

Abstract
The preferential dopamine D3 receptor agonist pramipexole (PRA) disrupts prepulse inhibition (PPI) of acoustic startle, an operational measure of sensorimotor gating, in rats. Drug effects on PPI are sensitive to numerous experimental variables; proceeding with in-depth analyses of drug effects without a clear understanding of these variables is inefficient. The present studies characterized the impact on PRA-induced PPI deficits by a range of experimental parameters. As shown previously, PRA reduced both PPI and startle magnitude beginning 5-15 min post-injection; PRA effects on PPI were statistically significant through 35 min post-injection, while those on startle magnitude were still significant 65 min post-injection. PRA-induced PPI deficits were evident under conditions that matched startle magnitude in vehicle and PRA conditions and were independent of PRA-induced changes in prepulse-elicited motor activity. Additionally, PRA-induced PPI deficits did not differ significantly between uni- vs. cross-modal stimuli or between male vs. female rats, with no robust effect of estrous phase in females. These findings demonstrate that PRA effects on PPI are observed across several different experimental conditions and are dissociable from changes in startle magnitude or prepulse-elicited responses. Recommendations are made regarding “optimal” experimental conditions for studying the neurobiology of PRA-induced changes in PPI in rats.
Keywords: Prepulse inhibition, pramipexole, dopamine, D3 receptor, rats
Introduction
PPI is an operational measure of sensorimotor gating that is disrupted in several neuropsychiatric disorders, including schizophrenia and Tourette Syndrome (Castellanos et al., 1996; Swerdlow et al., 2001, 2008b). PPI has been widely used in animal models to predict clinical efficacy of novel antipsychotic compounds. Recently, we reported that pramipexole (PRA) disrupts prepulse inhibition (PPI) of acoustic startle in rats (Weber et al., 2008, 2009). PRA is a non-ergot full agonist of dopamine D2 and D3 receptor subtypes, with selectivity for D3 over D2 receptors reported to be between 7:1 and 160:1 (Millan et al., 2002; Piercey et al., 1996; Svensson et al., 1994). The dopamine D3 receptor (D3R) is of particular interest in the pathophysiology and treatment of several neuropsychiatric disorders, including schizophrenia, Tourette Syndrome, substance dependence, and depression. The selectivity of PRA for D3 over D2 receptors makes it an important pharmacological tool for studying the effects of D3R activation.
PPI and PPI response to drug treatments are sensitive to many different experimental parameters. Historically, studies of the neurobiology of these drug effects often have preceded studies that clarified the optimal experimental parameters; this sometimes led to interpretative difficulties (Conti et al., 2009; Davis, 1988; Davis et al., 1990; Kinney et al., 1999; Palmer et al., 2000; Swerdlow et al., 1998, 2008b). A more efficient strategy is to clarify the parametric sensitivity of drug effects in advance of embarking on complex neurobiological studies. We previously reported PRA dose-response effects on PPI and reported that Sprague-Dawley and Long-Evans rat strains display differential sensitivity to the nonselective dopamine agonist apomorphine, but not PRA (Weber et al., 2008). We have also shown that PRA effects on PPI are sensitive to prepulse intervals (time between prepulse and pulse onset; Swerdlow et al., 2009). The studies reported here employ parametric approaches to explore the relationship between PPI-disruptive effects of PRA and time course, startle reduction, prepulse elicited reactions, stimulus modalities, estrous phase, and sex.
Methods
Experimental Animals
Adult Sprague-Dawley male (n = 56, 225-250 g; Harlan Laboratories, Livermore, CA) and female (n = 16, 175-200 g) rats were housed in same sex groups of 2-3 animals per cage, and maintained on a reversed light/dark schedule with water and food available ad libitum. Rats were handled within 2 d of arrival. Testing occurred during the dark phase. Males and females were housed in separate rooms and tested on different days. All experiments were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 85-23, revised 1985) and were approved by the UCSD Animal Subjects Committee (protocol #S01221).
Drugs
Pramipexole (PRA) was purchased from Toronto Research Chemicals (North York, Ontario, Canada). Drug doses are based on milligram/kilogram salts. PRA (0, 0.3, 1.0 mg/kg) was injected subcutaneously in a volume of 1 ml/kg body weight 15 min prior to behavioral testing, except in the time course experiment, in which there was no pretreatment time. Pseudorandom balanced dose orders were used.
