

Abstract
A structural and functional model of bacterial nitric oxide reductase (NOR) has been designed by introducing two glutamates (Glu) and three histidines (His) in sperm whale myoglobin. X-ray structural data indicate that the three His and one Glu (V68E) residues bind iron, mimicking the putative FeB site in NOR, while the second Glu (I107E) interacts with a water molecule and forms a hydrogen bonding network in the designed protein. Unlike the first Glu (V68E), which lowered the heme reduction potential by ∼110 mV, the second Glu has little effect on the heme potential, suggesting that the negatively charged Glu has a different role in redox tuning. More importantly, introducing the second Glu resulted in a ∼100% increase in NOR activity, suggesting the importance of a hydrogen bonding network in facilitating proton delivery during NOR reactivity. In addition, EPR and X-ray structural studies indicate that the designed protein binds iron, copper, or zinc in the FeB site, each with different effects on the structures and NOR activities, suggesting that both redox activity and an intermediate five-coordinate heme-NO species are important for high NOR activity. The designed protein offers an excellent model for NOR and demonstrates the power of using designed proteins as a simpler and more well-defined system to address important chemical and biological issues.
Keywords: biomimetic models, heme-copper oxidase, metalloprotein, protein design, protein engineering
Rational design of proteins that mimic both structure and function of more complex native enzymes has been a long sought-after goal, as the process is an ultimate test of our knowledge and an excellent means to develop advanced biocatalysts (1–3). Although designed proteins that model the structure of native enzymes have been known for a while (4–10), successful designs of proteins that mimic both the structure and function of native enzymes have been reported only recently (11–16). While being able to design such functional proteins is laudable, the impact of such an achievement would be greater if the designed proteins can be used to address fundamental issues in chemistry and biology that are difficult to tackle by other methods. One primary example is the roles of conserved glutamates and metal ions in bacterial nitric oxide reductase (NOR) (17–19).
NO is critical for all life (20). Bacterial denitrification is a crucial part of the nitrogen cycle in nature that involves a four-step, five-electron reduction of nitrate (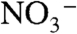 ) to dinitrogen (N2) (17, 19). Bacterial NOR is a membrane-bound protein that catalyzes one step of this process, namely, the two-electron reduction of NO to N2O (17, 19). With no crystal or solution structure available for bacterial NOR to date, sequence alignments and homology modeling (21, 22) have indicated that NOR is structurally homologous to the largest subunit (subunit I) of heme-copper oxidases (HCOs) (23), enzymes that catalyze reduction of O2 to water. The active sites of both NOR and HCO contain a proximal histidine-coordinated heme and a distal three histidine-coordinated metal center. However, the metal center in HCOs is occupied by a copper (called CuB), whereas a nonheme iron is present in NOR (called FeB) (23, 24). In addition, two conserved glutamates, shown by modeling to be close to the FeB site (21, 22), are found to be essential for NOR activity (24, 25). Some members of HCOs such as cytochrome cbb3 oxidase display NOR activity (26–28), although the activity is ∼50-fold lower than native NOR (26). Therefore, it is important to elucidate the structural features, specifically the roles of the conserved glutamates close to the FeB site and metal ions (copper vs. iron), responsible for the reduction of NO to N2O.
) to dinitrogen (N2) (17, 19). Bacterial NOR is a membrane-bound protein that catalyzes one step of this process, namely, the two-electron reduction of NO to N2O (17, 19). With no crystal or solution structure available for bacterial NOR to date, sequence alignments and homology modeling (21, 22) have indicated that NOR is structurally homologous to the largest subunit (subunit I) of heme-copper oxidases (HCOs) (23), enzymes that catalyze reduction of O2 to water. The active sites of both NOR and HCO contain a proximal histidine-coordinated heme and a distal three histidine-coordinated metal center. However, the metal center in HCOs is occupied by a copper (called CuB), whereas a nonheme iron is present in NOR (called FeB) (23, 24). In addition, two conserved glutamates, shown by modeling to be close to the FeB site (21, 22), are found to be essential for NOR activity (24, 25). Some members of HCOs such as cytochrome cbb3 oxidase display NOR activity (26–28), although the activity is ∼50-fold lower than native NOR (26). Therefore, it is important to elucidate the structural features, specifically the roles of the conserved glutamates close to the FeB site and metal ions (copper vs. iron), responsible for the reduction of NO to N2O.
To address these issues, biochemical and biophysical studies of native NOR and its variants have been carried out (24, 25, 29–37). For example, Richardson and coworkers investigated the effects of amino acid substitutions of the five conserved glutamates (E122 and E125 presumed to face the periplasm and E198, E202, and E267 located in the interior of the membrane, close to the catalytic site) in the catalytic subunit of Paracoccus denitrificans, NorB. The E122A, E125A, E198A, and E267A variants were inactive, indicating that these four glutamates are crucial for NOR activity (24, 25, 32, 33). On the other hand, Reimann et al. constructed a 3D model of NorB using homology modeling with the structures of HCOs as templates and suggested a plausible pathway consisting of these conserved glutamates for proton delivery (22). Despite these successes, the roles of the conserved glutamates and metal ions still remain to be fully elucidated, partly because of the difficulty in obtaining native NOR in high yield and the lack of a 3D structure. Even if these problems are resolved, it is still difficult to replace iron in the native FeB site with other metal ions, and spectroscopic studies of native NOR are often complicated by the presence of other metal cofactors (e.g., low-spin heme).
