

Abstract
Francisella tularensis is a highly virulent, intracellular pathogen that causes the disease tularaemia. A research surrogate for F. tularensis is Francisella novicida, which causes a tularaemia-like disease in mice, grows similarly in macrophages, and yet is unable to cause disease in humans. Both Francisella species contain a cluster of genes referred to as the Francisella pathogenicity island (FPI). Pathogenicity determinant protein A (PdpA), encoded by the pdpA gene, is located within the FPI and has been associated with the virulence of Francisella species. In this work we examined the properties of PdpA protein expression and localization as well as the phenotype of a F. novicida pdpA deletion mutant. Monoclonal antibody detection of PdpA showed that it is a soluble protein that is upregulated in iron-limiting conditions and undetectable in an mglA or mglB mutant background. Deletion of pdpA resulted in a strain that was highly attenuated for virulence in chicken embryos and mice.
INTRODUCTION
Francisella tularensis is a Category A bioterrorism threat and is the causative agent of the zoonotic disease tularaemia. The closely related bacterium Francisella novicida is unable to cause disease in healthy individuals; however, it causes a disease in mice that is very similar to human tularaemia and, as such, is a useful tool for studying Francisella virulence factors. Both F. tularensis and F. novicida grow vigorously in macrophages, with a life cycle that includes transient localization in a phagosome followed by growth in the host-cell cytosol (Anthony et al., 1991; Clemens et al., 2004; Fortier et al., 1994; Golovliov et al., 2003a; Proctor et al., 1975). Little information is available concerning the function of virulence factors that contribute to F. tularensis infection. Many of the genes that have been shown to be necessary for intracellular growth and virulence are located within the recently discovered Francisella pathogenicity island (FPI, see Fig. 1) (Nano et al., 2004; Nano & Schmerk, 2007). The FPI is about 28 kb in length and contains 16–19 genes, depending on the biotype and strain. Recent bioinformatics analysis indicates that several genes within the FPI encode products that are homologues of components of type VI secretion systems (T6SSs) (de Bruin et al., 2007; Ludu et al., 2008a). F. novicida contains one copy of the FPI whereas F. tularensis strains that have been examined contain two copies of the genomic island (Nano & Schmerk, 2007).
Fig. 1.

Deletion of pdpA. (a) The pdpA gene is the first cistron in a low-G+C operon of the Francisella pathogenicity island (FPI). A newly revised nomenclature is used to show the two convergent operons in the FPI. (b) Primers that amplify regions within the pdpA gene were used to screen wild-type (WT) F. novicida, the ΔpdpA mutant and the in cis complement ΔpdpA/SKX : : pdpA. (c) The exact nucleotides removed to create the ΔpdpA mutant are shown. The in-frame deletion preserves the last 21 nucleotides of pdpA, which contain the ribosome-binding site (shown in italics) of the downstream gene pdpB.
Disruptions of a number of genes within the FPI have been shown to severely reduce intracellular bacterial growth (de Bruin et al., 2007; Golovliov et al., 2003b; Gray et al., 2002; Lai et al., 2004; Lauriano et al., 2004; Nano et al., 2004; Santic et al., 2005; Tempel et al., 2006; Weiss et al., 2007). However, in most cases the phenotype of mutants could not be unequivocally demonstrated to be the result of the inactivation of a specific gene, since polarity effects were not studied nor genetic complementation performed. It is probable that most FPI genes contribute to intracellular growth and virulence, and deletion mutagenesis and complementation studies have provided strong evidence that iglA and iglC, two of the genes in the anmK-iglD operon (see Fig. 1) are absolutely required for intramacrophage growth.
