

Abstract
Identifying and designing physical systems for use as qubits, the basic units of quantum information, are critical steps in the development of a quantum computer. Among the possibilities in the solid state, a defect in diamond known as the nitrogen-vacancy (NV-1) center stands out for its robustness—its quantum state can be initialized, manipulated, and measured with high fidelity at room temperature. Here we describe how to systematically identify other deep center defects with similar quantum-mechanical properties. We present a list of physical criteria that these centers and their hosts should meet and explain how these requirements can be used in conjunction with electronic structure theory to intelligently sort through candidate defect systems. To illustrate these points in detail, we compare electronic structure calculations of the NV-1 center in diamond with those of several deep centers in 4H silicon carbide (SiC). We then discuss the proposed criteria for similar defects in other tetrahedrally coordinated semiconductors.
Keywords: semiconductor defects, spintronics, first-principles calculations
A quantum computer is a device that would exploit the rules of quantum mechanics to solve certain computational problems more efficiently than allowed by Boolean logic (1). Over the past two decades, qubits have been implemented in a wide variety of materials, including atoms (2), liquids (3), and solids such as superconductors (4), semiconductors (5), and ion-doped insulators (6). Recently, the diamond nitrogen-vacancy (NV-1) center has emerged as a leading qubit candidate because it is an individually addressable quantum system that may be initialized, manipulated, and measured with high fidelity at room temperature (7). Interestingly, even though these successes stem largely from the defect’s nature as a deep center (a point defect with highly localized electronic bound states confined to a region on the scale of a single lattice constant), no systematic effort has been made to identify other deep centers that might behave similarly. We outline the physical features that such deep centers and their hosts should exhibit and show how these criteria can be used to identify potential qubit candidates within a large class of defects structurally analogous to the diamond NV-1. To aid in the illustration of these points, we compare density functional theory (DFT) calculations of the diamond NV-1 with those of several defects found in 4H-SiC.
Searching for deep centers that behave like the diamond NV-1 is worthwhile for several reasons. From an engineering perspective, it is currently quite difficult to grow and fabricate devices from diamond. The discovery of a similar defect in a more technologically mature host material might allow for more sophisticated implementations of single- and multiqubit devices. Additionally, because deep centers and semiconductors as a whole exhibit a diverse set of physical characteristics, innovative areas of device functionality may potentially arise once the quantum properties of these defect systems are more fully explored. From a physics perspective, other deep centers with highly controllable quantum states might help to resolve outstanding questions regarding the structure and dynamical properties of the diamond NV-1 or of deep centers in general.
Defect and Host Criteria for NV-Like Systems
Structurally, the diamond NV-1 consists of a carbon vacancy and an adjacent substitutional nitrogen impurity. The bound states of this deep center are multiparticle states composed of six electrons: five contributed by the four atoms surrounding the vacancy, and one captured from the bulk. As shown in Fig. 1, the lowest energy bound state is a spin triplet (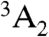 ) whose spin sublevels differ slightly in energy. The ms = 0 and -1 sublevels of this ground state can be chosen to function as the qubit state, and coherent rotations between the two sublevels may be induced by applying microwave radiation tuned to the energy splitting between them. A spin-conserving optical transition exists between the
) whose spin sublevels differ slightly in energy. The ms = 0 and -1 sublevels of this ground state can be chosen to function as the qubit state, and coherent rotations between the two sublevels may be induced by applying microwave radiation tuned to the energy splitting between them. A spin-conserving optical transition exists between the 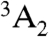 state and an excited-state triplet (3E) 1.945 eV higher in energy. In addition, there exists a spin-selective decay path between these two states that includes a nonradiative transition from 3E to an intermediate spin singlet (
state and an excited-state triplet (3E) 1.945 eV higher in energy. In addition, there exists a spin-selective decay path between these two states that includes a nonradiative transition from 3E to an intermediate spin singlet (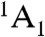 ). In combination, these transitions allow the center to be optically initialized and measured. That is, they allow the defect to be optically pumped into the ms = 0 sublevel of
). In combination, these transitions allow the center to be optically initialized and measured. That is, they allow the defect to be optically pumped into the ms = 0 sublevel of 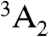 , and they cause the fluorescence intensity between 3E and
, and they cause the fluorescence intensity between 3E and 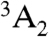 to be spin-dependent (8).
to be spin-dependent (8).
Fig. 1.

Multiplet structure of the NV-1 center in diamond. A 1.945-eV spin-conserving optical transition exists between the ground- (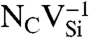 ) and excited- (3E) state triplets. Transitions from the ms = ± 1 sublevels of 3E to an intermediate spin singlet (
) and excited- (3E) state triplets. Transitions from the ms = ± 1 sublevels of 3E to an intermediate spin singlet (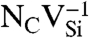 ) are much stronger than those from the ms = 0 sublevel. The spin-selective nature of this decay path can be used in conjunction with the 1.945 eV transition to optically polarize and measure the spin state of
) are much stronger than those from the ms = 0 sublevel. The spin-selective nature of this decay path can be used in conjunction with the 1.945 eV transition to optically polarize and measure the spin state of 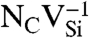 .