To overcome these limitations, a number of synthetic models of NOR using small organic molecules as ligands, have been made in which the nonheme FeB site can be replaced by a copper ion (17, 38–45). In addition, since these model systems lack additional metal-binding sites, spectroscopic studies are often simplified. Therefore, studies of these synthetic models have offered many insights. For example, Collman et al. showed that a fully reduced heme/nonheme FeB compound can react with two equivalents of NO leading to the formation of one equivalent of N2O and a bis-ferric product (41). On the other hand, Karlin and coworkers showed that a small heme/Cu complex can efficiently lead to reductive coupling of NO to N2O (43). However, it is also difficult to obtain the synthetic models in high yield due to the multiple steps required in chemical synthesis. Because of this limitation, no synthetic NOR model containing the two key conserved Glu residues (E198 and E267 in NOR) has been reported. It is also difficult to substitute different metal ions in the same metal-binding site without perturbing the site geometry and distances to the heme iron, as most ligands are not as rigid as those in native enzymes and different metal ions have different geometric and ligand donor set preferences.
We have recently designed a structural and functional protein model of bacterial NOR by engineering three histidines and one glutamate into the distal pocket of sperm whale myoglobin (swMb, L29H, F43H, H64, and V68E, named FeBMb) (14). Like synthetic models, this “bottom-up” approach complements the “top-down” approach of the study of native NOR in that it provides insights into whether certain “necessary” structural elements are enough to impart enzyme function. Thanks in part to recent advances in computational, molecular, and structural biology, the designed myoglobin protein model is much easier to synthesize and to crystallize than either native NOR or synthetic models. Since myoglobin has often been used for the development and calibration of numerous spectroscopic techniques (46–48), it is an ideal choice for spectroscopic studies. More importantly, the rigid protein network allows precise placement of either glutamate or metal ions in myoglobin to address their roles in NOR activity. Toward this goal, we have demonstrated that both the histidines and one of the glutamates are essential for iron binding and NO reduction activity (14). However, the role of the second Glu close to the FeB site and the role of different metal ions in the FeB site have not been addressed.
To address these important issues and to design even closer protein models of NOR, we introduced herein the second Glu to the second coordination sphere of the FeB site by mutating an Ile to a Glu (named I107E FeBMb). We show that the second Glu results in a ∼100% increase in NOR activity through hydrogen bonding interactions and that the two glutamates have dramatically different effects on the heme reduction potential. Additionally, by comparing the EPR, electrochemistry, X-ray structures, and NOR activity of iron, copper, and zinc derivatives of the designed protein, we have obtained deeper insights into the roles of metal ions in NOR.
Results
Structure and Function of Fe(II)-I107E FeBMb.
The X-ray crystal structures of heme-containing I107E FeBMb without metal ion in the FeB site and with Fe2+ in the FeB site are solved at 1.42-Å and 1.65-Å resolution, respectively (Fig. 1 A and B and Table S1). In the absence of metal ions in the FeB site, the structure shows a water molecule in the FeB site, which forms hydrogen bonds with NE2 atoms of all three His residues, both OE1 and OE2 atoms of E68, and the OE2 atom of E107 (Fig. 1A). Upon binding Fe2+, the Fe(II)-I107E FeBMb structure shows that Fe2+ is coordinated by three His, the OE2 atom of E68, and one water molecule. Notably, a water molecule bridges Fe2+ in the FeB site and the second glutamate (E107) with a distance of 2.32 Å to the OE2 atom of E107 (Fig. 1B).
Fig. 1.

Crystal structures of I107E FeBMb (A) (PDB ID code 3M38), Fe(II)-I107E FeBMb (B) (PDB ID code 3M39), Cu(II)-I107E FeBMb (C) (PDB ID code 3M3A), and Zn(II)-I107E FeBMb (D) (PDB ID code 3M3B). Water molecules, Fe(II), Cu(II), and Zn(II) are represented by red, green, orange, and gray spheres, respectively.
To probe the conformational changes of introducing the second Glu (E107), we performed a structural alignment of Fe(II)-I107E FeBMb and the previously reported Fe(II)-FeBMb (14). The comparison, shown in Fig. 2, indicates that both the polypeptide chain and the active site overlap well with each other. In addition, the two nonheme irons are located at similar positions with a 0.36-Å separation from each other. In contrast, E68 underwent a significant conformational rearrangement in the presence of E107. These observations suggest that the active site of FeBMb can be tuned by the formation of an extended hydrogen bonding network, resulting from the introduction of a second glutamate residue.
Fig. 2.

Overlay of Fe(II)-I107E FeBMb (cyan) (PDB ID code 3M39) with Fe(II)-FeBMb (orange) (PDB ID code 3K9Z).
The binding of Fe2+ to deoxy I107E FeBMb was further monitored by EPR (Fig. 3A). Since deoxy myoglobin contains Fe(II) heme that exhibits no EPR signals in X-band EPR (14), we added blue copper Cu(II)-azurin (49), a redox partner of native NOR (19), to oxidize both the reduced heme and nonheme irons in Fe(II)-I107E FeBMb to EPR-active Fe(III). Upon addition of Cu(II)-azurin, the oxidation of deoxy I107E FeBMb resulted in EPR signals at g = 6.12 and 5.56, typical of a high-spin heme-Fe(III). Upon addition of Fe2+, however, a decrease of the heme-Fe(III) EPR signals was observed, indicating that the Fe2+, when bound to the FeB site and oxidized by Cu(II)-azurin, is spin-coupled to heme-Fe(III). Such a spin coupling mimics that in NOR (35, 50–53), suggesting that I107E FeBMb models NOR closely, at least in this respect.
Fig. 3.

EPR spectra of deoxy I107E FeBMb (0.5 mM protein in 50 mM Bis-Tris, pH 7.0) with increasing concentrations of Fe2+ in the presence of wild-type Cu(II)-azurin (A), and oxidized I107E FeBMb with Cu2+ (B) or Zn2+ (C). Spectra were collected at 4 K, 5 mW power, and 9.05 GHz.
