Primer sequences for RPO, MIP-3α/CCL20, and IL-8 have been published previously.18,26 PCR controls were performed using water instead of cDNA. The data were statistically analyzed using the t-test (SPSS, v.14, Munich, Germany). The significance level was set at P ≤ 0.05.
Results
Whole-cell P. gingivalis-induced gene expression of MIP-3a/CCL20 is via PAR-2
GECs were transfected with siRNA specific for PAR-2, and transfection efficiency of the Alexa Fluor 488-tagged siRNA was monitored by fluorescence microscopy (Fig. 1A) and confirmed by real-time PCR (data not shown).18 The gene expression of MIP-3α/CCL20 was significantly up-regulated in response to P. gingivalis (P=0.006). This effect was completely abrogated when whole-cell P. gingivalis was pretreated with the protease inhibitor TLCK. Controls using blank bacteria medium did not influence the mRNA expression of MIP-3α/CCL20 (Fig. 1B). The gene expression of MIP-3α/CCL20 was significantly decreased in primary GECs transfected with siRNA specific for PAR-2 compared to non-siRNA when exposed to P. gingivalis (P=0.041; Fig. 1C).
Fig. 1.

Transfection of GECs with siRNA specific for PAR-2 and its effects on MIP-3α/CCL20 gene expression. (A) From left to right: phase contrast image, fluorescence image of the same cells, and merged images. Intracellular and perinuclear localization of siRNA. Fluorescence signals represent the intracellular, perinuclear localization of the transfected siRNA targeting the PAR-2 gene. Nearly all cells (>90%) have been transfected. Untransfected cells and the transfection agent without siRNA served as negative controls. Original magnification ×40. (B) Real-time PCR analysis of whole-cell P. gingivalis-induced (MOI of 50:1) MIP-3α/CCL20 gene expression in GECs. The gene expression of MIP-3α/CCL20 was significantly up-regulated in response to P. gingivalis (16 h of stimulation; *P=0.006). (C) SiRNA transfection for PAR-2 led to a significant reduction of the P. gingivalis-induced gene expression of MIP-3α/CCL20 (*P=0.041). Controls using non-silencing scrambled siRNA showed induction of MIP-3α/CCL20 gene expression similar to the level of untransfected primary GECs. The gene expression study was performed with triplicate samples derived from two different donors. *Significant difference (P<0.05). Error bars indicate SD values.
Activation of NF-κB/p65 following p. gingivalis stimulation
Next, the activation of NF-κB/p65 was tested in immortalized and primary GECs when exposed to whole cell P. gingivalis. Stimulation of GECs with P. gingivalis led to a time-dependent (15, 30, 45, and 60 min of stimulation) activation of the NF-κB/p65 complex (P<0.001; Fig. 2). Negative controls using the blank bacteria medium as well as the TLCK-pretreated P. gingivalis did not affect NF-κB/p65 activation in gingival epithelial cells (P>0.05; Fig. 2). The stimulation of GECs with IL-1β served as a positive control for the NF-κB/p65 activation, and a significantly increased activation for NF-κB/p65 was observed after 30 min of exposure to IL-1β (P<0.001; Fig. 2).
Fig. 2.