Apparatus
Startle chambers for rats (San Diego Instruments, San Diego, CA, USA) were housed in a sound-attenuated room, and consisted of a Plexiglas cylinder 8.2 cm in diameter resting on a 12.5 × 25.5 cm Plexiglas frame within a ventilated enclosure. Noise bursts were presented via a speaker mounted 24 cm above the cylinder. Visual stimuli consisted of flashes of incandescent white light delivered via a 15 W light bulb. The light bulb was mounted to the ceiling of the chamber in a corner of the startle chamber at a distance of approximately 22 cm from the center of the rat cylinder. Light flashes did not generate any audible sound. A piezoelectric accelerometer mounted below the Plexiglas frame detected and transduced motion from within the cylinder. Stimulus delivery was controlled by the SR-LAB microcomputer and interface assembly, which also digitized (0-4095), rectified, and recorded stabilimeter readings. One hundred 1-ms readings were collected beginning at stimulus onset. Startle amplitude was defined as the average of the 100 readings.
Startle testing procedure
Approximately 7-9 d after shipment arrival, rats were exposed to a short “matching” startle session. They were placed in the startle chambers for a 5 min acclimation period with a 70 dB(A) background noise, and then exposed to a total of 17 P-ALONE trials (40 ms – 120 dB(A) noise bursts) that were interspersed with 3 PREPULSE+PULSE trials in which P-ALONE was preceded 100 ms (onset-to-onset) by a 20 ms noise burst, 12 dB above background. Rats were assigned to drug dose groups based on average %PPI from the matching session to ensure similar baseline PPI levels between groups. Starting 2-5 d after the matching session, drug testing began. Inter-test interval was 4-7 d. Drug testing was done on multiple days with PRA as a within-subjects factor. Test sessions varied by experiment (described below), but included P-ALONE and PREPULSE+PULSE trial types presented in pseudorandom order. Interspersed between consecutive active trials were NOSTIM trials in which activity was recorded, but no stimulus was presented. Prepulses preceded pulses by 100 ms (onset-to-onset), and average ITI between active trials was 15 s.
Specific parameters in different test sessions are described below:
“Time Course Session”
The test session was divided into six 10-min blocks. Prepulses were 80 dB(A) (10 dB above background) and pulses were 120 dB(A). PREPULSE-ALONE active trials were also interspersed throughout the session and consisted of noise bursts 10 dB above background. Session duration was 65 min, including the initial 5 min acclimation period.
“Pulse Intensity Session”
Prepulses were 3, 5, or 10 dB above background and preceded either 105 dB(A) or 120 dB(A) pulses. Session duration was approximately 23 min.
“Cross-Modal Session”
10 dB acoustic or 40 ms light flashes preceded 120 dB(A). PREPULSE-ALONE active trials were also interspersed throughout the session and consisted of noise bursts 10 dB above background. Session duration was approximately 15 min.
“Sex Differences Session”
5, 10, or 15 dB prepulses preceded the 120 dB(A) pulse by 100 ms. Session duration was approximately 18.5 min.
Estrous phase determination
(Adapted from Marcondes et al., 2002) Vaginal secretion was collected with a cotton swab wetted with normal saline (NaCl 0.9%) and placed on glass slides. Unstained material was viewed under a light microscope without the use of a condenser lens, with 10× and 40× objective lenses. Based on cell type distribution, animals were classified as being in proestrus, estrus, metestrus, or diestrus. Vaginal lavage and estrous phase determination were performed for 8 d prior to matching session to reduce the stress of the procedure on the animals and ensure normal cycling and average cycle length of 4 d. Vaginal lavage was performed immediately after each startle testing session, and test sessions were spaced 4 d apart.
Data Analysis
PPI was defined as 100-[(startle amplitude on PREPULSE trials/startle amplitude on PULSE-ALONE trials) × 100], and was analyzed by mixed design ANOVAs. All data were inspected for the presence of “non-responders” defined by a mean startle response to PULSE-ALONE trials of < 10 units; none met this criteria. Other ANOVAs were used to assess responses on PULSE-ALONE, PREPULSE-ALONE, or NOSTIM trials. Where different prepulse intensities were present, data were collapsed across all prepulse intensities, unless otherwise noted. Post-hoc comparisons were conducted using Fisher's protected least significance difference (PLSD). Difference scores for PPI and startle magnitude were calculated by subtracting values from each rat on drug-treatment days from vehicle-treatment days. Correlations between difference scores for PPI scores and startle magnitude were conducted using Spearman Rank Correlation. Alpha was set at 0.05.