To probe the role of the second Glu (E107) in NO reduction activity, we measured the yield of N2O production by Fe(II)-I107E FeBMb with excess NO under one turnover conditions. We monitored N2O formation in the headspace of the solution using GC/MS and compared this result to that of Fe(II)-FeBMb, which lacks the second Glu (Fig. 4). Remarkably, Fe(II)-I107E FeBMb displays higher activity than Fe(II)-FeBMb. After ∼20 hr, ∼24% N2O was produced by Fe(II)-I107E FeBMb, in contrast to ∼10% yield for Fe(II)-FeBMb, strongly indicating that the second Glu plays an important role in NO reduction, likely facilitating proton uptake during NO reduction.
Fig. 4.

Time-dependent N2O production by Fe(II)-I107E FeBMb (▴) and Fe(II)-FeBMb (●) with ∼50 eq. NO under single turnover conditions. The yield was determined by a comparison of the ratio of NO∶N2O peaks from the GC/MS chromatograms.
Other Metal Ions Binding to I107E FeBMb.
To find out if the resting state of the protein, i.e., oxidized or met I107E FeBMb, can bind other metal ions, Cu2+ or Zn2+ was titrated into met I107E FeBMb and monitored by EPR spectroscopy (Fig. 3 B and C). In the absence of metal ions, met I107E FeBMb exhibited high-spin heme signals at g = 6.03, 5.08, and 1.98 (Fig. 3B, black line). Upon addition of 2 eq of Cu2+, the signals at g = 6.03 and 5.08 decreased and a broad peak around g = 2.95 increased, probably due to spin coupling between heme-Fe(III) and Cu2+ in the FeB site. In contrast, addition of Zn2+, a metal ion with no unpaired electrons [i.e., incapable of spin coupling to heme-Fe(III)], produced an increase in the high-spin heme signals at g = 5.88 and 5.60 (Fig. 3C), indicating that the interaction between E68 and heme iron was weakened after metal binding.
The X-ray crystal structures of I107E FeBMb with Cu2+ or Zn2+ in the FeB site were solved at 1.37-Å and 1.60-Å resolution, respectively (Fig. 1 C and D and Table S1). Compared to Fe(II)-I107E FeBMb (Fig. 1B), a similar binding site was observed for Cu(II)-I107E FeBMb (Fig. 1C), where H29, H43, and H64 coordinate to Cu2+ with distances of 2.09, 2.10, and 2.04 Å, respectively, slightly shorter than the corresponding distances in the Fe2+ structure. In comparison to Fe(II)-I107E FeBMb, the water bridging the Cu2+ and the second Glu (E107) is shifted toward Cu2+ in the FeB site (2.03 Å) with respect to E107 (3.04 Å). Interestingly, this bridging water molecule was not observed in Zn(II)-I107E FeBMb (Fig. 1D), but the two O atoms of E68 coordinate to Zn2+ with similar distances (2.26 Å for OE1 and 2.29 Å for OE2). The longer distance between OE1 of E68 and heme iron in the Zn-bound structure (2.68 Å) in comparison to the Cu- and Fe-bound structures, is likely the result of a weaker interaction, which is also supported by an observed increase of the high-spin heme signals in the EPR spectra upon Zn2+ binding (Fig. 3C). These results suggest I107E FeBMb is capable of incorporating different metal ions into its designed FeB site, offering an excellent opportunity to compare the role of these metal ions in the same protein scaffold.
Effect of Glutamates and Metal Ions on the Redox Potential of I107E FeBMb.
Since EPR and X-ray structural studies indicate metal binding to I107E FeBMb, we used spectroelectrochemistry to measure the effects of glutamates and metal ions on the heme reduction potential. When there is no metal ion in the FeB site, the I107E FeBMb displays a reduction potential of -134 ± 3 mV vs. the normal hydrogen electrode (NHE) (Fig. S1A), similar to that of FeBMb (-158 ± 4 mV) without the I107E mutation (14). In the presence of Cu2+, I107E FeBMb has a reduction potential (-137 ± 2 mV) (Fig. S1B) almost identical to that of the same protein in the absence of metal ions in the FeB site, indicating that copper binding to the FeB site has little effect on the reduction potential of the heme iron. This observation is similar to that observed for Cu2+ binding to CuBMb (Cu(II)-CuBMb, 80 mV vs. CuBMb, 77 mV) (54). On the other hand, the presence of Fe2+ and Zn2+ increased the reduction potential of I107E FeBMb from -134 ± 3 mV to -64 ± 3 mV vs. NHE (Fig. S1C) and -105 ± 2 mV vs. NHE (Fig. S1D), respectively. The different effects of Cu2+, Fe2+, and Zn2+ on the reduction potential of I107E-FeBMb indicate that these metal ions in the FeB site may play different roles through different coordination properties.
NOR Activity of I107E FeBMb in the Presence of Different Metal Ions.
The NO reduction activity of I107E FeBMb in the presence of Fe2+, Cu+, or Zn2+ was monitored by GC/MS under single turnover conditions. When Fe(II)-I107E FeBMb was exposed to excess NO, N2O could be observed to form with increased yield over time (Fig. S2). Similarly, N2O formation was observed for Cu(I)-I107E FeBMb, indicating that Fe or Cu binding to the FeB site results in comparable NOR activities. It should be noted that because of the high solubility of N2O (∼25 mM in water at room temperature), GC/MS cannot be used to quantify the rates of NO reduction under these conditions. In contrast, no N2O formation was observed with redox inactive Zn2+, which demonstrates that redox active Fe2+ or Cu+ in the FeB site plays a crucial role in NO reduction.