.
Two features of the diamond NV-1 help to distinguish it from other solid state qubit systems. First, the center’s highly localized bound states are well isolated from sources of decoherence. At room temperature, the ground state can exhibit extremely long spin coherence times of up to 1.8 ms (9), which is close to the regime needed for quantum error correction, given that manipulation rates greater than 200 MHz have been demonstrated (10, 11). Second, the structure of the defect’s excited-state manifold allows the defect to be optically initialized and measured with high fidelity under ambient conditions. Many other solid state systems are initialized via thermal equilibration and therefore require cryogenic operating temperatures (12–14). And, whereas other systems can be initialized optically (15) or can operate at room temperature (16), they currently can be measured with high fidelity only in an ensemble.
To reproduce these two features, there are several criteria that a candidate deep center and its host should meet. Specifically, centers should exhibit the following five characteristics (for simplicity, we restrict discussion of these characteristics to centers for which, like the diamond NV-1, spin can be treated as a good quantum number):
-
(D1)
A bound state that is suitable for use as a qubit. This state must be paramagnetic and long-lived, and an energy splitting must exist between at least two of the state’s spin sublevels. If the qubit state is to be manipulated via electron spin resonance, the size of this energy splitting must fall within an appropriate range of the radio frequency spectrum.
-
(D2)
An optical pumping cycle that polarizes the qubit state. This cycle will most likely consist of an optical transition from the ground state to an excited state, followed by a spin-selective decay path that includes one or more nonradiative transitions between states of differing spin multiplicity.
-
(D3)
Luminescence to or from the qubit state that varies by qubit sublevel in some differentiable way, whether by intensity, wavelength, or other property. If fluorescence from an excited state is used to probe the qubit, the fluorescent transition should be spin-conserving. In addition, the strength of this fluorescent transition, which depends on the lifetime of the excited state, should be large enough to enable efficient, high fidelity measurement of individual defect qubit states.
-
(D4)
Optical transitions that do not introduce interference from the electronic states of the host. All optical transitions used to prepare and measure the qubit state must be lower in energy than the energy required to transfer an electron into (out of) the center from (to) the electronic states of the host.
-
(D5)
Bound states that are separated from each other by energies large enough to avoid thermal excitation between them. If the energy difference between two bound states is too small, thermal excitations may couple states and destroy spin information.
In addition, an ideal crystalline host will have the following qualities, the final three of which should help to reduce decoherence in the defect:
-
(H1)
A wide band gap, so that it can accommodate a deep center that will satisfy requirement D4 above.
-
(H2)
Small spin-orbit coupling, in order to avoid unwanted spin flips in the defect bound states.
-
(H3)
Availability as high-quality, bulk, or thin-film single crystals, in order to avoid imperfections or paramagnetic impurities that could affect the deep center’s spin state.
-
(H4)
Constituent elements with naturally occurring isotopes of zero nuclear spin, so that spin bath effects may be eliminated from the host via isotopic engineering (9).
It is relatively easy to identify hosts that satisfy criteria H1–H4, and this is discussed in detail in Identifying Candidate Host Materials in SI Text. However, it is not as simple to predict whether a given defect will satisfy criteria D1–D5. Nevertheless, certain aspects of a defect’s electronic structure can be predicted in a straightforward manner by using first-principles calculations. For example, the spin of a defect bound state can be calculated, determining whether the defect is paramagnetic or not, which is a major component of criterion D1. However, it is difficult to accurately compute the energy splittings between the spin sublevels of a bound state. In the case of criterion D2, the defect-induced gap levels obtained via first-principles calculations can be used to predict whether a paramagnetic defect will possess internal optical transitions. In addition, the energies of these optical transitions can be calculated by using constrained DFT, and excitonic effects can be included through the Bethe–Salpeter formalism (17). An explicit characterization of nonradiative decay paths, on the other hand, is much more challenging and is generally beyond the reach of current first-principles methods (18). The application of first-principles calculations to criterion D3 depends on the desired method of qubit measurement. In cases like the diamond NV-1, where the intensity of luminescence to the qubit state should vary by spin sublevel, first-principles predictions are again limited by the challenging nature of nonradiative transitions. In cases where the wavelength of luminescence is meant to vary by spin sublevel, the small magnitude of the spin sublevel splittings (in the microwave range for qubits manipulated via electron spin resonance) renders it difficult to quantitatively evaluate luminescence energies on the basis of first-principles calculations. Still, in combination with perturbation theory, first-principles calculations can provide information about the ordering of sublevels, and this can help guide experimental identification of observed optical transitions. Additionally, the excited-state lifetime discussed in criterion D3 can be characterized by calculating the magnitudes of the dipole matrix elements associated with the internal optical transitions. Finally, the properties associated with criteria D4 and D5 can be studied explicitly by analyzing the energies of calculated defect-induced gap levels, both relative to each other and relative to the band edges of the host material.