Regulation of FPI gene expression has been linked to several factors. MglA is the best studied of the Francisella global regulators (Baron & Nano, 1998; Brotcke et al., 2006; Charity et al., 2007). Studies of gene regulation in Francisella have revealed that FPI-encoded mRNA species are depressed in an mglA mutant. The MglA protein has been shown to bind RNA polymerase complexes, but its direct role in affecting mRNA levels of FPI genes has not yet been established. Low iron concentrations also increase the level of mRNA for several FPI-encoded mRNA transcripts (Deng et al., 2006). Mass spectroscopy analysis has shown that IglA, IglB, IglC, IglD and PdpB are all increased in low-iron conditions (Deng et al., 2006; Lenco et al., 2007). Immunoblot analysis has shown that both iron concentration and MglA play a role in IglA expression. It is clear that other regulatory proteins, such as FevR, SspA and PmrA, also affect expression of FPI genes (Brotcke & Monack, 2008; Charity et al., 2007; Mohapatra et al., 2007).
The pdpA gene is 2.46 kb in size, representing one of the largest ORFs in the FPI (Nano & Schmerk, 2007). A gene replacement in pdpA was made as part of the initial characterization of the FPI, and the resulting mutant was unable to grow in macrophages and was avirulent in mice (Nano et al., 2004). We now know that the replacement mutant had polar effects on downstream genes, resulting in decreased expression of PdpB and increased attenuation in chicken embryos (this work). It is likely that the in trans complementing plasmid recreated the intact operon by integration into the chromosome. In this paper we examine the expression and characteristics of the PdpA protein as well as the phenotype of a non-polar deletion mutant of pdpA.
METHODS
Bacterial strains and growth conditions.
The bacterial strains and plasmids used in this study are listed in Table 1. Escherichia coli strains were grown using Luria Bertani (LB) broth or agar supplemented with 250 μg ampicillin ml−1, 30 μg kanamycin ml−1 or 100 μg erythromycin ml−1, as required. F. novicida and F. tularensis strains were grown using trypticase soy agar or broth supplemented with 0.1 % (w/v) cysteine (TSAC, TSBC). When necessary, 30 μg erythromycin ml−1 or 15 μg kanamycin ml−1 was added. For negative selection in deletion mutagenesis experiments, sucrose was added to TSAC to a final concentration of 10 % (w/v).
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Characteristics | Source or reference |
|---|---|---|
| Strains | ||
| U112 | Wild-type F. novicida | ATCC 15482 |
| JLO | U112 with deletion in FTN_1390, where SKX vector inserts; identical growth and virulence with respect to U112 | Ludu et al. (2008b) |
| GB2 | U112 with point mutation in global virulence regulator, mglA | Baron & Nano (1998) |
| GB6 | U112, mglB : : mTn10KmR | Baron & Nano (1998) |
| SC92 | U112, O-antigen gene : : EmR | Cowley et al. (2000) |
| F. tularensis subsp. holarctica LVS | Live vaccine strain, type B biotype | ATCC 29684 |
| F. tularensis subsp. tularensis B38 | Attenuated type A biotype strain | ATCC 6223 |
| ΔpdpA | JLO with a deletion of pdpA | This study |
| ΔpdpA/SKX : : pdpA | ΔpdpA complemented with the integrating pJL-SKX : : pdpA construct | This study |
| ΔpdpA-LPS | ΔpdpA, O-antigen gene : : EmR from SC92 | This study |
| NZ9 | U112, pdpA : : EmR gene replacement mutant. | Laboratory strain |
| ΔpdpA/SKX : : pdpA-LPS | ΔpdpA/SKX : : pdpA, O-antigen gene : : EmR from SC92 | This study |
| E. coli DH5α | F−supE44 Δ(lacZYA–argF)U169 (φ80lacZΔM15) hsdR17 ( |
Invitrogen |
| E. coli BL21 (DE3) pLysS | B F–dcm ompT hsdS ( |
Stratagene |
| Plasmids | ||
| pWSK29 | Low-copy cloning vector, Apr | Wang & Kushner (1991) |
| pJL-SKX | pWSK29 with integrating SKX cassette, Kmr, Ampr | Ludu et al. (2008b) |
| pJL-ES-X | pWSK29 containing an ermCsacB cassette with flanking XhoI restriction sites | Ludu et al. (2008b) |
| pWSK29 : : ΔpdpA | A clone containing the fused flanking regions of pdpA; used to create the F. novicida ΔpdpA mutant | This study |
| pJL-SKX : : pdpA | An integrating complementation construct containing the full pdpA gene and its predicted promoter region (522 bp upstream of pdpA start) | This study |
| pET 28a | Expression vector with 6×His and T7 tags | Invitrogen |
| pET 28a : : pdpA-C | Clone expressing amino acids 405–783 of PdpA with a carboxyterminal 6×His tag. Used to create the monoclonal antibody against PdpA | This study |
Mutagenesis and complementation.