To gain deeper insight into the process of NO reduction, EPR studies were further performed to monitor the initial process of NO reduction. In the absence of metal ions, the EPR spectrum of ferrous I107E FeBMb-NO shows hyperfine splitting resulting from bound NO and the proximal histidine, indicating the formation of a six-coordinate ferrous heme-NO species (Fig. 5, top line). After incubation of Fe(II)-I107E FeBMb with excess NO, a distinct three-line hyperfine structure appears at 15 min (Fig. 5A), suggesting the formation of a five-coordinate ferrous heme-NO species as a result of cleavage of the proximal His-Fe heme bond (55). A three-line hyperfine structure was also observed for Cu+ and Zn2+, except that the signal intensity is lower with Cu(I)-I107E FeBMb-NO (Fig. 5B) and more pronounced in Zn(II)-I107E FeBMb-NO (Fig. 5C). The lower intensity of the three-line hyperfine structure for Cu(I)-I107E FeBMb-NO suggests the major species formed is a six-coordinate ferric heme-NO complex, which is EPR silent (41). These differences further suggest that the metal ion in the FeB site plays a key role in formation of the intermediates, thereby tuning NOR activity.
Fig. 5.

EPR spectra of deoxy I107E FeBMb (0.5 mM) with no metal bound in the presence of NO after 5 min (top line), with 2 eq Fe2+ (A), Cu+ (B), or Zn2+ (C) incubated with excess NO (∼200 eq) for 1, 5, and 15 min. Spectra were collected in 50 mM Bis-Tris pH 7.0 at 30 K, 0.2 mW power, and 9.05 GHz.
Discussion
Using Rationally Designed Proteins to Address Important Issues in Chemistry and Biology.
Important issues such as the roles of the conserved glutamates and nonheme FeB in NOR have been previously addressed using biochemical and biophysical studies or biomimetic modeling (24, 25, 27–37, 45, 56, 57). As a complementary approach, rational protein design, using small, easy-to-produce and well-characterized proteins such as myoglobin, offers a powerful method with which to gain insights into more complex native enzymes such as NOR (14). Similar to synthetic models (41, 43), the metal ion at the putative FeB site in the protein model can be substituted freely. Better yet, Glu residues can be placed at precise locations in the protein, including the secondary coordination sphere, due to its rigid network. By carefully choosing a suitable protein template, rational protein design could be generally applied to address other important issues in chemistry and biology.
The Roles of Glutamates.
Although two conserved glutamates (E198 and E267) are known to be crucial for NOR activity (24, 25), their roles are not well defined (18, 19). In a previous study (14), we demonstrated that one Glu, E68, is important for both iron binding and NOR activity of FeBMb. The crystal structures of both Fe(II)-FeBMb and Fe(II)-I107E FeBMb show that one O atom of E68 directly coordinates to FeB (Fig. 2). In synthetic models of NOR, it has also been found that the presence of a glutamic acid mimic significantly increases the stability of iron binding to the FeB site (40). Furthermore, a theoretical study by Blomberg et al. (58) showed that a model with an FeB coordinated by three histidines, one glutamate, and one water molecule provides an energetically feasible reaction mechanism of NO reduction. However, the structural model of NOR constructed recently by Reimann et al. (22) shows that the closest conserved Glu (E267) still has its carboxylate O atom 7 Å away from FeB, which suggests that Glu may not bind to FeB in native NOR. One interesting finding from our study is that the Glu (E68) underwent a significant conformational rearrangement in the presence of another Glu (E107) (Fig. 2). Therefore, the FeBMb provides a viable model of NOR that is consistent with Blomberg’s model, but cannot rule out Reimann’s model due to possible conformation changes.
While the role of the first Glu is still uncertain until a 3D structure of NOR in its active form is available, the role of the second Glu is even less defined. We address this question by introducing a second Glu (E107) to FeBMb. The crystal structures shown in Fig. 1 indicate that E107 interacts with a water molecule and forms a hydrogen bonding network in both Fe(II)-I107E FeBMb and Cu(II)-I107E FeBMb. Interestingly, although a similar water molecule was observed in the active site of Fe(II)-FeBMb (Fig. 2), activity assay data indicate that the presence of E107 in Fe(II)-I107E FeBMb increases NOR activity by ∼100% (Fig. 4), suggesting that the second Glu may potentionally play a role in providing one of the protons during reduction of NO to N2O. Although free Glu outside a protein has a pKa ∼ 4.3, the studies of native NOR showed that the pKa of its Glu close to the active site has a value of ∼6.6 (22, 25, 33). The hydrogen binding network in our protein models may contribute to the fine-tuning of the Glu pKa to be more neutral, similar to that in NOR. Moreover, it is interesting that Cu(I)-I107E FeBMb also shows NOR activity, which provides an interesting protein model of HCOs with NOR function (26–28), even though Glu residues are not conserved in native HCOs.
Additionally, spectroelectrochemical studies showed that the reduction potential of I107E FeBMb with no metal ion in the FeB site is similar to that of FeBMb, but much lower than that of CuBMb (77 mV) (54), which contains the same three His, but no Glu in the metal-binding site above the heme. Since both FeBMb and I107E FeBMb contain the V68E mutation that has been shown to decrease the heme reduction potential of native myoglobin from 59 mV to -137 mV (59), it is likely that the introduction of a negatively charged Glu close to the heme group is what is responsible for the dramatically lower heme redox potential. A conserved glutamate, predicted to be located near the catalytic heme b3 in NOR, was proposed to be responsible for a ∼260-mV decrease in reduction potential (60 mV) in comparison to the other two heme centers, heme b (345 mV) and heme c (310 mV) (60). Our FeBMb and I107E FeBMb models mimic this feature of NOR. Notably, although introduction of the first Glu (E68) lowered the heme potential by ∼110 mV (14), introduction of the second Glu via the I107E mutation did not result in a significant difference in the heme reduction potential, suggesting that the effect of the two conserved Glu residues in NOR on heme reduction potential is not additive, with the effects highly dependent on the location of the Glu.
The Roles of Metal Ions.