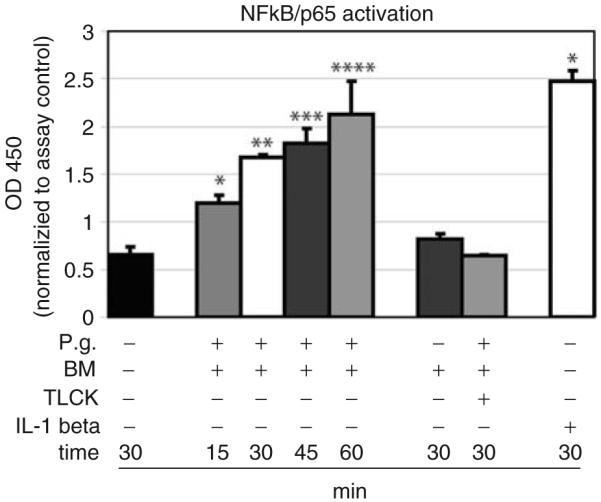
NF-κB/p65 was activated in GECs when exposed to whole-cell P. gingivalis. In response to P. gingivalis, a time-dependent increase in the NF-κB/p65 activation was demonstrated (15 min, 30 min, 45 min, and 60 min; * to **** P<0.001). Stimulation with IL-1β served as a positive control for NF-κB/p65 activation (P<0.001). Both P. gingivalis pretreated with the protease inhibitor TLCK and blank bacteria medium did not activate NF-κB/p65 in gingival epithelial cells. Triplicate experiments were performed on OKF6/TERT-2 and primary GECs from one donor. *Significant difference (P<0.05). Error bars indicate SD values.
P. gingivalis-induced MIP-3α/CCL20 gene expression is PLC and p38/MAPK dependent
To investigate intracellular signaling in response to P. gingivalis, the effects of specific inhibitors for PLC, PI3K, JNK I, and p38/MAPK on MIP-3α/CCL20 gene expression were analyzed in both immortalized and primary GECs. Pre-incubation of OKF6/TERT-2 cells and GECs with U73122, an inhibitor of PLC, led to a significantly decreased mRNA expression of MIP-3α/CCL20 compared to untreated cells in response to P. gingivalis (P=0.016; Fig. 3A). In contrast, preincubation using the corresponding control inhibitor U73343 (control for U73122) did not affect the gene expression of MIP-3α/CCL20 when compared with untreated cells stimulated with P. gingivalis (Fig. 3A).
Fig. 3.

Analysis of the effect of inhibiting PLC, PI3K, JNK I in P. gingivalis-induced MIP-3α/CCL20 gene expression using real-time PCR. (A) In primary GECs pretreated with the specific inhibitor for PLC (U73122), gene expression of MIP-3α/CCL20 was partially, but significantly, blocked after stimulation with P. gingivalis (*P=0.016). (B) Pre-incubation with the inhibitor for PI3K did not affect P. gingivalis-induced MIP-3α/CCL20 gene expression. (C) Pre-incubation with the inhibitor for JNK I did not influence P. gingivalis-induced MIP-3α/CCL20 gene expression. Triplicate experiments were performed on OKF6/TERT-2 and primary GECs from two different donors. *Significant difference (P<0.05). Error bars indicate SD values.
Pre-incubation of OKF6/TERT-2 cells and GECs with inhibitors for both PI3K and JNK I did not alter the gene expression of MIP-3α/CCL20 when exposed to whole-cell P. gingivalis (Fig. 3B,C). Different dilutions for inhibitors of PI3K (20 μM, 40 μM, 80 μM) and JNK I (1 μM, 4 μM) were used, and the results were similar (data not shown).
Cells treated with the specific inhibitor for p38/MAPK exhibited significant reduced MIP-3α/CCL20 gene expression in a dose-dependent manner in OKF6/TERT-2 cells (1 μM, P=0.029; 3 μM, P=0.007; 9 μM, P=0.004; Fig. 4A). In primary GECs, the p38/MAPK inhibitor resulted in nearly complete inhibition of MIP-3α/CCL20 gene expression upon P. gingivalis exposure (9 μM, P=0.023; Fig. 4B).
Fig. 4.

Analysis of the effect of inhibiting p38/MAPK in P. gingivalis-induced MIP-3α/CCL20 gene expression using real-time PCR. MIP-3α/CCL20 gene expression was analyzed using real-time PCR. (A) GECs (OKF6/TERT-2) pre-incubated with the pharmacological inhibitor for p38/MAPK showed a dose-dependent decrease in MIP-3α/CCL20 gene expression when exposed to P. gingivalis (1 μM, 3 μM, and 9 μM; *P=0.029, **P=0.007, and ***P=0.009, respectively). (B) The same conditions as in (A) was performed on primary GECs. The p38/MAPK inhibitor blocked the P. gingivalis-induced MIP-3α/CCL20 gene expression (*P=0.023). Controls, using the inhibitor without subsequent bacterial stimulation showed no effect on the MIP-3α/CCL20 gene expression. Triplicate experiments were performed on OKF6/TERT-2 and primary GECs from three different donors. *Significant difference (P<0.05). Error bars indicate SD values.