To create the pdpA deletion mutant strain the upstream and downstream regions flanking the pdpA gene were joined using fusion overlap PCR and the resulting amplicon was cloned into pWSK29 to create pWSK29 : : ΔpdpA. The last 22 bp of pdpA were left to preserve the ribosome-binding site of pdpB (see Fig. 1). The insert from pWSK29 : : ΔpdpA was excised using XhoI, ligated to the XhoI-XhoI sacB-EmR cassette from pJL-ES-X, and the ligation reaction was used to transform F. novicida JL0. The integration and excision of the sacB-EmR cassette was used to create a markerless deletion of the pdpA gene as previously described (Ludu et al., 2008a). The F. novicida JL0 strain was used as the wild-type strain in order to maintain an isogenic background for mutants and complemented strains, as the integrating plasmid used for complementation inserts at the non-essential locus FTN_1390 that is deleted in the JL0 strain. The ΔpdpA mutant was complemented using the kanamycin integration vector pJL-SKX (Ludu et al., 2008b) that inserts into the chromosome via a double crossover recombination event. The full-length pdpA gene and the promoter region, 522 bp upstream of pdpA, were ligated to pJL-SKX. The vector was then linearized and used to transform the pdpA deletion mutants, creating in cis complementation strains. The sequence of the genomic regions in the pdpA deletions has been deposited with GenBank and assigned the number EU810409. The sequences of the primers used in all the genetic constructs will be made available upon request.
SDS-PAGE and immunoblotting.
All SDS-PAGE was carried out using standard techniques. The Bradford (Bio-Rad) protein assay was used to determine sample protein concentration and normalize the amount of protein loaded in each lane. Once separated, the proteins were transferred to an Immobilon-FL PVDF membrane (Millipore) and then blocked in 5 % skim milk in PBS, pH 7.4. Mouse monoclonal antibody against PdpA was added at a 1 : 2000 dilution, whereas mouse monoclonal antibodies against PdpB and IglB were used at a 1 : 4000 dilution. Rabbit polyclonal antibody against MglB was used at a 1 : 5000 dilution. Bound antibody was detected using IRDye800-conjugated goat anti-mouse or IRDye800-conjugated goat anti-rabbit antibody (Rockland Immunochemicals) and visualized, and the integrated intensity of fluorescence quantified, using the LiCor Odyssey imaging software version 2.1. The anti-PdpA monoclonal antibody was produced by immunizing mice with recombinant C-terminal portion (aa 405–783) of PdpA expressed from pET28a. The anti-PdpB and anti-IglB antibodies were also produced by immunizing mice with recombinant protein expressed from pET28a. The mouse anti-PdpA hybridoma developed in this research, and the hybridomas producing anti-IglB and anti-PdpB, have been deposited with the American Type Culture Collection's Biodefence and Emerging Infections Resources program.
Iron-limitation studies.
F. novicida strains were grown overnight in TSBC at 37 °C without shaking. Then 3 ml of overnight culture was added to 30 ml fresh TSBC and allowed to grow for 1 h at 37 °C while shaking at 200 r.p.m. After 1 h the cultures were split into three flasks. One flask was left untreated and deferoxamine mesylate salt (Desferal) was added to the other two flasks to a final concentration of 1 mM. FeSO4 (1 mM) was also added to one of the Desferal-containing flasks. Bacterial cultures were grown for a further 2 h. Cells were harvested by centrifugation at 1000 g for 15 min and resuspended in PBS, pH 7.4, containing protease inhibitors (Sigma). Cell suspensions were sonicated with five 30 s pulses and then prepared for SDS-PAGE and Western blotting as described above.