Results
Time course of effects
Rats treated with vehicle or PRA 1.0 mg/kg were immediately placed in startle chambers after injections for a 5-min acclimation, and responses were then measured over the course of a 60 min test session, divided into six 10-minute blocks. Repeated measures ANOVA of %PPI showed a significant main effect of PRA 1.0 mg/kg (F=80.61, df 1,7; p<0.0001), but no effect of time block (F=1.00, df 5,35; NS), and no PRA × block interaction (F=1.27, df 5, 35; NS) (Fig. 1A). Fisher's PLSD revealed that %PPI was significantly lower in PRA-treated rats during the first, second, and third 10-min blocks (p<0.025, 0.001, 0.0005, respectively). %PPI of PRA-treated rats was significantly lower during the third block compared to the first block (p<0.05) and the fourth block (p<0.05).
Figure 1. Time course of PRA effects on %PPI (A) and startle magnitude (B).

A: PRA 1.0 mg/kg significantly reduced PPI during the first 30 minutes of testing, with maximal effects during the third 10-minute block. (*, p<0.03) B: Startle-reducing effects of PRA persist across 60 min. This difference was significant during blocks 1,2,3, and 6. A decrease in startle magnitude during blocks 4 and 5 of the vehicle condition likely account for the lack of significance of PRA effects during these time periods.
ANOVA of startle response on P-ALONE trials revealed significant main effects of PRA (F=21.69, df 1,7; p<0.003), block (F=6.14, df 5,35; p<0.005), and a significant PRA × block interaction (F=5.24, df 5,35; p<0.002) (Fig. 1B). Fisher's PLSD showed that startle magnitude was significantly lower in PRA treated rats during blocks 1, 2, 3, and 6 (p<0.002, 0.0001, 0.0005, 0.02, respectively). Although the difference in startle magnitude between PRA- and vehicle-treated rats failed to reach significance during blocks 4 and 5, this was likely due to a decrease in startle magnitude in the vehicle group during these blocks. Indeed, post-hoc tests did not reveal a significant difference between any blocks among PRA-treated rats, but among vehicle-treated rats, startle magnitude for blocks 4 and 5 were significantly lower than that of block 1 (p<0.04) and block 2 (p<0.006).
PRA effects on PPI using weak and intense pulses
To match startle magnitude between vehicle and PRA treatment groups, animals were tested in a session that included trials with 120 dB(A) and 105 dB(A) pulse intensities, presented alone or preceded by a prepulse. ANOVA of %PPI revealed significant main effects of PRA dose (F=14.11, df 1,15; p<0.002) and pulse intensity (F=5.24, df 1,15; p<0.04), and a significant PRA × pulse intensity interaction (F=6.26, df 1,15; p<0.03) (Fig. 2A). This interaction reflected significant PPI-disruptive effects of PRA with 120 dB(A) pulses, but not with 105 dB(A) pulses.
Figure 2. Effects of PRA on %PPI in trials matched for startle amplitude.
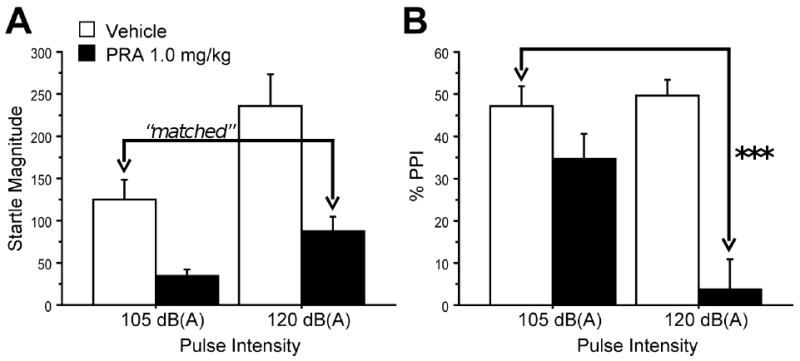
A: Startle magnitude was matched between vehicle-treated rats after “low intensity” 105 dB(A) pulses and PRA-treated rats after “high intensity” 120 dB(A) pulses. B: Under conditions in which startle magnitude was matched, PRA 1.0 mg/kg significantly reduced PPI (***, p<0.0001)
As expected, there was also a significant main effect of prepulse intensity (F=11.43, df 2,30; p<0.0005), but no significant PRA × prepulse intensity or pulse intensity × prepulse intensity interactions (F=2.91, df 2,30; NS and F=3.19, df 2,30; NS, respectively). Data were collapsed across prepulse intensity for post-hoc analyses of PPI. ANOVA of startle response to P-ALONE revealed significant main effects of PRA (F=27.84, df 1,15; p<0.0001), pulse intensity (F=37.45, df 1,15; p<0.0001), and a significant PRA × pulse intensity interaction (F=6.33, df 1,15; p<0.03) (Fig. 2B). Importantly, Fisher's PLSD revealed that startle magnitude was not significantly different between vehicle-treated rats after “low intensity” pulses and PRA-treated rats after “high-intensity” pulses (Fig. 2A). Post-hoc comparisons of %PPI in these two conditions with matched startle magnitude showed that PPI was significantly reduced in PRA- vs. vehicle-treated rats (p<0.0001) (Fig. 2B).