The roles of metal ions in NOR are another important question as iron is found in the native FeB site and HCO employs copper at the corresponding CuB site. With different metal ions in the FeB site, the crystal structures clearly show the heme and nonheme dinuclear center existing in different local environments (Fig. 1). Although a similar hydrogen bond network is formed in both Fe(II)-I107E FeBMb and Cu(II)-I107E FeBMb, the conformation of E68 and E107 with respect to the nonheme metal center and heme iron is different from each other. Moreover, the coordination geometry differs significantly with Zn2+ in the FeB site. A hydrogen bond is absent from the Zn crystal structure, but both the O atoms of E68 act as metal-binding ligands. These observations demonstrate that the identity of the metal ion in the FeB site can tune the active site through their interactions with the His and Glu ligands, resulting in formation of different coordination geometries with different hydrogen bonds.
In addition to structural fine-tuning, the metal ion at the FeB site can also tune the heme iron reduction potential in I107E FeBMb. Spectroelectrochemical studies showed that the binding of Fe2+ or Zn2+ results in an increase in the heme reduction potential by ∼70 mV and ∼30 mV, respectively (Fig. S1). In the case of Cu(II)-I107E FeBMb, the crystal structure shows that OE1 of E68 is closer to the heme iron (2.07 Å) (Fig. 1C) than its metal-free form (2.15 Å) (Fig. 1A). The stronger interaction from the negatively charged E68 could offset the effect of positively charged Cu2+ binding, resulting in similar reduction potentials observed for Cu(II)-I107E FeBMb and I107E FeBMb.
In a previous study (61), EPR data showed that during NO reduction, the binding of Cu+ to the CuB site of CuBMb can weaken the proximal heme Fe-His bond, while complete cleavage of the heme Fe-His bond occurred when Zn2+ was bound to CuBMb-NO. In this study, we observed that a five-coordinate heme-NO species was formed with Fe2+, Cu+, or Zn2+ bound to the FeB site of I107E FeBMb (Fig. 5). Significantly, a five-coordinate heme-NO species has also been observed for both NOR (30, 31, 35) and the member of the HCO family with the highest NO reduction activity, cytochrome cbb3 oxidases (26, 62). However, this species was not observed for Fe(II)-FeBMb-NO and Cu(I)-FeBMb-NO, which lack the second Glu (E107). In both these cases, the proximal heme Fe-His bond was only weakened, as indicated by a decrease of the nine-line hyperfine splitting signals in the EPR spectra (Fig. S3). These observations suggest that formation of a five-coordinate heme-NO species may play an important role in NOR reactivity.
Conclusions
We have successfully designed a structural and functional model of NOR, by introducing a second glutamate in the vicinity of the FeB site, named I107E FeBMb. This protein model mimics native NOR more closely by bearing the structural feature of three histidines and two glutamates in the FeB site, as predicted for native NOR. We have demonstrated that the two glutamates can play different roles in NO reduction activity; namely, one acts as a ligand to FeB (E68), and the other acts as a proton transfer group (E107). Furthermore, by substituting different metal ions into the nonheme metal site, we have demonstrated that FeB plays crucial roles in fine-tuning the active site by donating electrons and by mediating the formation of a five-coordinate heme-NO intermediate during NO reduction. In the absence of a crystal structure for native NOR, this study offers an ideal protein model and provides valuable structural as well as mechanistic information for native NOR.
Materials and Methods
Protein Preparation.
I107E FeBMb (swMb L29H/F43H/V68E/I107E) was constructed, expressed, and purified using the procedure described previously (14). The purity and identity were confirmed by SDS-PAGE and electrospray ionized MS: observed: 17,392 ± 1 Da; calculated: 17,391 Da.
EPR Spectroscopy.
EPR spectra were recorded on a Bruker ESP 300 equipped with an Oxford liquid helium cryostat and an ITC4 temperature controller. The samples of met I107E FeBMb, Cu(II)-, or Zn(II)-I107E FeBMb were prepared as described previously (14). The samples of NO-bound deoxy I107E FeBMb, Cu(I)-, Fe(II)-, or Zn(II)-I107E FeBMb were prepared by injecting 1 mL of purified NO gas into the EPR tube containing 300 μL of protein (0.5 mM, 10% glycerol, in 50 mM Bis-Tris pH 7.0). The samples were then flash frozen in liquid N2 after incubation for 1, 5, or 15 min. The molar extinction coefficient of the Soret band of I107E FeBMb at 406 nm (175 mM-1·cm-1), calculated using the standard hemochromagen method (63), was used to determine protein concentration. The metal sources of Cu(I), Cu(II), Zn(II), and Fe(II) were [(CH3CN)4Cu]PF6, CuSO4, ZnSO4·7H2O, and FeCl2, respectively.
Spectroelectrochemical Measurements.
Protein reduction potentials were measured using an optically transparent thin layer electrode as previously described (64). The potential of the working electrode was applied in the positive direction for deoxy I107E FeBMb with Fe2+ and in the negative direction for metal free and with Cu2+ or Zn2+. Other procedures are the same as described previously (54).
X-Ray Crystallographic Studies.
Fe(II)-I107E FeBMb was crystallized anaerobically in a glove box at room temperature using the conditions described for Fe(II)-FeBMb (14). I107E FeBMb, Cu(II)-, or Zn(II)-I107E FeBMb were crystallized aerobically. Diffraction-quality crystals were soaked in a cryoprotectant solution of 30% PEG 400 and flash frozen in liquid nitrogen. Diffraction data were collected at the Brookhaven National Lab Synchrotron Light Source X12C beamline. The crystal structure was solved using the same method as for Fe(II)-FeBMb (14).
NOR Activity Assay.