Even though some aspects of D1–D5 are difficult to evaluate by using standard first-principles methods, potential qubit candidates can still be identified by observing that a close relationship exists between the atomic configuration of a defect and its electronic structure (19). In tetrahedrally coordinated semiconductors, vacancies and vacancy-related complexes similar to the NV-1 center in diamond are likely to possess bound states with comparable physical properties. Within this group of defects, one can then use first-principles calculations to determine which members are compatible with criteria D1–D5. In the next section, we demonstrate in detail how this assessment can be made, by using as an example the results of calculations performed for several defects in diamond and SiC.
Formation Energies, Defect-Level Diagrams, and Configuration-Coordinate Diagrams
DFT calculations have become an indispensable tool for studying the properties of defects. Recent advances using hybrid functionals, which incorporate some degree of the Hartree–Fock exchange interaction, have led to very accurate descriptions of defect states by overcoming the well-known band-gap problem of traditional DFT. We apply this methodology to defect systems analogous to the NV-1 defect in diamond. Specifically, we discuss defect formation energies and configuration-coordinate diagrams for defect excitations, as well as the arrangement of the defect-induced gap levels, which we discuss in terms of “defect-level diagrams” (DLDs).
One of the most important quantities that can be extracted from first-principles calculations is the formation energy (Ef) of a defect. Ef provides information on the overall stability of a given defect, as well as the relative stabilities between different atomic configurations and charge states. Ef determines the defect concentration through a Boltzmann relation (20):
where NS is the number of possible defect sites. Strictly speaking, this expression is valid only in equilibrium; however, formation energies are informative even when defects are created in nonequilibrium processes, such as ion implantation. Specifically, the magnitude of Ef still provides an indicator of which defects are most likely to form. Once a defect is formed, the relative stability of different charge states for a given defect is always determined by the dependence of Ef on the Fermi level, whatever the creation process of the defect.
The NV Center in Diamond.
As explained in Methods, Ef depends on the charge state of the defect and on the Fermi level (εF), which is referenced to the valence-band maximum (VBM) of the bulk host material. Fig. 2 shows Ef for the NV center in diamond, as well as for the carbon vacancy (VC), and substitutional nitrogen (NC). For each defect, only the charge state with the lowest Ef is included at each value of εF. For a given εF, the charge state of a defect is equal to the slope of that defect’s Ef curve at that point. The kinks in the Ef curves correspond to charge-state-transition levels, i.e., εF values where the charge state of the defect changes. Fig. 2 thus shows the range of εF for which each charge state of the defect is stable; both the isolated vacancy and the NV center can be stable in the +1, 0, -1, or -2 charge states. Determining which charge states are stable, and under what conditions, is crucial to evaluating whether these vacancy-related defects satisfy criterion D1. This determination is crucial because each charge state will correspond to a different spin configuration, with some having paramagnetic ground states and others not.
Fig. 2.

Formation energy, Ef, as a function of Fermi level, εF. Ef was calculated for various defects in (A) diamond and (B) 4H-SiC (in C-rich conditions). The shaded areas show the range of stability of NV-1 in diamond, and 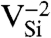 (Blue),
(Blue), 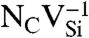 (Green), and
(Green), and 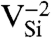 (Purple) in SiC.
(Purple) in SiC.
The ground-state spin of each charge state can be determined by considering the defect’s electronic structure. The electronic structure of vacancy-related centers in tetrahedrally coordinated semiconductors can be understood in terms of atomic sp3 orbitals and the corresponding single-particle levels. In an environment with tetrahedral symmetry, the four degenerate sp3 dangling-bond (DB) orbitals neighboring a vacancy are split into a low-energy symmetric a1 level and three degenerate t2 levels (as seen in Fig. 3A for the -2 charge state of VC, which is stable in N-doped diamond). Because of the high symmetry of the isolated vacancy, this level structure does not lead to a ground-state triplet. However, a ground-state triplet can be achieved by placing an impurity atom next to the vacancy, thus shifting the a1 level [becoming a1(1)] and splitting the degeneracy of the t2 levels into a1(2), ex, and ey levels (21); Fig. 3B shows the DLD for NV-1.
Fig. 3.

Defect-level diagrams for vacancy-related complexes. These diagrams show the single-particle defect states for (A) the 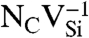 and (B) the NV-1 in diamond, as well as for (C) the
and (B) the NV-1 in diamond, as well as for (C) the 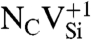 and (D) the
and (D) the 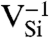 in 4H-SiC. The spin-majority (spin-minority) channel is denoted by upward- (downward-) pointing arrows.
in 4H-SiC. The spin-majority (spin-minority) channel is denoted by upward- (downward-) pointing arrows.