PRA effects on motor responses to prepulses
PREPULSE-ALONE trials were interspersed throughout the time course and cross-modal test sessions described above in order to measure prepulse-elicited responses (PPER). ANOVA of response to PREPULSE-ALONE during the time course session did not reveal any significant main effects of PRA (F=1.720, df 1,7; NS) or time block (F<1, df 5,35; NS), and no PRA × block interaction (F<1, df 5,35; NS) (mean (SEM) PPER, vehicle vs. PRA: 3.88 (2.02) vs. 0.58 (0.25)). There was a high degree of variability in the vehicle group during blocks 1-3 (Average response (SEM) for Block 1=8.583 (8.019), Block 2=6.000 (5.117), Block 3=7.875 (7.875)), due to a single outlier value in each of these blocks from one of two different animals. Responses for these same two animals during NOSTIM, PULSE-ALONE, and PREPULSE+PULSE trials were not outside the range of the rest of the group. ANOVA of response to acoustic PREPULSE-ALONE during the cross-modal test also failed to reveal a significant main effect of PRA (F<1, df 2,30; NS) (mean (SEM) PPER, vehicle vs. PRA 0.3 mg/kg vs. PRA 1.0 mg/kg: 1.70 (0.78) vs. 1.19 (0.56) vs. 1.89 (0.45)).
Cross-modal PPI
ANOVA of %PPI revealed significant main effects of PRA dose (0, 0.3, 1.0 mg/kg) (F=8.59, df 2,30; p<0.002) and prepulse modality (F=13.46, df 1,15; p<0.003), but no significant PRA × prepulse modality interaction (F=2.84, df 2,30; NS) (Fig. 3). Fisher's PLSD showed a significant effect of both doses of PRA on acoustic PPI (PRA 0.3 mg/kg p<0.004, PRA 1.0 mg/kg p<0.0001), and a significant effect of 1.0 mg/kg PRA on visual PPI (p<0.03). Effects of the lower dose of PRA failed to reach significance for visual PPI, and inspection of the data (Fig. 3) suggested more robust effects of PRA on PPI when unimodal acoustic stimuli were used.
Figure 3. Effects of PRA on PPI with uni- and cross-modal stimuli.

PPI with acoustic or visual (light) prepulses showed sensitivity to PRA (0, 0.3, 1.0 mg/kg). (*, p<0.03; **, p<0.004; ***, p<0.0001)
ANOVA of startle magnitude to PULSE-ALONE revealed a significant main effect of PRA (F=11.18, df 2,30; p<0.0005) (data not shown). Difference scores between vehicle treatment day and drug treatment day (either 0.3 or 1.0 mg/kg PRA) were calculated for startle magnitude and acoustic and visual PPI scores. Difference scores for PPI and startle magnitude were compared through Spearman Rank Correlation, and no significant correlations between PRA effects on startle magnitude and PPI were found (range of ρ=0.03-0.30, all NS). Thus, drug effects on PPI were separable from drug effects on startle magnitude.