Controls using the inhibitors alone at highest concentration without subsequent stimulation exhibited no effect on the gene expression of MIP-3α/CCL20 in both immortalized and primary GECs (Figs 3A-C and 4A,B).
P. gingivalis-induced MIP-3α/CCL20 gene expression is via NF-κB/p65
Further, the influence of the transcription factors NFAT and/or NF-κB/p65 on signaling for MIP-3α/CCL20 gene expression was tested in gingival epithelial cells when exposed to whole-cell P. gingivalis. No influence of the NFAT inhibitor VIVIT peptide on the gene expression of MIP-3α/CCL20 was observed after treatment with P. gingivalis with both primary gingival epithelial cells and OKF6/TERT-2 cells, although various concentrations of inhibitor were used (1 μM, 2 μM, and 4 μM; Fig. 5A). In contrast, the P. gingivalis-induced gene expression of MIP-3α/CCL20 was blocked (P=0.02) when cells were pretreated with an inhibitor for NF-κB (MG132, 10 μM; Fig. 5B). In order to verify the finding that MIP-3α/CCL20 gene expression was dependent on pathways mediated via NF-κB, a second pharmacological inhibitor for NF-κB/p65 (JSH 23) was tested at various concentrations (5 μM, 10 μM, and 30 μM). A dose-dependent decrease (P<0.001) of P. gingivalis-induced MIP-3α/CCL20 gene expression was observed with nearly complete inhibition at the highest concentration (Fig. 5C). In control experiments, the inhibitors at maximum concentrations without bacterial challenge did not affect MIP-3α/CCL20 gene expression in OKF6/TERT-2 or primary GECs (Fig. 5A–C).
Fig. 5.

Analysis of the effect of inhibiting NFAT and NF-κB in P. gingivalis-induced MIP-3α/CCL20 gene expression using real-time PCR. (A) Pre-incubation with the inhibitor for NFAT did not influence P. gingivalis-induced MIP-3α/CCL20 gene expression. (B) In GECs pretreated with the specific inhibitor for NF-κB (MG132), gene expression of MIP-3α/CCL20 was blocked after stimulation with P. gingivalis (*P=0.02). (C) The results showed a dose-dependent decrease of the P. gingivalis-induced MIP-3α/CCL20 gene expression in gingival epithelial cells (5 μM, 10 μM, and 30 μM; *P=0.01, **P<0.001, and ***P=0.006, respectively). Triplicate experiments were performed on OKF6/TERT-2 and primary GECs from two to three different donors. *Significant difference (P<0.05). Error bars indicate SD values.
P. gingivalis-induced IL-8 gene expression was p38/MAPK- and NF-κB/p65-dependent
The P. gingivalis-induced gene expression was significantly reduced when cells were pretreated with a specific inhibitor for p38/MAPK (P<0.001; Fig. 6A). Pretreatment of GECs with various concentrations of the NF-κB inhibitor JSH 23 (5 μM, 10 μM, and 30 μM) led to a dose-dependent reduction of P. gingivalis-induced IL-8 gene expression (P=0.013, P=0.019, and P=0.012, respectively; Fig. 6B). Control experiments using the inhibitors alone did not affect gene expression of IL-8 in GECs (Fig. 6A,B).
Fig. 6.