The NV-1 defect is stable for εF between 2.78 and 5.14 eV, consistent with the likely position of εF in N-doped diamond [N being a deep donor located ∼1.9 eV below the conduction-band minimum (CBM) (22)]. The associated single-particle eigenvalues are listed in Table 1; their occupation determines the spin state (i.e., a spin-one triplet). The location of the defect-induced gap levels illustrates the defect’s compliance with D4 and D5, because they are well isolated from the bulk bands, with a relatively large spacing between occupied and unoccupied levels. As shown by the green dashed arrow in Fig. 3B, we can remove an electron from the spin-minority channel of the a1(2) level and place it into one of the ei spin-minority levels, keeping the ground-state atomic configuration fixed. The corresponding absorption energy of 2.27 eV is shown in the configuration-coordinate diagram of Fig. 4A. If we subsequently allow the atomic positions to relax, maintaining the excited-state triplet electronic configuration, we obtain a zero-phonon line (ZPL) energy of 2.02 eV and a Frank–Condon shift of 0.26 eV, both in good agreement with experiment (23) as well as with recent calculations (24). This discussion illustrates how the computationally accessible properties relevant to criterion D2 can be obtained.
Table 1.
Defect-level energies for various vacancy and NV centers in diamond and SiC (underlined values indicate the level is occupied)
| Material |
Defect |
No. of els. |
Spin majority |
Spin minority |
||||||
|
a1(1) |
a1(2) |
ex |
ey |
a1(1) |
a1(2) |
ex |
ey |
|||
| Diamond | V-2 | 6 | −1.46 | 2.66 | 2.66 | 2.66 | −0.09 | 4.67 | 4.67 | 4.67 |
| Diamond | NV-1 | 6 | −2.04 | 1.31 | 2.33 | 2.33 | −1.39 | 2.06 | 5.07 | 5.07 |
| 4H SiC | 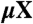 |
6 | −0.41 | 0.06 | 0.29 | 0.33 | −0.72 | 0.93 | 2.35 | 2.51 |
| 4H SiC | 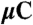 |
6 | −0.11 | 0.41 | 0.89 | 1.23 | 0.29 | 1.66 | 2.66 | 2.90 |
| 4H SiC | 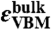 |
5 | −0.11 | 0.43 | 0.43 | 0.67 | 0.53 | 2.66 | 2.67 | 3.14 |
| 4H SiC | 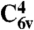 |
4 | −0.41 | 0.14 | 0.20 | 2.42 | −0.47 | 1.86 | 2.69 | 2.82 |
Fig. 4.

Configuration-coordinate diagrams for spin-conserving triplet excitation. Excitation cycles for (A) the NV-1 center in diamond and (B) the 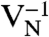 center in SiC are shown. Absorption, emission, and ZPL transitions are indicated, along with their energies.
center in SiC are shown. Absorption, emission, and ZPL transitions are indicated, along with their energies.
Defect Centers in SiC.
SiC shares many similarities with diamond, and recent experimental evidence indicates that it may also harbor deep centers suitable for quantum computing (25–28). We focus on the 4H polytype because large single crystals are readily available, and because its band gap (3.27 eV) is larger than that of 3C-SiC (2.39 eV) and 6H-SiC (3.02 eV) (29). Fig. 2B shows Ef for the silicon and carbon vacancies (VSi and VC), as well as for substitutional nitrogen (NSi and NC). Ef is more than 4 eV larger for NSi than for NC, so N has an extremely strong energetic preference to sit on a C site. This large energy difference implies that only nitrogen-vacancy centers composed of a NC and a VSi will form in SiC. According to Fig. 2B, NCVSi is stable in the 0, -1, and -2 charge states. Similar to the diamond NV-1 defect, six electrons are confined to the 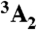 defect, which is stable for εF between 1.60 and 2.83 eV. The levels for the corresponding DLD are listed in Table 1. Note that the degeneracy of the ei levels is lifted because of the lower symmetry of the crystal structure.
defect, which is stable for εF between 1.60 and 2.83 eV. The levels for the corresponding DLD are listed in Table 1. Note that the degeneracy of the ei levels is lifted because of the lower symmetry of the crystal structure.
The calculated configuration-coordinate diagram for 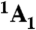 is shown in Fig. 4B. Comparing these numbers with the diamond NV-1 (Fig. 4A), we see that the vertical transitions are about half as large in the
is shown in Fig. 4B. Comparing these numbers with the diamond NV-1 (Fig. 4A), we see that the vertical transitions are about half as large in the 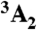 center, whereas the relaxation energies are more than 75% smaller. The difference in transition energies can be attributed to the larger lattice constant of SiC compared with diamond: Although the vacancy is surrounded by C atoms in both materials, the larger lattice constant of SiC leads to a smaller overlap among the sp3 DB orbitals and therefore to a smaller splitting between a1(2) and ei levels.
center, whereas the relaxation energies are more than 75% smaller. The difference in transition energies can be attributed to the larger lattice constant of SiC compared with diamond: Although the vacancy is surrounded by C atoms in both materials, the larger lattice constant of SiC leads to a smaller overlap among the sp3 DB orbitals and therefore to a smaller splitting between a1(2) and ei levels.