Effects of PRA on PPI across the estrous cycle
Some females displayed prolonged cycle length, but all showed progression through different phases of the cycle during this observation period. Test sessions were spaced 4 days apart to minimize the amount of phase variability for each rat between test days. However, estrous phase was determined after each PPI testing session, and not all cells of a dose × estrous phase comparison were filled. Due to phase cycles that were slightly more or less than 4 days long, many rats were in the same phase for only two out of three drug testing days. This precluded a definitive within-subject assessment of PRA sensitivity across the estrous cycle. Qualitatively, within a given estrous phase, rats tested at two different doses of PRA had reduced %PPI with the higher dose relative to the lower dose. As there was a significant effect of prepulse intensity (F=12.44, df 2,30; p<0.0002) but no prepulse intensity × PRA dose interaction (F=2.10, df 4,60; NS), data were collapsed across prepulse intensity for further statistical analyses. Inspection of the data for rats that were in the same estrous phase for two different test days revealed a significant effect of PRA higher dose vs. lower dose (F=14.10, df 1,9; p<0.005), but no effect of estrous phase (F<1, df 2,9; NS) and no dose × phase interaction (F<1, df 2,9; NS). This lack of estrous phase effect or dose × phase interaction was true whether the lower dose was the vehicle or PRA 0.3 mg/kg condition. A similar analysis of startle magnitude between conditions of higher vs. lower PRA doses showed a significant effect of PRA dose (F=10.85, df 1,9; p<0.01), but no significant effect of estrous phase (F<1, df 2,9; NS) and no dose × phase interaction (F<1, df 2,9; NS). Again, there was no significant effect of estrous phase or dose × phase interaction whether the lower dose was vehicle or PRA 0.3 mg/kg.
Sex Differences
Data from age-matched male rats were combined with data from the estrous cycle study to assess sex differences in PRA effects on PPI (Fig. 4). ANOVA of %PPI showed a significant main effect of PRA dose (0, 0.3, 1.0 mg/kg) (F=33.07, df 2,60; p<0.0001), but no significant effect of sex (F=2.77, df 1,30; NS) and no PRA × sex interaction (F=1.33, df 2,60; NS). There was a significant effect of prepulse intensity (F=33.39, df 2,60; p< 0.0001) but no prepulse intensity × PRA dose interaction (F=1.59, df 4,120; NS). Data were collapsed across prepulse intensity for post-hoc analyses. As had been found in females, post-hoc tests in males indicated that both 0.3 and 1.0 mg/kg PRA had a significant effect on PPI (p<0.002, 0.0001, respectively). However, post-hoc tests in females revealed a significant difference between the low and high doses of PRA (p<0.002), while these two conditions were not significantly different in males. Therefore, females exhibited a step-wise decrease in PPI with increasing doses of PRA, while males achieved a maximal effect with the lower dose of PRA.
Figure 4. PRA effects on %PPI (A) and startle magnitude (B) in male and female rats.

A: Both sexes exhibited dose-dependent reductions in PPI after treated with PRA (0, 0.3, 1.0 mg/kg), though the difference between PPI in low- and high-dose conditions was significant in females but not males. (**, p<0.003; ***, p<0.0001) B: PRA (0, 0.3, 1.0 mg/kg) reduced startle magnitude in both males and females. There was a significant effect of sex (p<0.02), with lower startle magnitude in females than males with both active drug doses.
ANOVA of startle magnitude across male and female groups revealed a significant main effect of PRA dose (F=18.22, df 2,30; p<0.0001). In contrast to PPI data, there was a significant main effect of sex (F=7.19, df 1,30; p<0.02), but no PRA × sex interaction (F<1, df 2,60; NS). ANOVAs comparing startle magnitude at each drug condition indicated that the sex difference was due to significantly lower startle magnitude in females compared to males in both active drug conditions, but not in the vehicle condition (Vehicle: F=1.35, df 1,30; NS; PRA 0.3 mg/kg: F=19.40, df 1,30; p=0.0001; PRA 1.0 mg/kg: F=13.30, df 1,30; p=0.001).
Again, PPI and startle magnitude difference scores were calculated for each rat based on vehicle and PRA treatment days. Spearman Rank Correlations, split by sex, did not indicate any correlation between PRA effects on PPI and startle magnitude at either dose (range of ρ= -0.17-0.28, all NS), suggesting that drug effects on PPI were separable from drug effects on startle magnitude.
Discussion
Compared to the D2 receptor, relatively little is known about the behavioral and cellular/molecular effects of D3 receptor activation or inhibition. The recent emergence of drugs like PRA, which are preferential for D3 versus D2 receptors, has facilitated the study of D3 receptor systems in behavioral models. DA agonist effects on PPI are sensitive to many experimental variables, such as stimulus parameters (Mansbach et al., 1988; Weber and Swerdlow, 2008; Swerdlow et al., 2009), rat strain (Conti et al., 2009; Swerdlow et al., 2004; Weber et al., 2008; Weber and Swerdlow, 2008), sex (Lehmann et al., 1999; Swerdlow et al., 2008a) and estrous phase (Kinkead 2008; Koch, 1998), but most studies of the neurobiology of DA agonist effects on PPI proceeded without the benefit of knowing about these modifying experimental variables. The current studies were designed to “fill in the gaps” regarding parametric effects on PRA-induced PPI deficits in rats in advance of more detailed studies of the neurobiology of D3R effects on PPI.