Analysis of the effect of inhibiting p38/MAPK and NF-κB/p65 in P. gingivalis-induced IL-8 gene expression using real-time PCR. (A) The p38/MAPK inhibitor blocked the P. gingivalis-induced IL-8 gene expression (*P<0.001). (B) Gingival epithelial cells pre-incubated with the pharmacological inhibitor for NF-κB/p65 (JSH 23) showed a dose-dependent decrease in the IL-8 gene expression when exposed to P. gingivalis (5 μM, 10 μM, and 30 μM; *P=0.013, P=**P=0.019, and ***P=0.012, respectively). Triplicate experiments were and performed on OKF6/TERT-2 and primary GECs from three different donors. *Significant difference (P<0.05). Error bars indicate SD values.
Discussion
Gingival epithelial cells provide a number of different mechanisms, such as the synthesis of antimicrobial peptides, that help resist bacterial infection of the gingival tissue.2 Previously, the response of GECs to the cell-free supernatant from P. gingivalis regarding gene expression of the two related antimicrobial peptides hBD-2 and MIP-3α/CCL20 has been investigated. Also, it has been shown that the gene expression of both hBD-2 and MIP-3α/CCL20 was inducible by P. gingivalis proteases, and this induction was mediated via the proteinase-activated receptor PAR-2 in GECs.18 In previously published studies, it has been demonstrated that activation of PAR-2 leads to NF-κB activation in keratinocytes.30-34 In addition, it has been implicated that stimulation with P. gingivalis activates NF-κB-mediated pathways in keratinocytes, macrophages, and monocytes.35,36 The results of this study show a time-dependent increase of the NF-κB/p65 activity in response to P. gingivalis in immortalized and primary GECs, and suggest similar responses between human dermal keratinocytes and GECs.36
Although the involvement of PAR-2 in P. gingivalis-induced MIP-3α/CCL20 mRNA expression was shown, little is known regarding the involvement of PLC in signaling transduction pathways following P. gingivalis infection. For vascular endothelial cells, it was shown that LPS from P. gingivalis not only activates PARs, but also induces IL-8 expression via PLC.37 To our knowledge, there is no data available showing involvement of PLC in MIP-3α/CCL20 gene expression in GECs. The results of this present study demonstrated that the inhibitor for PLC (U73122) partially blocked the P. gingivalis-induced MIP-3α/CCL20 mRNA expression suggesting that not only PLC but also other components of the intracellular signaling cascade are involved.
Furthermore, it has been shown that primary epithelial cells infected with P. gingivalis utilize PI3K-mediated pathways to promote cell survival.38 In this context, it was conceivable that PI3K may be involved in MIP-3α/CCL20 gene expression assuming that the antimicrobial and/or chemokine-like activities of MIP-3α/CCL20 provide cell protective functions following bacterial infection. In this study, however, the specific inhibitor for PI3K (LY294002) did not decrease or block the P. gingivalis-induced MIP-3α/CCL20 gene expression.
In epithelial cells, JNK I is involved in mediating the induction of hBD-2 gene expression.19 Since both hBD-2 and MIP-3α/CCL20 are structurally related peptides with similar functions,6,8,39,40 it was of interest to test if JNK I was part of the P. gingivalis-initiated cascade leading to MIP-3α/CCL20 mRNA expression in GECs. The results of the present study showed that the inhibitor for JNK I failed to decrease or block the MIP-3α/CCL20 gene expression in GECs when exposed to P. gingivalis. As JNK I has been shown to be involved in hBD-2 transduction,19 the results suggest that the JNK I may be a regulative step that divides signaling for both related peptides hBD-2 and MIP-3α/CCL20.
In studying the role of p38/MAPK in MIP-3α/CCL20 mRNA induction, a dose-dependent decrease of the MIP-3α/CCL20 mRNA in OKF6/TERT-2 cells was observed, whereas in primary GECs, the P. gingivalis-induced MIP-3α/CCL20 gene expression was almost completely blocked. These results accord with a recently published study on airway epithelial cells, where the expression of MIP-3α/CCL20 was also p38/MAPK dependent.41 Further differences between OKF6/TERT-2 and primary GECs will be discussed below.