Subcellular fractionation of F. novicida.
F. novicida U112 was added to J774A.1 macrophages at an m.o.i. of 1000 : 1 (bacterium to macrophage). Infected monolayers were incubated for 1 h at 37 °C, 5 % CO2 in complete Dulbecco's Modified Eagle Medium (cDMEM) supplemented with 10 % (v/v) fetal bovine serum (FBS) and 4 mM l-glutamine. Macrophages were washed five times in sterile PBS (Invitrogen) and fresh growth medium was added. The infection was allowed to progress for 18 h, at which point the macrophages were lysed by adding deoxycholic acid to a final concentration of 0.1 % (w/v). Bacteria were separated from the macrophage lysate by centrifugation at 2000 g for 15 min. The bacterial pellet was resuspended in PBS and sonicated to lyse cells. Any unbroken cells were removed by centrifugation at 10 000 g for 10 min. A sample was taken at this point as the total protein fraction. The cell lysate was ultracentrifuged at 100 000 g, 4 °C (Beckman ultramicrocentrifuge TLA-100.3) for 2 h to pellet the membranes. The supernatant representing the soluble proteins was removed and ultracentrifuged once more for 30 min at 100 000 g, 4 °C to minimize membrane protein contamination. The pellet representing the membrane-associated proteins was washed once in PBS and ultracentrifuged once more for 30 min at 100 000 g, 4 °C. The membrane-associated protein pellet was then treated with 1 % (w/v) sodium lauroyl sarcosinate (Sarkosyl) and left at room temperature for 30 min. The inner membrane (Sarkosyl-soluble) and outer membrane (Sarkosyl-insoluble) fractions were separated by ultracentrifugation at 100 000 g at 4 °C for 2 h. The fractions were then processed for SDS-PAGE and immunoblotting as described above. To estimate the amount of inner-membrane protein associated with each fraction, the NADH oxidase activity was determined using the method described by Osborn et al. (1972). Fraction purity was determined by comparing relative enzyme activity per milligram of protein per fraction.
Chicken embryo infections.
Fertilized White Leghorn chicken embryos were incubated for 7 days prior to infection. After incubation, embryos were infected with the appropriate strains of F. novicida in a range of doses as described by Nix et al. (2006). After infection, embryo death was monitored over a period of 6 days; the results of one inoculating dose are presented.
Mouse infections.
For in vivo infections, 6- to 8-week-old male specific-pathogen-free BALB/cByJ mice were purchased from the Jackson Laboratory. Animals were housed in sterile micro-isolator cages in a barrier environment at the Center for Biologics Evaluation and Research. Mice were fed autoclaved food and water ad libitum, and all experiments were performed under Institutional Animal Care and Use Committee guidelines. Mice were given 0.1 ml of appropriately diluted bacteria intradermally at the base of the tail; actual doses of inoculated bacteria were simultaneously determined by plate count. All materials used in animals, including bacteria, were diluted in PBS (BioWhittaker) containing <0.01 ng endotoxin ml−1.
Graphing and statistics.
The Prism GraphPad v4.03 software was used to create graphs and determine statistical values. For comparison of different survival curves the log-rank test was used to generate P-values.
RESULTS
Detection of PdpA with monoclonal antibody
The pdpA gene is the first in an AT-rich, 12-cistron operon in the FPI (Fig. 1); it is 2463 bp in length, and encodes a predicted 820 aa (95 kDa) protein. The deduced protein shows greater than 97 % amino acid identity across all sequenced strains of Francisella. The PdpA protein lacks a sec-associated signal peptide, and does not show significant similarity to other proteins by standard blastp analysis. However, iterated blast analysis (psi-blast) suggests the presence of a eukaryotic F-box motif, which is often found in ubiqitin–ligase complex-associated proteins. Analysis of PdpA using HHpredict software also detects several protein domains associated with the ubiquitin-proteasome pathway.