It is interesting to also consider isolated vacancies in SiC. 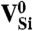 in SiC can support a spin-conserving triplet excitation because the broken tetrahedral symmetry of the host splits the t2 levels (Fig. 3 C and D). Our calculated formation energies in Fig. 2B show that the 0, -1, and -2 charge states are all stable. Similar to the diamond NV-1 or SiC
in SiC can support a spin-conserving triplet excitation because the broken tetrahedral symmetry of the host splits the t2 levels (Fig. 3 C and D). Our calculated formation energies in Fig. 2B show that the 0, -1, and -2 charge states are all stable. Similar to the diamond NV-1 or SiC 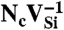 defects, six electrons are bound to
defects, six electrons are bound to 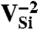 , which is stable in n-type material for Fermi levels within 0.3 eV of the CBM. The DLD in Fig. 3C (energies in Table 1) shows that the spin-minority a1(2) level lies above the spin-majority ei levels, which raises the possibility that VSi may also exhibit a ground-state triplet when occupied with four electrons instead of six. This result is indeed borne out by explicit calculations, as shown in Fig. 3D. In principle, a similar situation could occur by removing two electrons from the
, which is stable in n-type material for Fermi levels within 0.3 eV of the CBM. The DLD in Fig. 3C (energies in Table 1) shows that the spin-minority a1(2) level lies above the spin-majority ei levels, which raises the possibility that VSi may also exhibit a ground-state triplet when occupied with four electrons instead of six. This result is indeed borne out by explicit calculations, as shown in Fig. 3D. In principle, a similar situation could occur by removing two electrons from the 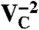 defect; however,
defect; however, 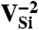 is not a stable charge state, as evident from Fig. 2B. However, the close proximity of the spin-majority a1(2) level to the VBM is cause for concern in light of criterion D4. Finally, we note that
is not a stable charge state, as evident from Fig. 2B. However, the close proximity of the spin-majority a1(2) level to the VBM is cause for concern in light of criterion D4. Finally, we note that 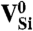 forms a ground-state quartet (i.e., spin 3/2; see Table 1), and therefore this defect does not allow for spin-conserving triplet excitations.
forms a ground-state quartet (i.e., spin 3/2; see Table 1), and therefore this defect does not allow for spin-conserving triplet excitations.
Discovering NV Analogs in Other Material Systems
Moving beyond SiC, it is important to establish some general guidelines and procedures for identifying defects that may be analogous to the NV center in diamond. For the purposes of this discussion, we will focus on tetrahedrally coordinated compound semiconductors, considering both cation and anion vacancies.
Cation Vacancies.
In cation vacancies, the defect levels are determined by interacting anion sp3 DBs. Because anion DBs lie close to the VBM (30) (Fig. 5B), the t2 vacancy levels will tend to be located in the lower half of the band gap. To satisfy criterion D4, these t2 levels should be well separated from the VBM. The t2 levels will be split by (i) Jahn–Teller distortions, (ii) the presence of an impurity, and/or (iii) crystal-field splitting in hosts with lower than cubic symmetry. This splitting should be sufficiently large to satisfy criterion D5, but small enough to satisfy D4, and to avoid pushing the a1(2) level too close to the VBM. The energy position of the anion DB orbitals and the splitting between the a1 and t2 vacancy orbitals (ΔCV, Fig. 5B) are therefore important quantities for identifying defect systems for quantum computing applications, and in Trends in Defect-Level Splitting in SI Text we address how the choice of host and defect center impacts their value.
Fig. 5.

Development of defect-level structure in tetrahedrally coordinated compound semiconductors. Atomic sp3 DBs (A) interact to form a1 and t2 levels in an ideal vacancy (B), with the t2 levels splitting further in vacancy complexes (C).
Further design flexibility is added by placing an impurity next to the vacancy, thus creating a complex. The energy of the impurity’s sp3 orbital relative to that of the host anion DB orbital affects the splitting between the a1(2) and ei orbitals (δCV, Fig. 5C). If the impurity DB orbital is significantly lower in energy than the host anion DB, the splitting δCV will be large. Too large a δCV value (relative to the band gap) is undesirable, because it might push the a1(2) level close to or below the VBM. An attractive interaction is needed between the vacancy and impurity in order for the complex to form. Therefore, because cation vacancies tend to be negatively charged, we should choose impurities that act as donors, i.e., elements to the right of the host anion in the periodic table. An example of such a defect is the self-activation center in ZnSe, which is a complex formed by a Zn vacancy and a donor impurity. In the positive charge state (which would be stable in p-type material), this defect gives rise to a ground-state triplet with six electrons (see Electron Counting for Defects in SI Text). It remains to be determined whether this defect satisfies all the other proposed criteria.
Anion Vacancies.