In order to test the initial hypothesis, two different inhibitors for NF-κB (MG132 and JSH 23) were used to elucidate the role of NF-κB in the signaling pathway mediating MIP-3α/CCL20 gene expression. In addition, an inhibitor for another nuclear factor, NFAT, (VIVIT peptide) was used as a control. It is known that gene expression of the related antimicrobial peptide hBD-2 is mediated via the NFB pathway in epithelial cells when exposed to P. gingivalis.19,42 In both OKF6/TERT-2 and primary GECs, pretreatment with MG132 and JSH23 blocked gene expression of MIP-3α/CCL20, whereas the inhibitor for NFAT did not affect the MIP-3α/CCL20 mRNA. This finding confirmed our hypothesis that gene expression of MIP-3α/CCL20 is mediated via NF-κB in P. gingivalis-stimulated GECs.
Although the data were consistent among OKF6/TERT-2 and GECs, the magnitude of the MIP-3α/CCL20 mRNA induction differed not only between the immortalized cell line and primary GECs, but also between cells derived from different donors of primary GECs. Nevertheless, the MIP-3α/CCL20 gene expression pattern following P. gingivalis stimulation was reproducible throughout the triplicate experiments. These differences suggest distinct cellular functions regarding immune responses that vary from immortalized to primary GECs. While OKF6/TERT-2 cell tend to response generally with a higher magnitude (up to 100-fold) of MIP-3α/CCL20 gene expression, primary GECs responded more moderately (up to 25-fold). It is possible that primary GECs have certain control mechanisms that prevent the cell from over-reacting to pathogenic bacteria, whereas immortalized GECs may lack comparable regulatory mechanisms. Although OKF6/TERT-2 cells represent a reasonable model for studies on innate immune responses following bacterial stimulation, additional experiments on primary GECs are necessary to confirm and deliberate data derived from OKF6/TERT-2 cells.
Conclusions
This study showed up-regulation of MIP-3α/CCL20 mRNA expression in GECs when exposed to the periodontal pathogen P. gingivalis. Gene expression of MIP-3α/CCL20 was dependent on pathways including PAR-2, PLC, p38/MAPK, and NF-κB in immortalized as well as in primary GECs. The gene expression of the antimicrobial peptides hBD-2 and CCL20 is inducible by P. gingivalis2,18,19 mediated by similar intracellular pathways including the activation of NF-κB.42 Hence, gingival epithelial cells provide a sophisticated innate defense mechanism by producing hBD-2 and MIP-3α/CCL20 in response to P. gingivalis, which in turn may be interpreted as a parallel immunological pathway at the cellular level. Future studies may lead to a better understanding of the cross-talk between pathogenic bacteria and host cells. Individual variations in the antimicrobial host response may give an explanation for inter-individual differences in the susceptibility to periodontal disease.
Acknowledgements
The authors thank P. Braham (Department of Periodontology, University of Washington) for helping with bacterial cell culture. This study was funded by NIDCR grant R01DE16961.