PRA demonstrated significant effects on PPI during the first 30 min after systemic administration, with maximal effects during the third 10-minute block. The PRA-induced reduction in startle magnitude remained relatively constant across the one-hour test. In order to separate the startle-reducing effects from PRA-induced PPI deficits, startle magnitude was matched between vehicle- and PRA-treated rats by using startling pulses with 120 dB(A) and105 dB(A) intensities. Startle magnitudes in response to 105 dB pulses in vehicle-treated rats were very comparable to startle magnitudes in response to 120 dB pulses in PRA-treated rats; under these conditions of matched startle magnitude, PRA still significantly reduced PPI, suggesting that PRA-induced PPI deficits are independent of PRA effects on startle magnitude. This conclusion is further supported by a lack of correlation between startle magnitude and PPI difference scores, at two doses of PRA with uni- and cross-modal stimuli, as well as with acoustic stimuli in both males and females. The present studies also revealed that the PPI-disruptive effects of PRA were most evident when startle pulses were intense (120 dB) vs. weak (105 dB), despite the fact that post-vehicle levels of PPI were comparable with 120 vs. 105 dB pulses. PRA effects on PPI were also dissociated from those on prepulse-elicited reactions, which have been suggested by some to be relevant to drug-induced changes in PPI (Yee & Feldon, 2009).
PRA-induced PPI deficits were evident using both acoustic and visual (light) prepulses with acoustic startle pulses. This is important because it confirms that the PPI-disruptive effects of PRA are mediated at a point in the nervous system that receives integrated visual and auditory information, rather than within primary sensory circuitry. Dose-response effects were observed in both uni- and cross-modal conditions. Apomorphine, a non-selective D1 and D2-like receptor agonist, has also been found to decrease PPI with both acoustic and visual prepulses (Campeau and Davis 1995, Weber and Swerdlow 2008), and PPI deficits in both schizophrenia and Tourette Syndrome patients are observed across sensory modalities as well (Braff et al., 1992; Castellanos et al., 1996; Swerdlow et al., 2001). Differences in the magnitude of PRA-induced PPI deficits between uni- and cross-modal conditions may reflect psychometric differences between the salience of the acoustic prepulse and light flash prepulse. Nonetheless, these findings suggest that under the present test conditions, unimodal acoustic stimuli may be most sensitive for mechanistic studies of the D3 regulation of PPI.
To date, research on the effects of PRA on PPI in rats has only been conducted in males. PPI is sexually dimorphic in humans (Kumari et al., 2003; Swerdlow et al. 1999) and in some strains of rats (e.g. Lehman et al., 1999; Swerdlow et al. 2008a). Studies of estrous cycle phase effects on PPI have had inconsistent results (Adams et al., 2008; Bubenikova et al., 2005; Kinkead et al., 2008; Koch, 1998). Here, we report that PRA-induced PPI deficits can be observed in females as well as males. Furthermore, no significant sex differences in PRA-induced PPI deficits were found. Thus, either male or female rats appear to be suitable for studies of the D3 regulation of PPI, and the preliminary evidence here suggests that estrous phase may not be a confounding factor.
The D3 agonist, pramipexole, appears to be a tool suited for probing the neurobiology of the D3 regulation of sensorimotor gating. The present studies suggest that the PPI-disruptive effects of PRA have a rapid onset and at least a 30 minute duration, and can be separated experimentally from changes in startle magnitude and prepulse-elicited motor reactions. Optimal experimental conditions for studying the PPI-disruptive effects of PRA would appear to include intense startle pulses and unimodal acoustic stimuli, with comparable magnitudes of PRA effects in male and female SD rats, that do not appear to vary across the estrous cycle in females. Added to the findings of past studies of the impact of rat strain (Weber et al., 2008) and prepulse interval (Swerdlow et al., 2009), the present parametric analyses provide a fairly broad platform of experimental variables needed for conducting informed studies of the D3 regulation of PPI in rats.
Acknowledgments
The authors gratefully acknowledge the assistance of Maria Bongiovanni in manuscript preparation, and Alex Yang and Ishita Desai for their expert technical assistance. Research was supported by awards from the Tourette Syndrome Association (MW), VISN22 MIRECC (MW), NARSAD (MW), and by T32AG000216-18 (WC), MH068366 (NRS) and MH042228 (NRS).