Anion vacancies are less likely to lead to triplet configurations, because the cation DBs that give rise to their defect levels tend to be located in the upper part of the band gap (30) (Fig. 5B) and the vacancy orbital splitting (ΔAV) will tend to push the t2 orbitals close to or above the CBM. This result is indeed what occurs for an oxygen vacancy in ZnO, for which only the a1(1) level lies within the band-gap (31). To avoid this, the semiconductor needs to have a large enough band gap, the cation sp3 DB orbitals need to be well below the CBM, and the vacancy orbital splitting (ΔAV) needs to be small. These criteria are met in AlN, in which the VN has t2 levels within the band-gap. The arguments about further splitting of the levels are similar to our discussion of cation vacancies. Regarding the choice of impurity, because anion vacancies tend to act as donors, one might think that acceptor-type impurities might be the best choice, in order to maximize attraction. However, electron counting then reveals that a level occupation similar to that of the diamond NV-1 cannot be achieved because this requires that the anion vacancy (or complex) be in a negative charge state. But in AlN, 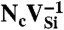 is stable if εF is in the upper part of the band gap, and a donor impurity will fulfill the requirements of (i) being attracted to the vacancy and (ii) supplying additional electrons to achieve the desired orbital occupation.
is stable if εF is in the upper part of the band gap, and a donor impurity will fulfill the requirements of (i) being attracted to the vacancy and (ii) supplying additional electrons to achieve the desired orbital occupation.
Beyond NV Analogs
The world of deep centers is vast, and only one small subset has been discussed here in detail. Future work is needed to determine which other classes of deep centers are compatible with the defect and host criteria that have been outlined. For example, many isolated substitutional or interstitial impurities act as deep centers (19), but no such center satisfying D1–D5 has been identified so far. In the octahedrally coordinated hosts MgO and CaO, optical spin polarization has been reported in vacancy-related complexes with D4h symmetry (32, 33), but more exploration is required to determine what other features these centers have in common with the NV-1 center in diamond. Still other classes of deep centers become open to investigation if the stipulation that spin be a good quantum number is removed. In this case, optical selection rules are relaxed, and alternative mechanisms of optical polarization may then be possible (34).
Methods
The following expression gives Ef for the NV center in diamond in charge state q:
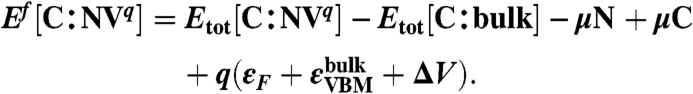 |
The Etot[C∶X] terms are the total energies of the diamond supercell with the NVq defect and of the bulk supercell. The 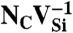 terms are the chemical potential references used for N and C. For diamond,
terms are the chemical potential references used for N and C. For diamond, 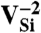 is simply the energy per C atom in the crystal. εF is the Fermi level, referenced to the VBM in the bulk,
is simply the energy per C atom in the crystal. εF is the Fermi level, referenced to the VBM in the bulk, 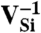 ; the ΔV term is used to align the bulk VBM to that of the defect supercell (20, 35).
; the ΔV term is used to align the bulk VBM to that of the defect supercell (20, 35).
The first-principles calculations were performed by using supercells of 64 atoms for C in the diamond structure and 96 atoms for SiC in the 4H polytype (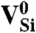 space group), with finite-size corrections for the charged-defect calculations (35). Projector augmented wave pseudopotentials were used as implemented in the Vienna Ab-initio Simulation Package (36, 37). We used a 400-eV plane-wave cutoff and a 2 × 2 × 2 special-k-point mesh to carry out integration over the Brillouin zone. The hybrid functional calculations (including atomic relaxations) were performed within the HSE06 formalism (38, 39). The calculated band gaps are 5.36 eV for diamond and 3.17 eV for 4H-SiC. Defects in 4H-SiC can occur on two possible inequivalent Si or C sites; tests indicate the corresponding energies differ by less than 0.1 eV. The results reported here are for the hexagonal site. For the NV defect in SiC, there are two choices for the position of the substitutional N atom: one associated with the single longer bulk Si-C bond length (along the c axis), and the other associated with the three shorter bulk Si-C bond lengths. For our calculations, we chose the site associated with the shorter Si-C bond. All defect excitation energies were calculated by using constrained DFT, by removing an electron out of an occupied defect level and placing it into an unoccupied defect level. We note that transitions between internal defect levels are likely to be more accurately calculated than defect-to-band transitions (40). For the purposes of assessing our criteria, this trend is advantageous, because information about defect-to-band transitions is used only qualitatively in determining whether such transitions are suppressed.