References
- 1.Dale BA. Periodontal epithelium: a newly recognized role in health and disease. Periodontol 2000. 2002;30:70–78. doi: 10.1034/j.1600-0757.2002.03007.x. [DOI] [PubMed] [Google Scholar]
- 2.Chung WO, Dommisch H, Yin L, Dale BA. Expression of defensins in gingiva and their role in periodontal health and disease. Curr Pharm Des. 2007;13:3073–3083. doi: 10.2174/138161207782110435. [DOI] [PubMed] [Google Scholar]
- 3.Lehrer RI. Primate defensins. Nat Rev Microbiol. 2004;2:727–738. doi: 10.1038/nrmicro976. [DOI] [PubMed] [Google Scholar]
- 4.Yang D, Chen Q, Hoover DM, et al. Many chemokines including CCL20/MIP-3alpha display antimicrobial activity. J Leukoc Biol. 2003;74:448–455. doi: 10.1189/jlb.0103024. [DOI] [PubMed] [Google Scholar]
- 5.Yang D, Chertov O, Bykovskaia SN, et al. Beta-defensins: linking innate and adaptive immunity through dendritic and T cell CCR6. Science. 1999;286:525–528. doi: 10.1126/science.286.5439.525. [DOI] [PubMed] [Google Scholar]
- 6.Yang D, Chen Q, Chertov O, Oppenheim JJ. Human neutrophil defensins selectively chemoattract naive T and immature dendritic cells. J Leukoc Biol. 2000;68:9–14. [PubMed] [Google Scholar]
- 7.Hoover DM, Boulegue C, Yang D, et al. The structure of human macrophage inflammatory protein-3alpha /CCL20. Linking antimicrobial and CC chemokine receptor-6-binding activities with human beta-defensins. J Biol Chem. 2002;277:37647–37654. doi: 10.1074/jbc.M203907200. [DOI] [PubMed] [Google Scholar]
- 8.Schutyser E, Struyf S, Van Damme J. The CC chemokine CCL20 and its receptor CCR6. Cytokine Growth Factor Rev. 2003;14:409–426. doi: 10.1016/s1359-6101(03)00049-2. [DOI] [PubMed] [Google Scholar]
- 9.Yang D, Biragyn A, Kwak LW, Oppenheim JJ. Mammalian defensins in immunity: more than just microbicidal. Trends Immunol. 2002;23:291–296. doi: 10.1016/s1471-4906(02)02246-9. [DOI] [PubMed] [Google Scholar]
- 10.Socransky SS, Haffajee AD, Cugini MA, Smith C, Kent RL., Jr Microbial complexes in subgingival plaque. J Clin Periodontol. 1998;25:134–144. doi: 10.1111/j.1600-051x.1998.tb02419.x. [DOI] [PubMed] [Google Scholar]
- 11.Lamont RJ, Jenkinson HF. Life below the gum line: pathogenic mechanisms of Porphyromonas gingivalis. Microbiol Mol Biol Rev. 1998;62:1244–1263. doi: 10.1128/mmbr.62.4.1244-1263.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holt SC, Kesavalu L, Walker S, Genco CA. Virulence factors of Porphyromonas gingivalis. Periodontol 2000. 1999;20:168–238. doi: 10.1111/j.1600-0757.1999.tb00162.x. [DOI] [PubMed] [Google Scholar]
- 13.Sandros J, Papapanou PN, Nannmark U, Dahlen G. Porphyromonas gingivalis invades human pocket epithelium in vitro. J Periodontal Res. 1994;29:62–69. doi: 10.1111/j.1600-0765.1994.tb01092.x. [DOI] [PubMed] [Google Scholar]
- 14.Rudney JD, Chen R, Sedgewick GJ. Intracellular Actinobacillus actinomycetemcomitans and Porphyromonas gingivalis in buccal epithelial cells collected from human subjects. Infect Immun. 2001;69:2700–2707. doi: 10.1128/IAI.69.4.2700-2707.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lamont RJ, Chan A, Belton CM, Izutsu KT, Vasel D, Weinberg A. Porphyromonas gingivalis invasion of gingival epithelial cells. Infect Immun. 1995;63:3878–3885. doi: 10.1128/iai.63.10.3878-3885.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hintermann E, Haake SK, Christen U, Sharabi A, Quaranta V. Discrete proteolysis of focal contact and adherens junction components in Porphyromonas gingivalis-infected oral keratinocytes: a strategy for cell adhesion and migration disabling. Infect Immun. 2002;70:5846–5856. doi: 10.1128/IAI.70.10.5846-5856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Belton CM, Izutsu KT, Goodwin PC, Park Y, Lamont RJ. Fluorescence image analysis of the association between Porphyromonas gingivalis and gingival epithelial cells. Cell Microbiol. 1999;1:215–223. doi: 10.1046/j.1462-5822.1999.00022.x. [DOI] [PubMed] [Google Scholar]