NO reduction was monitored by GC/MS. The protein was reduced to the deoxy form by excess dithionite that was removed with a size-exclusion column (PD-10). Then 2 eq metal, Cu(I), Fe(II), or Zn(II), was added to the protein solution (0.6 mM, 3 mL in 50 mM Bis-Tris buffer, pH 7.0). The samples were prepared anaerobically in a glove box. Purified NO gas was injected into the head space of the reaction flask with the molar ratio of NO∶protein ≈ 50∶1. Other procedures are the same as described previously (14, 61).
Supplementary Material
Acknowledgments.
We thank Dr. Mark J. Nilges for help with EPR analysis, and Furong Sun and Beth D. Eves for aiding in GC/MS data collection. This work was supported by NIH Grant GM062211.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.pdb.org (PDB ID codes 3M38, 3M39, 3M3A, and 3M3B).
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1000526107/-/DCSupplemental.
References
- 1.Lu Y, Yeung N, Sieracki N, Marshall NM. Design of functional metalloproteins. Nature. 2009;460:855–862. doi: 10.1038/nature08304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nanda V, Koder RL. Designing artificial enzymes by intuition and computation. Nat Chem. 2010;2:15–24. doi: 10.1038/nchem.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gray HB. Biological inorganic chemistry at the beginning of the 21st century. Proc Natl Acad Sci USA. 2003;100:3563–3568. doi: 10.1073/pnas.0730378100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Regan L, DeGrado WF. Characterization of a helical protein designed from first principles. Science. 1988;241:976–978. doi: 10.1126/science.3043666. [DOI] [PubMed] [Google Scholar]
- 5.Hecht MH, Richardson JS, Richardson DC, Ogden RC. De novo design, expression, and characterization of Felix: A four-helix bundle protein of native-like sequence. Science. 1990;249:884–891. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- 6.Robertson DE, et al. Design and synthesis of multi-heme proteins. Nature. 1994;368:425–432. doi: 10.1038/368425a0. [DOI] [PubMed] [Google Scholar]
- 7.Lu Y, Valentine JS. Engineering metal-binding sites in proteins. Curr Opin Struct Biol. 1997;7:495–500. doi: 10.1016/s0959-440x(97)80112-1. [DOI] [PubMed] [Google Scholar]
- 8.Reedy CJ, Gibney BR. Heme protein assemblies. Chem Rev. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- 9.Watanabe Y, Hayashi T. Functionalization of myoglobin. Prog Inorg Chem. 2005;54:449–493. [Google Scholar]
- 10.Ghosh D, Pecoraro VL. Probing metal-protein interactions using a de novo design approach. Curr Opin Chem Biol. 2005;9:97–103. doi: 10.1016/j.cbpa.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 11.Kaplan J, DeGrado WF. De novo design of catalytic proteins. Proc Natl Acad Sci USA. 2004;101:11566–11570. doi: 10.1073/pnas.0404387101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kang SG, Saven JG. Computational protein design: Structure, function and combinatorial diversity. Curr Opin Chem Biol. 2007;11:329–334. doi: 10.1016/j.cbpa.2007.05.006. [DOI] [PubMed] [Google Scholar]
- 13.Jiang L, et al. De novo computational design of retro-aldol enzymes. Science. 2008;319:1387–1391. doi: 10.1126/science.1152692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yeung N, et al. Rational design of a structural and functional nitric oxide reductase. Nature. 2009;462:1079–1082. doi: 10.1038/nature08620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Koder RL, et al. Design and engineering of an O2 transport protein. Nature. 2009;458:305–309. doi: 10.1038/nature07841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinisch T, Ward TR. Design strategies for the creation of artifical metalloenzymes. Curr Opin Chem Biol. 2010;14:184–199. doi: 10.1016/j.cbpa.2009.11.026. [DOI] [PubMed] [Google Scholar]
- 17.Wasser IM, de Vries S, Moënne-Loccoz P, Schröder I, Karlin KD. Nitric oxide in biological denitrification: Fe/Cu metalloenzyme and metal complex NOx redox chemistry. Chem Rev. 2002;102:1201–1234. doi: 10.1021/cr0006627. [DOI] [PubMed] [Google Scholar]
- 18.Yeung N, Lu Y. One heme, diverse functions: Using biosynthetic myoglobin models to gain insights into heme-copper oxidases and nitric oxide reductases. Chem Biodivers. 2008;5:1437–1454. doi: 10.1002/cbdv.200890134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watmough NJ, Field SJ, Hughes RJL, Richardson DJ. The bacterial respiratory nitric oxide reductase. Biochem Soc Trans. 2009;37:392–399. doi: 10.1042/BST0370392. [DOI] [PubMed] [Google Scholar]
- 20.Cary SPL, Winger JA, Derbyshire ER, Marletta MA. Nitric oxide signaling: No longer simply on or off. Trends Biochem Sci. 2006;31:231–239. doi: 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 21.Sakurai N, Sakurai T. Genomic DNA cloning of the region encoding nitric oxide reductase in paracoccus halodenitrificans and a structure model relevant to cytochrome oxidase. Biochem Biophys Res Commun. 1998;243:400–406. doi: 10.1006/bbrc.1998.8106. [DOI] [PubMed] [Google Scholar]
- 22.Reimann J, Flock U, Lepp H, Honigmann A, Ädelroth P. A pathway for protons in nitric oxide reductase from paracoccus denitrificans. Biochim Biophys Acta. 2007;1767:362–373. doi: 10.1016/j.bbabio.2007.03.006. [DOI] [PubMed] [Google Scholar]