space group), with finite-size corrections for the charged-defect calculations (35). Projector augmented wave pseudopotentials were used as implemented in the Vienna Ab-initio Simulation Package (36, 37). We used a 400-eV plane-wave cutoff and a 2 × 2 × 2 special-k-point mesh to carry out integration over the Brillouin zone. The hybrid functional calculations (including atomic relaxations) were performed within the HSE06 formalism (38, 39). The calculated band gaps are 5.36 eV for diamond and 3.17 eV for 4H-SiC. Defects in 4H-SiC can occur on two possible inequivalent Si or C sites; tests indicate the corresponding energies differ by less than 0.1 eV. The results reported here are for the hexagonal site. For the NV defect in SiC, there are two choices for the position of the substitutional N atom: one associated with the single longer bulk Si-C bond length (along the c axis), and the other associated with the three shorter bulk Si-C bond lengths. For our calculations, we chose the site associated with the shorter Si-C bond. All defect excitation energies were calculated by using constrained DFT, by removing an electron out of an occupied defect level and placing it into an unoccupied defect level. We note that transitions between internal defect levels are likely to be more accurately calculated than defect-to-band transitions (40). For the purposes of assessing our criteria, this trend is advantageous, because information about defect-to-band transitions is used only qualitatively in determining whether such transitions are suppressed.
Supplementary Material
Acknowledgments.
We are grateful to G.D. Fuchs, F.J. Heremans, and D.M. Toyli for useful discussions. This work was supported by Army Research Office, Air Force Office of Scientific Research, and the National Science Foundation Materials Research Science and Engineering Centers Program. It made use of the California NanoSystems Institute Computing Facility and TeraGrid (Texas Advanced Computing Center and San Diego Supercomputer Center) with support from the National Science Foundation.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/cgi/content/full/1003052107/DCSupplemental.
References
- 1.Childs AM, van Dam W. Quantum algorithms for algebraic problems. Rev Mod Phys. 2010;82:1–52. [Google Scholar]
- 2.Monroe C. Quantum information processing with atoms and photons. Nature. 2002;416:238–246. doi: 10.1038/416238a. [DOI] [PubMed] [Google Scholar]
- 3.Cory DG, et al. NMR based quantum information processing: Achievements and prospects. Fortschr Phys. 2000;48:875–907. [Google Scholar]
- 4.Devoret MH, Martinis JM. Implementing qubits with superconducting integrated circuits. Quantum Inf Process. 2004;3:163–203. [Google Scholar]
- 5.Hanson R, Awschalom DD. Coherent manipulation of single spins in semiconductors. Nature. 2008;453:1043–1049. doi: 10.1038/nature07129. [DOI] [PubMed] [Google Scholar]
- 6.Bertaina S, et al. Rare-earth solid-state qubits. Nat Nanotechnol. 2007;2:39–42. doi: 10.1038/nnano.2006.174. [DOI] [PubMed] [Google Scholar]
- 7.Neumann P, et al. Multipartite entanglement among single spins in diamond. Science. 2008;320:1326–1329. doi: 10.1126/science.1157233. [DOI] [PubMed] [Google Scholar]
- 8.Manson NB, Harrison JP, Sellars MJ. Nitrogen-vacancy center in diamond: Model of the electronic structure and associated dynamics. Phys Rev B. 2006;74:104303. [Google Scholar]
- 9.Balasubramanian G, et al. Ultralong spin coherence time in isotopically engineered diamond. Nat Mater. 2009;8:383–387. doi: 10.1038/nmat2420. [DOI] [PubMed] [Google Scholar]
- 10.DiVincenzo DP. The physical implementation of quantum computation. Fortschr Phys. 2000;48:771–783. [Google Scholar]
- 11.Fuchs GD, Dobrovitski VV, Toyli DM, Heremans FJ, Awschalom DD. Gigahertz dynamics of a strongly driven single quantum spin. Science. 2009;326:1520–1522. doi: 10.1126/science.1181193. 10.1126/science.1181193. [DOI] [PubMed] [Google Scholar]
- 12.Kane BE. A silicon-based nuclear spin quantum computer. Nature. 1998;393:133–137. [Google Scholar]
- 13.Petta JR, et al. Coherent manipulation of coupled electron spins in semiconductor quantum dots. Science. 2005;309:2180–2184. doi: 10.1126/science.1116955. [DOI] [PubMed] [Google Scholar]
- 14.Berezovsky J, Mikkelsen MH, Stoltz NG, Coldren LA, Awschalom DD. Picosecond coherent optical manipulation of a single electron spin in a quantum dot. Science. 2008;320:349–352. doi: 10.1126/science.1154798. [DOI] [PubMed] [Google Scholar]