Although we previously used a genetic approach to demonstrate that PdpA plays a role in virulence, there has been no biochemical evidence demonstrating the expression of PdpA. To detect the product of the pdpA gene we developed a monoclonal antibody against a recombinant fragment of PdpA, and used it in Western immunoblots of F. novicida and F. tularensis extracts. As can be seen in Fig. 2(a), the PdpA band in F. novicida often runs as a distorted band of relative molecular mass of approximately 90. In the F. tularensis subsp. holarctica strain LVS or the F. tularensis subsp. tularensis strain B38 the PdpA band migrates as a tight band at a relative molecular mass of approximately 82. F. novicida has an O-antigen structure and banding pattern that is different from that found in F. tularensis strains and this may account for the distortion of the PdpA band that occurs in F. novicida but not in F. tularensis extracts.
Fig. 2.

Detection and subcellular localization of PdpA in Francisella. (a) Reactivity of anti-PdpA monoclonal antibody with cell extracts of F. novicida and two F. tularensis subspecies. The O-antigen in F. novicida is thought to cause aberrant migration of PdpA. The migration of an 83 kDa molecular mass marker is shown on the right. (b) Effect of deletion of pdpA and mutation of O-antigen gene. An O-antigen mutation from the previously described mutant SC92 (‘WT-LPS’) was transferred to the ΔpdpA mutant and the complemented strain ΔpdpA/SKX : : pdpA, and Western blots with anti-PdpA and anti-PdpB monoclonal antibodies are shown. For the top part of (b) the number at the right indicates actual migration of a molecular mass marker, and for the bottom part, the number indicates a calculated relative molecular mass. (c) A Western blot using anti-PdpB antibody shows that the pdpA : : EmR gene replacement mutant NZ9 expresses greatly reduced levels of PdpB compared to wild-type (WT). (d) Bacteria isolated from a J774A.1 macrophage infection were fractionated to separate soluble and membrane-associated proteins. PdpA was found to localize with the soluble proteins, as was the transcriptional regulator MglB. PdpB localized to the Sarkosyl-soluble protein fraction, indicating its association with the inner membrane of F. novicida. NADH oxidase activity was determined in each fraction as a measure of the relative amount of inner-membrane protein. The number to the right of the top panel of (d) represents the actual migration of the molecular mass marker, whereas the numbers in the two bottom panels represent calculated relative molecular masses. The amount of sample loaded in each lane was normalized by protein content. Results are representative of duplicate experiments.
To add confidence that the reactive band seen in immunoblots was truly PdpA, we created a deletion mutant that eliminated codons 1–813 of pdpA (Fig. 1b, c); the last seven codons were not deleted in order to preserve the ribosome-binding site of pdpB. The deletion mutation was verified by PCR analysis (Fig. 1b) and by sequencing of the genomic region of the ΔpdpA mutant. We also complemented the ΔpdpA mutation by inserting pdpA and the region encompassing 522 bp upstream of pdpA into an integrating vector, pJL-SKX, which inserts into the chromosome at approximately 507 kb from the pdpA locus. As mentioned, we reasoned that the distortion of the PdpA band was due to its co-migration with several LPS bands that are present in F. novicida but not in F. tularensis at the approximate relative molecular mass where PdpA is found. Hence, we also introduced a genetic lesion that diminishes the amount of O-antigen produced in F. novicida into the ΔpdpA mutant and the ΔpdpA/SKX : : pdpA complemented strains. Immunoblot analysis of the resulting O-antigen-defective strains (Fig. 2b) showed that PdpA is not made in the ΔpdpA strain but is made in the wild-type and the complemented strain (both with O-antigen mutations). These results also show that the reduction in the amount of O-antigen results in PdpA migrating as a sharper band. In order to ascertain if the ΔpdpA lesion affected expression of genes downstream of pdpA, we assessed the expression levels of PdpB, which is encoded by the cistron immediately downstream of pdpA. As Fig. 2(b) shows, the level of PdpB was essentially identical in the wild-type, the ΔpdpA and the ΔpdpA/SKX : : pdpA strains. We also assessed the expression of PdpA and PdpB in ΔpdpA and ΔpdpA/SKX : : pdpA strains that had the wild-type form of the O-antigen and found essentially identical results as in Fig. 2(b) except that the PdpA band was usually distorted (data not shown). Unlike the markerless pdpA mutant constructed in this study, the pdpA : : EmR allelic replacement mutant, NZ9, demonstrated marked polarity effects on PdpB (Fig. 2c).