Footnotes
Reprint requests should be addressed to Dr. Neal R. Swerdlow.
Interests to declare: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams AL, Hudson A, Ryan CL, Doucette TA. Effects of estrous stage and time of day on prepulse inhibition in female rats. J Neurosci Methods. 2008;173:295–8. doi: 10.1016/j.jneumeth.2008.06.014. [DOI] [PubMed] [Google Scholar]
- Braff DL, Grillon C, Geyer MA. Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry. 1992;49:206–15. doi: 10.1001/archpsyc.1992.01820030038005. [DOI] [PubMed] [Google Scholar]
- Bubenikova V, Votava M, Horacek J, Palenicek T. Relation of sex and estrous phase to deficits in prepulse inhibition of the startle response induced by ecstasy (MDMA) Behav Pharmacol. 2005;16:127–30. doi: 10.1097/00008877-200503000-00009. [DOI] [PubMed] [Google Scholar]
- Campeau S, Davis M. Involvement of the central nucleus and basolateral complex of the amygdala in fear conditioning measured with fear-potentiated startle in rats trained concurrently with auditory and visual conditioned stimuli. J Neurosci. 1995;15:2301–11. doi: 10.1523/JNEUROSCI.15-03-02301.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellanos FX, Fine EJ, Kaysen D, Marsh WL, Rapoport JL, Hallett M. Sensorimotor gating in boys with Tourette's syndrome and ADHD: preliminary results. Biol Psychiatry. 1996;39:33–41. doi: 10.1016/0006-3223(95)00101-8. [DOI] [PubMed] [Google Scholar]
- Conti LH, Sutherland JE, Muhlhauser CM. Interaction between the effects of corticotropin-releasing factor and prepulse parameters on prepulse inhibition in two inbred rat strains and the F1 generation of a cross between them. Behav Brain Res. 2009;200:165–72. doi: 10.1016/j.bbr.2009.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M. Apomorphine, d-amphetamine, strychnine and yohimbine do not alter prepulse inhibition of the acoustic startle reflex. Psychopharmacology. 1988;95:151–6. doi: 10.1007/BF00174500. [DOI] [PubMed] [Google Scholar]
- Davis M, Mansbach RS, Swerdlow NR, Campeau S, Braff DL, Geyer MA. Apomorphine disrupts the inhibition of acoustic startle induced by weak prepulses in rats. Psychopharmacology. 1990;102:1–4. doi: 10.1007/BF02245735. [DOI] [PubMed] [Google Scholar]
- Kinkead B, Yan F, Owens MJ, Nemeroff CB. Endogenous neurotensin is involved in estrous cycle related alterations in prepulse inhibition of the acoustic startle reflex in female rats. Psychoneuroendocrinology. 2008;33:178–87. doi: 10.1016/j.psyneuen.2007.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney GG, Wilkinson LO, Saywell KL, Tricklebank MD. Rat strain differences in the ability to disrupt sensorimotor gating are limited to the dopaminergic system, specific to prepulse inhibition, and unrelated to changes in startle amplitude or nucleus accumbens dopamine receptor sensitivity. J Neurosci. 1999;19:5644–53. doi: 10.1523/JNEUROSCI.19-13-05644.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch M. Sensorimotor gating changes across the estrous cycle in female rats. Physiol Behav. 1998;64:625–8. doi: 10.1016/s0031-9384(98)00098-5. [DOI] [PubMed] [Google Scholar]
- Kumari V, Gray JA, Gupta P, Luscher S, Sharma T. Sex differences in prepulse inhibition of the acoustic startle response. Pers Individ Dif. 2003;35:733–42. [Google Scholar]
- Lehmann J, Pryce CR, Feldon J. Sex differences in the acoustic startle response and prepulse inhibition in Wistar rats. Behav Brain Res. 1999;104:113–7. doi: 10.1016/s0166-4328(99)00058-3. [DOI] [PubMed] [Google Scholar]
- Mansbach RS, Geyer MA, Braff DL. Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology. 1988;94:507–14. doi: 10.1007/BF00212846. [DOI] [PubMed] [Google Scholar]
- Marcondes FK, Bianchi FJ, Tanno AP. Determination of the estrous cycle phases of rats: some helpful considerations. Braz J Biol. 2002;62:609–14. doi: 10.1590/s1519-69842002000400008. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Maiofiss L, Cussac D, Audinot V, Boutin JA, Newman-Tancredi A. Differential actions of antiparkinson agents at multiple classes of monoaminergic receptor. I. A multivariate analysis of the binding profiles of 14 drugs at 21 native and cloned human receptor subtypes. J Pharmacol Exp Ther. 2002;303:791–804. doi: 10.1124/jpet.102.039867. [DOI] [PubMed] [Google Scholar]