- 18.Dommisch H, Chung WO, Rohani MG, et al. Protease-activated receptor 2 mediates human beta-defensin 2 and CC chemokine ligand 20 mRNA expression in response to proteases secreted by Porphyromonas gingivalis. Infect Immun. 2007;75:4326–4333. doi: 10.1128/IAI.00455-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung WO, Dale BA. Innate immune response of oral and foreskin keratinocytes: utilization of different signaling pathways by various bacterial species. Infect Immun. 2004;72:352–358. doi: 10.1128/IAI.72.1.352-358.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shpacovitch VM, Brzoska T, Buddenkotte J, et al. Agonists of proteinase-activated receptor 2 induce cytokine release and activation of nuclear transcription factor kappaB in human dermal microvascular endothelial cells. J Invest Dermatol. 2002;118:380–385. doi: 10.1046/j.0022-202x.2001.01658.x. [DOI] [PubMed] [Google Scholar]
- 21.Ye RD. Regulation of nuclear factor kappaB activation by G-protein-coupled receptors. J Leukoc Biol. 2001;70:839–848. [PubMed] [Google Scholar]
- 22.Uehara A, Sugawara Y, Sasano T, Takada H, Sugawara S. Proinflammatory cytokines induce proteinase 3 as membrane-bound and secretory forms in human oral epithelial cells and antibodies to proteinase 3 activate the cells through protease-activated receptor-2. J Immunol. 2004;173:4179–4189. doi: 10.4049/jimmunol.173.6.4179. [DOI] [PubMed] [Google Scholar]
- 23.Dickson MA, Hahn WC, Ino Y, et al. Human keratinocytes that express hTERT and also bypass a p16(INK4a)-enforced mechanism that limits life span become immortal yet retain normal growth and differentiation characteristics. Mol Cell Biol. 2000;20:1436–1447. doi: 10.1128/mcb.20.4.1436-1447.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fujimura S, Hirai K, Shibata Y, Nakayama K, Nakamura T. Comparative properties of envelope-associated arginine-gingipains and lysine-gingipain of Porphyromonas gingivalis. FEMS Microbiol Lett. 1998;163:173–179. doi: 10.1111/j.1574-6968.1998.tb13042.x. [DOI] [PubMed] [Google Scholar]
- 25.Holzhausen M, Spolidorio LC, Ellen RP, et al. Protease-activated receptor-2 activation: a major role in the pathogenesis of Porphyromonas gingivalis infection. Am J Pathol. 2006;168:1189–1199. doi: 10.2353/ajpath.2006.050658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chung WO, Hansen SR, Rao D, Dale BA. Protease-activated receptor signaling increases epithelial antimicrobial peptide expression. J Immunol. 2004;173:5165–5170. doi: 10.4049/jimmunol.173.8.5165. [DOI] [PubMed] [Google Scholar]
- 27.Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Desloges N, Schubert C, Wolff MH, Rahaus M. Varicella-zoster virus infection induces the secretion of interleukin-8. Med Microbiol Immunol. 2008;197:277–284. doi: 10.1007/s00430-007-0060-3. [DOI] [PubMed] [Google Scholar]
- 29.Wang Y, Yang J, Gao Y, Dong LJ, Liu S, Yao Z. Reciprocal regulation of 5-alpha-dihydrotestosterone, interleukin-6 and interleukin-8 during proliferation of epithelial ovarian carcinoma. Cancer Biol Ther. 2007;6:864–871. doi: 10.4161/cbt.6.6.4093. [DOI] [PubMed] [Google Scholar]