Solubility of PdpA
Some of the genes in the FPI encode homologues of components of T6SSs, and have been shown to be membrane associated. To test the possible role of PdpA as a membrane-bound component of a possible T6SS, we analysed the localization of PdpA in F. novicida cells that had infected macrophages. We reasoned that the localization of PdpA during an infection would provide information that might suggest a function. The data presented in Fig. 2(d) show that PdpA localizes to the soluble bacterial fraction, and co-localizes with the gene regulatory protein MglB. We had previously shown that PdpB localizes to the inner membrane in broth-grown cells (Ludu et al., 2008a), and it localized to the same fraction in macrophage-grown cells (Fig. 2d). Outer-membrane proteins Tul4 and FopA were only detected in the Sarkosyl-insoluble fraction using anti-F. novicida antibody (data not shown). Similar results were obtained when we analysed the localization of PdpA in broth-grown F. novicida (data not shown), with the majority of PdpA localizing to the soluble fraction.
We previously showed that inactivation of FPI genes iglA, dotU and pdpB, which are homologues of canonical components of T6SSs, abolished transport of IglC to the outer membrane (Ludu et al., 2008a). We found that IglC transport in the ΔpdpA background was unaffected (data not shown), suggesting that PdpA does not play a role in T6SS-mediated secretion. However, these results should be regarded cautiously, since we have so little information about possible T6SS-mediated secretion in general, and especially in Francisella. Attempts to visualize localization of PdpA in the macrophage cytosol were unproductive (data not shown).
PdpA levels affected by mglAB background and iron concentration
Previous work has shown that FPI mRNA expression is depressed when MglA is absent and is increased by low iron concentration (Brotcke et al., 2006; Charity et al., 2007; Deng et al., 2006; Lauriano et al., 2003), but none of these works examined protein expression levels. In order to assess the effect of these factors on PdpA protein expression, we examined protein production by immunoblot analysis. The expression of PdpA was much reduced in both mglA (GB2) and mglB (GB6) genetic backgrounds as compared to expression in the wild-type strain of F. novicida (Fig. 3a). A similar effect was seen on the expression of PdpB and IglB in the mglA and mglB backgrounds (Fig. 3a). Addition of the chelator Desferal to cultures of F. novicida resulted in an increase in PdpA expression in F. novicida (Fig. 3b), presumably due to the reduction in the amount of available iron. IglB, IglC and PdpB expression responded to the addition of Desferal essentially the same as did PdpA (Fig. 3b). The effect of the chelator on protein expression was reversed by the addition of 1 mM FeSO4, which resulted in PdpA, PdpB, IglB and IglC levels that were below those found in untreated cultures (Fig. 3b). Expression levels of a cross-reactive control protein remained constant regardless of the addition of chelator or excess iron. The levels of PdpB, IglB and IglC in the ΔpdpA strain responded essentially identically to chelator or iron conditions as in the wild-type F. novicida strain (data not shown).
Fig. 3.

Regulation of PdpA expression by iron and transcriptional regulators. (a) Levels of PdpA, PdpB and IglB protein expression were determined in mglA and mglB mutant backgrounds. (b) Expression of these proteins, as well as IglC, was also determined in varying iron conditions. The relative level of fluorescence signal generated by reactivity of anti-PdpA, anti-PdpB, anti-IglB, or anti-IglC monoclonal antibodies, calculated as described in Methods, is indicated. The amount of sample loaded to the wells of each polyacrylamide gel was normalized by protein content. The reactivity of a non-specific, cross-reactive protein band is shown in the bottom panel. Results are representative of experiments performed in triplicate.