- 23.Pereira MM, Sousa FL, Verissimo AF, Teixeira M. Looking for the minimum common denominator in haem-copper oxygen reductases: Towards a unified catalytic mechanism. Biochim Biophys Acta. 2008;1777:929–934. doi: 10.1016/j.bbabio.2008.05.441. [DOI] [PubMed] [Google Scholar]
- 24.Butland G, Spiro S, Watmough NJ, Richardson DJ. Two conserved glutamates in the bacterial nitric oxide reductase are essential for activity but not assembly of the enzyme. J Bacteriol. 2001;183:189–199. doi: 10.1128/JB.183.1.189-199.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flock U, Lachmann P, Reimann J, Watmough NJ, Äedelroth P. Exploring the terminal region of the proton pathway in the bacterial nitric oxide reductase. J Inorg Biochem. 2009;103:845–850. doi: 10.1016/j.jinorgbio.2009.02.008. [DOI] [PubMed] [Google Scholar]
- 26.Forte E, et al. The cytochrome cbb3 from pseudomonas stutzeri displays nitric oxide reductase activity. Eur J Biochem. 2001;268:6486–6490. doi: 10.1046/j.0014-2956.2001.02597.x. [DOI] [PubMed] [Google Scholar]
- 27.Huang Y, Reimann J, Lepp H, Drici N, Ädelroth P. Vectorial proton transfer coupled to reduction of O2 and NO by a heme-copper oxidase. Proc Natl Acad Sci USA. 2008;105:20257–20262. doi: 10.1073/pnas.0805429106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hayashi T, et al. Accommodation of two diatomic molecules in cytochrome bo3: Insights into NO reductase activity in terminal oxidases. Biochemistry. 2009;48:883–890. doi: 10.1021/bi801915r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moënne-Loccoz P. Spectroscopic characterization of heme iron-nitrosyl species and their role in NO reductase mechanisms in diiron proteins. Nat Prod Rep. 2007;24:610–620. doi: 10.1039/b604194a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Moënne-Loccoz P, de Vries S. Structural characterization of the catalytic high-spin heme b of nitric oxide reductase: A resonance raman study. J Am Chem Soc. 1998;120:5147–5152. [Google Scholar]
- 31.Sakurai T, Sakurai N, Matsumoto H, Hirota S, Yamauchi O. Roles of four iron centers in paracoccus halodenitrificans nitric oxide reductase. Biochem Biophys Res Commun. 1998;251:248–251. doi: 10.1006/bbrc.1998.9451. [DOI] [PubMed] [Google Scholar]
- 32.Thorndycroft FH, Butland G, Richardson DJ, Watmough NJ. A new assay for nitric oxide reductase reveals two conserved glutamate residues form the entrance to a proton-conducting channel in the bacterial enzyme. Biochem J. 2007;401:111–119. doi: 10.1042/BJ20060856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Flock U, et al. Defining the proton entry point in the bacterial respiratory nitric-oxide reductase. J Biol Chem. 2008;283:3839–3845. doi: 10.1074/jbc.M704615200. [DOI] [PubMed] [Google Scholar]
- 34.Hendriks JHM, Jasaitis A, Saraste M, Verkhovsky MI. Proton and electron pathways in the bacterial nitric oxide reductase. Biochemistry. 2002;41:2331–2340. doi: 10.1021/bi0121050. [DOI] [PubMed] [Google Scholar]
- 35.Kumita H, et al. NO reduction by nitric-oxide reductase from denitrifying bacterium pseudomonas aeruginosa: Characterization of reaction intermediates that appear in the single turnover cycle. J Biol Chem. 2004;279:55247–55254. doi: 10.1074/jbc.M409996200. [DOI] [PubMed] [Google Scholar]
- 36.Pinakoulaki E, Varotsis C. Resonance raman spectroscopy of nitric oxide reductase and cbb3 heme-copper oxidase. J Phys Chem B. 2008;112:1851–1857. doi: 10.1021/jp077295o. [DOI] [PubMed] [Google Scholar]
- 37.Kapetanaki SM, et al. Ultrafast ligand binding dynamics in the active site of native bacterial nitric oxide reductase. Biochim Biophys Acta. 2008;1777:919–924. doi: 10.1016/j.bbabio.2008.03.012. [DOI] [PubMed] [Google Scholar]
- 38.Wasser IM, et al. Synthesis and spectroscopy of m-oxo (O2-)-bridged Heme/Non-heme diiron complexes: Models for the active site of nitric oxide reductase. Inorg Chem. 2004;43:651–662. doi: 10.1021/ic0348143. [DOI] [PubMed] [Google Scholar]
- 39.Wasser IM, Huang H, Moënne-Loccoz P, Karlin KD. Heme/Non-heme diiron(II) complexes and O2, CO, and NO adducts as reduced and substrate-bound models for the active site of bacterial nitric oxide reductase. J Am Chem Soc. 2005;127:3310–3320. doi: 10.1021/ja0458773. [DOI] [PubMed] [Google Scholar]
- 40.Collman JP, Yan Y, Lei J, Dinolfo PH. Active-site models of bacterial nitric oxide reductase featuring tris-histidyl and glutamic acid mimics: Influence of a carboxylate ligand on FeB binding and the heme Fe/FeB redox potential. Inorg Chem. 2006;45:7581–7583. doi: 10.1021/ic0609150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collman JP, et al. A functional nitric oxide reductase model. Proc Natl Acad Sci USA. 2008;105:15660–15665. doi: 10.1073/pnas.0808606105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collman JP, et al. Intermediates involved in the two electron reduction of NO to N2O by a functional synthetic model of heme containing bacterial NO reductase. J Am Chem Soc. 2008;130:16498–16499. doi: 10.1021/ja807700n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang J, Schopfer MP, Sarjeant Amy AN, Karlin KD. Heme-copper assembly mediated reductive coupling of nitrogen monoxide (*NO) J Am Chem Soc. 2009;131:450–451. doi: 10.1021/ja8084324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Liu J, Naruta Y, Tani F. A functional model of the cytochrome c oxidase active site: Unique conversion of a heme-m-peroxo-CuII intermediate into heme-superoxo/CuI. Angew Chem Int Edit. 2005;44:1836–1840. doi: 10.1002/anie.200462582. [DOI] [PubMed] [Google Scholar]