- Palmer AA, Dulawa SC, Mottiwala AA, Conti LH, Geyer MA, Printz MP. Prepulse startle deficit in the Brown Norway rat: a potential genetic model. Behav Neurosci. 2000;114:374–88. doi: 10.1037//0735-7044.114.2.374. [DOI] [PubMed] [Google Scholar]
- Piercey MF, Walker EL, Feldpausch DL, Camacho-Ochoa M. High affinity binding for pramipexole, a dopamine D3 receptor ligand, in rat striatum. Neurosci Lett. 1996;219:138–40. [PubMed] [Google Scholar]
- Svensson K, Carlsson A, Huff RM, Kling-Petersen T, Waters N. Behavioral and neurochemical data suggest functional differences between dopamine D2 and D3 receptors. Eur J Pharmacol. 1994;263:235–43. doi: 10.1016/0014-2999(94)90718-8. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Breier M, Mora AB, Ko D, Shoemaker JM. A novel rat strain with enhanced sensitivity to the effects of dopamine agonists on startle gating. Pharmacol Biochem Behav. 2008a;88:280–90. doi: 10.1016/j.pbb.2007.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Geyer MA, Hartman PL, Sprock J, Auerbach PP, Cadenhead K, et al. Sex differences in sensorimotor gating of the human startle reflex: all smoke? Psychopharmacology. 1999;146:228–32. doi: 10.1007/s002130051111. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Karban B, Ploum Y, Sharp R, Geyer MA, Eastvold A. Tactile prepuff inhibition of startle in children with Tourette's syndrome: in search of an “fMRI-friendly” startle paradigm. Biol Psychiatry. 2001;50:578–85. doi: 10.1016/s0006-3223(01)01164-7. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Lelham SA, Sutherland Owens AN, Chang WL, Sassen SD, Talledo JA. Pramipexole effects on startle gating in rats and normal men. Psychopharmacology. 2009;205:689–98. doi: 10.1007/s00213-009-1577-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow NR, Shoemaker JM, Auerbach PP, Pitcher L, Goins J, Platten A. Heritable differences in the dopaminergic regulation of sensorimotor gating. II. Temporal, pharmacologic and generational analyses of apomorphine effects on prepulse inhibition. Psychopharmacology. 2004;174:452–62. doi: 10.1007/s00213-003-1480-4. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Varty GB, Geyer MA. Discrepant findings of clozapine effects on prepulse inhibition of startle: is it the route or the rat? Neuropsychopharmacology. 1998;18:50–6. doi: 10.1016/S0893-133X(97)00110-3. [DOI] [PubMed] [Google Scholar]
- Swerdlow NR, Weber M, Qu Y, Light GA, Braff DL. Realistic expectations of prepulse inhibition in translational models for schizophrenia research. Psychopharmacology. 2008b;199:331–88. doi: 10.1007/s00213-008-1072-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Chang WL, Breier M, Ko D, Swerdlow NR. Heritable strain differences in sensitivity to the startle gating-disruptive effects of D2 but not D3 receptor stimulation. Behav Pharmacol. 2008;19:786–95. doi: 10.1097/FBP.0b013e32831c3b2b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Chang WL, Durbin JP, Park PE, Luedtke RR, Mach RH, et al. Using prepulse inhibition to detect functional D3 receptor antagonism: effects of WC10 and WC44. Pharmacol Biochem Behav. 2009;93:141–7. doi: 10.1016/j.pbb.2009.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber M, Swerdlow NR. Rat strain differences in startle gating-disruptive effects of apomorphine occur with both acoustic and visual prepulses. Pharmacol Biochem Behav. 2008;88:306–11. doi: 10.1016/j.pbb.2007.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee BK, Feldon J. Distinct forms of prepulse inhibition disruption distinguishable by the associated changes in prepulse-elicited reaction. Behav Brain Res. 2009;204:387–95. doi: 10.1016/j.bbr.2008.11.049. [DOI] [PubMed] [Google Scholar]