Virulence properties of the ΔpdpA mutant
Our previous work and the studies of others suggest that PdpA plays a role in virulence. However, these previous studies were unable to measure expression of proteins encoded by genes downstream of pdpA or failed to carry out genetic complementation; most studies failed to carry out either of these controls. Hence, we felt it was important to test the virulence of a ΔpdpA mutant that lacked translational polarity and could be complemented with a single copy of pdpA. We first performed a series of infections with different inoculating doses in chicken embryos and followed these with smaller experimental infections of mice. The data presented in Fig. 4 show that, at the inoculating dose of 103 c.f.u., wild-type F. novicida killed 100 % of the chicken embryos by day 4 post-infection. In contrast, the ΔpdpA strain failed to kill 50 % of the embryos by 6 days after the start of the infection. Complementation of the ΔpdpA mutation with a single copy of wild-type pdpA resulted in a strain, ΔpdpA/SKX : : pdpA, that was equally virulent as the wild-type strain in the chicken embryo infection assay. Similar results were found in mouse infection experiments. Infection with 105 wild-type F. novicida killed 5/5 BALB/cByJ male mice but infection of 10 mice with 107 ΔpdpA failed to kill any mice. As we have shown before (Ludu et al., 2008a), infections of chicken embryos with F. novicida mutants are largely predictive of the outcome of infection of mice with the same mutants. However, mice appear to be considerably more resistant to killing, especially by attenuated F. novicida.
Fig. 4.
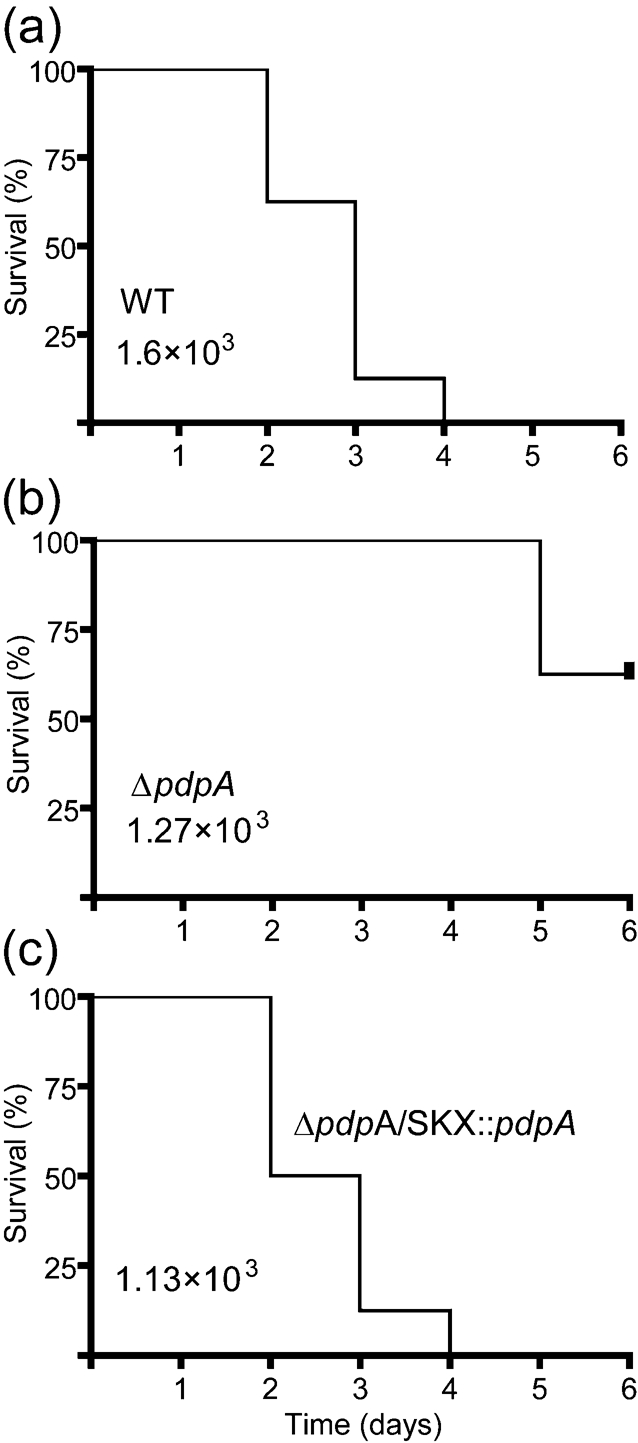
Attenuation of the ΔpdpA mutant in chicken embryos. Seven-day-old chicken embryos were infected with 103 c.f.u. of wild-type (WT) (a), ΔpdpA (b) and ΔpdpA/SKX : : pdpA (c) strains of F. novicida. Time to death of the embryos was monitored over a period of 6 days. All experiments were done at three separate inoculating doses, and the experiments included at least seven embryos per infective dose per strain; the survival curves presented are representative of three separate trials. The statistical difference between the WT and ΔpdpA strain survival curve was measured by the log-rank test and yielded a P-value of <0.001. The P-value for a comparison of ΔpdpA vs ΔpdpA/SKX : : pdpA was <0.001.
DISCUSSION
Recent study of F. tularensis has yielded surprisingly little advancement concerning the characterization and function of virulence-associated proteins. Although many virulence factors have been discovered, particularly within the FPI, several of these proteins have proven to be very difficult to study due to toxicity issues and low expression levels. This research represents the first examples of PdpA protein characterization and has investigated the virulence of a non-polar, markerless, in-frame, pdpA deletion mutant.