- 30.Macfarlane SR, Sloss CM, Cameron P, Kanke T, McKenzie RC, Plevin R. The role of intracellular Ca2+ in the regulation of proteinase-activated receptor-2 mediated nuclear factor kappa B signalling in keratinocytes. Br J Pharmacol. 2005;145:535–544. doi: 10.1038/sj.bjp.0706204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshida N, Katada K, Handa O, et al. Interleukin-8 production via protease-activated receptor 2 in human esophageal epithelial cells. Int J Mol Med. 2007;19:335–340. doi: 10.3892/ijmm.19.2.335. [DOI] [PubMed] [Google Scholar]
- 32.Goon Goh F, Sloss CM, Cunningham MR, Nilsson M, Cadalbert L, Plevin R. G-protein-dependent and -independent pathways regulate proteinase-activated receptor-2 mediated p65 NFkappaB serine 536 phosphorylation in human keratinocytes. Cell Signal. 2008;20:1267–1274. doi: 10.1016/j.cellsig.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 33.Vliagoftis H, Schwingshackl A, Milne CD, et al. Proteinase-activated receptor-2-mediated matrix metalloproteinase-9 release from airway epithelial cells. J Allergy Clin Immunol. 2000;106:537–545. doi: 10.1067/mai.2000.109058. [DOI] [PubMed] [Google Scholar]
- 34.Bretschneider E, Kaufmann R, Braun M, et al. Evidence for proteinase-activated receptor-2 (PAR-2)-mediated mitogenesis in coronary artery smooth muscle cells. Br J Pharmacol. 1999;126:1735–1740. doi: 10.1038/sj.bjp.0702509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foster N, Cheetham J, Taylor JJ, Preshaw PM. VIP inhibits Porphyromonas gingivalis LPS-induced immune responses in human monocytes. J Dent Res. 2005;84:999–1004. doi: 10.1177/154405910508401106. [DOI] [PubMed] [Google Scholar]
- 36.Brozovic S, Sahoo R, Barve S, et al. Porphyromonas gingivalis enhances FasL expression via up-regulation of NFkappaB-mediated gene transcription and induces apoptotic cell death in human gingival epithelial cells. Microbiology. 2006;152:797–806. doi: 10.1099/mic.0.28472-0. [DOI] [PubMed] [Google Scholar]
- 37.Inomata M, Into T, Ishihara Y, Nakashima M, Noguchi T, Matsushita K. Arginine-specific gingipain A from Porphyromonas gingivalis induces Weibel–Palade body exocytosis and enhanced activation of vascular endothelial cells through protease-activated receptors. Microbes Infect. 2007;9:1500–1506. doi: 10.1016/j.micinf.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 38.Yilmaz O, Jungas T, Verbeke P, Ojcius DM. Activation of the phosphatidylinositol 3-kinase/Akt pathway contributes to survival of primary epithelial cells infected with the periodontal pathogen Porphyromonas gingivalis. Infect Immun. 2004;72:3743–3751. doi: 10.1128/IAI.72.7.3743-3751.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Duits LA, Ravensbergen B, Rademaker M, Hiemstra PS, Nibbering PH. Expression of beta-defensin 1 and 2 mRNA by human monocytes, macrophages and dendritic cells. Immunology. 2002;106:517–525. doi: 10.1046/j.1365-2567.2002.01430.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biragyn A, Ruffini PA, Leifer CA, et al. Toll-like receptor 4-dependent activation of dendritic cells by beta-defensin 2. Science. 2002;298:1025–1029. doi: 10.1126/science.1075565. [DOI] [PubMed] [Google Scholar]
- 41.Marcet B, Horckmans M, Libert F, Hassid S, Boeynaems JM, Communi D. Extracellular nucleotides regulate CCL20 release from human primary airway epithelial cells, monocytes and monocyte-derived dendritic cells. J Cell Physiol. 2007;211:716–727. doi: 10.1002/jcp.20979. [DOI] [PubMed] [Google Scholar]
- 42.Chung WO, Dale BA. Differential utilization of nuclear factor-kappaB signaling pathways for gingival epithelial cell responses to oral commensal and pathogenic bacteria. Oral Microbiol Immunol. 2008;23:119–126. doi: 10.1111/j.1399-302X.2007.00398.x. [DOI] [PMC free article] [PubMed] [Google Scholar]