-
45.Ghiladi RA, et al. Heme-copper/dioxygen adduct formation relevant to cytochrome c oxidase: Spectroscopic characterization of
 . J Biol Inorg Chem. 2005;10:63–77. doi: 10.1007/s00775-004-0609-1. [DOI] [PubMed] [Google Scholar]
. J Biol Inorg Chem. 2005;10:63–77. doi: 10.1007/s00775-004-0609-1. [DOI] [PubMed] [Google Scholar] - 46.Sage JT. Myoglobin and CO: Structure, energetics, and disorder. J Biol Inorg Chem. 1997;2:537–543. [Google Scholar]
- 47.Sigman JA, Kim HK, Zhao X, Carey JR, Lu Y. The role of copper and protons in heme-copper oxidases: Kinetic study of an engineered heme-copper center in myoglobin. Proc Natl Acad Sci USA. 2003;100:3629–3634. doi: 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Davydov R, Hoffman BM. EPR and ENDOR studies of fe(II) hemoproteins reduced and oxidized at 77 K. J Biol Inorg Chem. 2008;13:357–369. doi: 10.1007/s00775-007-0328-5. [DOI] [PubMed] [Google Scholar]
- 49.Marshall NM, et al. Rationally tuning the reduction potential of a single cupredoxin beyond the natural range. Nature. 2009;462:113–116. doi: 10.1038/nature08551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Girsch P, de Vries S. Purification and initial kinetic and spectroscopic characterization of NO reductase from paracoccus denitrificans. Biochim Biophys Acta. 1997;1318:202–216. doi: 10.1016/s0005-2728(96)00138-7. [DOI] [PubMed] [Google Scholar]
- 51.Sakurai N, Sakurai T. Isolation and characterization of nitric oxide reductase from paracoccus halodenitrificans. Biochemistry. 1997;36:13809–13815. doi: 10.1021/bi971070u. [DOI] [PubMed] [Google Scholar]
- 52.Hendriks J, et al. The active site of the bacterial nitric oxide reductase is a dinuclear iron center. Biochemistry. 1998;37:13102–13109. doi: 10.1021/bi980943x. [DOI] [PubMed] [Google Scholar]
- 53.Moënne-Loccoz P, et al. Nitric oxide reductase from paracoccus denitrificans contains an oxo-bridged Heme/Non-heme diiron center. J Am Chem Soc. 2000;122:9344–9345. [Google Scholar]
- 54.Zhao X, Yeung N, Wang Z, Guo Z, Lu Y. Effects of metal ions in the CuB center on the redox properties of heme in heme-copper oxidases: Spectroelectrochemical studies of an engineered heme-copper center in myoglobin. Biochemistry. 2005;44:1210–1214. doi: 10.1021/bi0479151. [DOI] [PubMed] [Google Scholar]
- 55.Decatur SM, et al. Trans effects in nitric oxide binding to myoglobin cavity mutant H93G. Biochemistry. 1996;35:4939–4944. doi: 10.1021/bi951661p. [DOI] [PubMed] [Google Scholar]
- 56.Pervitsky D, Immoos C, van der Veer W, Farmer PJ. Photolysis of the HNO adduct of myoglobin: Transient generation of the aminoxyl radical. J Am Chem Soc. 2007;129:9590–9591. doi: 10.1021/ja073420y. [DOI] [PubMed] [Google Scholar]
- 57.Berto TC, Praneeth VKK, Goodrich LE, Lehnert N. Iron-porphyrin NO complexes with covalently attached N-donor ligands: Formation of a stable six-coordinate species in solution. J Am Chem Soc. 2009;131:17116–17126. doi: 10.1021/ja904368n. [DOI] [PubMed] [Google Scholar]
- 58.Blomberg LM, Blomberg MRA, Siegbahn PEM. Reduction of nitric oxide in bacterial nitric oxide reductase-a theoretical model study. Biochim Biophys Acta. 2006;1757:240–252. doi: 10.1016/j.bbabio.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 59.Varadarajan R, Zewert TE, Gray HB, Boxer SG. Effects of buried ionizable amino acids on the reduction potential of recombinant myoglobin. Science. 1989;243:69–72. doi: 10.1126/science.2563171. [DOI] [PubMed] [Google Scholar]
- 60.Grönberg KLC, et al. A low-redox potential heme in the dinuclear center of bacterial nitric oxide reductase: Implications for the evolution of energy-conserving heme-copper oxidases. Biochemistry. 1999;38:13780–13786. doi: 10.1021/bi9916426. [DOI] [PubMed] [Google Scholar]
- 61.Zhao X, Yeung N, Russell BS, Garner DK, Lu Y. Catalytic reduction of NO to N2O by a designed heme copper center in myoglobin: Implications for the role of metal ions. J Am Chem Soc. 2006;128:6766–6767. doi: 10.1021/ja058822p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pinakoulaki E, Stavrakis S, Urbani A, Varotsis C. Resonance raman detection of a ferrous five-coordinate nitrosylheme b3 complex in cytochrome cbb3 oxidase from pseudomonas stutzeri. J Am Chem Soc. 2002;124:9378–9379. doi: 10.1021/ja0271633. [DOI] [PubMed] [Google Scholar]
- 63.Morrison M, Horie S. Determination of heme a concentration in cytochrome preparations by hemochromogen method. Anal Biochem. 1965;12:77–82. doi: 10.1016/0003-2697(65)90144-2. [DOI] [PubMed] [Google Scholar]
- 64.Taboy CH, Bonaventura C, Crumbliss AL. Anaerobic oxidations of myoglobin and hemoglobin by spectroelectrochemistry. Method Enzymol. 2002;353:187–209. doi: 10.1016/s0076-6879(02)53048-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.