- 15.Wesenberg JH, Mølmer K, Rippe L, Kröll S. Scalable designs for quantum computing with rare-earth-ion-doped crystals. Phys Rev A. 2007;75:012304. [Google Scholar]
- 16.Nellutla S, et al. Coherent manipulation of electron spins up to ambient temperatures in Cr5+(S = 1/2) doped K3NbO8. Phys Rev Lett. 2007;99:137601. doi: 10.1103/PhysRevLett.99.137601. [DOI] [PubMed] [Google Scholar]
- 17.Ma Y, Rohlfing M. Quasiparticle band structure and optical spectrum of CaF2. Phys Rev B. 2007;75:205114. [Google Scholar]
- 18.Gali A, Fyta M, Kaxiras E. Ab initio supercell calculations on nitrogen-vacancy center in diamond: Electronic structure and hyperfine tensors. Phys Rev B. 2008;77:155206. [Google Scholar]
- 19.Lannoo M, Bourgoin J. Point Defects in Semiconductors I: Theoretical Aspects. New York: Springer–Verlag; 1981. pp. 9–22.pp. 68–92. [Google Scholar]
- 20.Van de Walle CG, Neugebauer J. First-principles calculations for defects and impurities: Applications to III-nitrides. J Appl Phys. 2004;95:3851–3879. [Google Scholar]
- 21.Lenef A, Rand SC. Electronic structure of the N-V center in diamond: Theory. Phys Rev B. 1996;53:13441–13455. doi: 10.1103/physrevb.53.13441. [DOI] [PubMed] [Google Scholar]
- 22.Heremans FJ, Fuchs GD, Wang CF, Hanson R, Awschalom DD. Generation and transport of photoexcited electrons in single-crystal diamond. Appl Phys Lett. 2009;94:152102. [Google Scholar]
- 23.Davies G, Hamer MF. Optical studies of the 1.945 eV vibronic band in diamond. Proc R Soc Lond Ser-A. 1976;348:285–298. [Google Scholar]
- 24.Gali A, Janzén E, Deak P, Kresse G, Kaxiras E. Theory of spin-conserving excitation of the N-V- center in diamond. Phys Rev Lett. 2009;103:186404. doi: 10.1103/PhysRevLett.103.186404. [DOI] [PubMed] [Google Scholar]
- 25.Son NT, Zolnai Z, Janzén E. Silicon vacancy related TV2a center in 4H-SiC. Phys Rev B. 2003;68:205211. [Google Scholar]
- 26.Mizuochi N, et al. Continuous-wave and pulsed EPR study of the negatively charged silicon vacancy with S = 3/2 and C3v symmetry in n-type 4H-SiC. Phys Rev B. 2002;66:235202. [Google Scholar]
- 27.Orlinski SB, Schmidt J, Mokhov EN, Baranov PG. Silicon and carbon vacancies in neutron-irradiated SiC: A high-field electron paramagnetic resonance study. Phys Rev B. 2003;67:125207. [Google Scholar]
- 28.Baranov PG, et al. Spin polarization induced by optical and microwave resonance radiation in a Si vacancy in SiC: A promising subject for the spectroscopy of single defects. JETP Lett. 2007;86:202–206. [Google Scholar]
- 29.Madelung O. Semiconductors: Data Handbook. 3rd Ed. New York: Springer–Verlag; 2004. [Google Scholar]
- 30.Harrison W. Electronic Structure and the Properties of Solids. Mineola, NY: Dover; 1989. [Google Scholar]
- 31.Janotti A, Van de Walle CG. Oxygen vacancies in ZnO. Appl Phys Lett. 2005;87:122102. [Google Scholar]
- 32.Tanimoto DH, Ziniker WM, Kemp JO. Optical spin polarization in M-like centers in CaO. Phys Rev Lett. 1965;14:645–647. [Google Scholar]
- 33.Henderson B. Optical pumping cycle of exchange-coupled F+-center pairs in MgO and CaO. J Phys C Solid State. 1976;9:2185–2195. [Google Scholar]
- 34.Geschwind S. In: Electron Paramagnetic Resonance. Geswchind S, editor. New York: Plenum; 1972. pp. 372–386. [Google Scholar]
- 35.Freysoldt C, Neugebauer J, Van de Walle CG. Fully ab initio finite-size corrections for charged supercell calculations. Phys Rev Lett. 2009;102:016402. doi: 10.1103/PhysRevLett.102.016402. [DOI] [PubMed] [Google Scholar]
- 36.Kresse G, Furthmuller J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys Rev B. 1996;54:11169. doi: 10.1103/physrevb.54.11169. [DOI] [PubMed] [Google Scholar]
- 37.Kresse G, Joubert J. From ultrasoft pseudopotentials to the projector augmented-wave method. Phys Rev B. 1999;59:1758–1775. [Google Scholar]
- 38.Heyd J, Scuseria GE, Ernzerhof M. Hybrid functionals based on a screened Coulomb potential. J Chem Phys. 2003;118:8207–8215. [Google Scholar]
- 39.Marsman M, Paier J, Stroppa A, Kresse G. Hybrid functionals applied to extended systems. J Phys Condens Mater. 2008;20:064201. doi: 10.1088/0953-8984/20/6/064201. [DOI] [PubMed] [Google Scholar]
- 40.Alkauskas A, Broqvist P, Pasquarello A. Defect energy levels in density functional calculations: Alignment and band gap problem. Phys Rev Lett. 2008;101:046405. doi: 10.1103/PhysRevLett.101.046405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.