Immediately downstream of pdpA lies the pdpB gene, and the pdpB ribosome-binding site overlaps the 3′ end of pdpA. This tight translational coupling of pdpA and pdpB, and between most other adjacent FPI genes, is consistent with mRNA expression studies that indicate that FPI genes are controlled by the same global regulators, including MglA, SspA, PmrA and FevR, or by the same environmental cues, such as low iron concentration (Brotcke et al., 2006; Brotcke & Monack, 2008; Charity et al., 2007; Deng et al., 2006; Lenco et al., 2007; Mohapatra et al., 2007). Data presented in this work on the effect of MglA and iron concentration on expression of PdpA and other FPI-encoded proteins provide evidence that expression extends to the protein level. The MglB protein has been presumed to be involved in expression, and in this work we provided evidence supporting this idea, as PdpA, PdpB and IglB expression was virtually undetectable in an mglB mutant background.
It is also apparent that the allelic replacement of pdpA has polar effects on expression of the downstream gene pdpB, as there is considerably less PdpB protein present in strain NZ9 (ΔpdpA : : EmR). This polarity may account for the increased attenuation of NZ9 in chicken embryos compared to ΔpdpA (data not shown).
One central biological question regarding the FPI-encoded proteins involves their possible role in secretion or as effector proteins that interact with host-cell components. Using macrophage-grown F. novicida we found PdpA in the bacterial cytoplasm. Although macrophage-grown F. novicida cells are more difficult to analyse than broth-grown cells, we reasoned that they would provide better evidence of the biologically relevant localization of PdpA than analysis of broth-grown F. novicida. Localization of PdpA to the bacterial cytoplasm may lead one to believe that the protein is not a component of the T6SS, as most proteins involved in assembling a secretion apparatus are membrane associated. However, this is not always the case; T6SS proteins have been shown to localize to both membrane and cytoplasmic fractions (Wu et al., 2008) so our results have to be interpreted carefully. As a soluble protein, PdpA may function as a chaperone component of the T6SS or may be a secreted effector protein. We believe that PdpA might be a secreted effector protein as it does not appear to play a role in secretion as defined by our recently described criteria (Ludu et al., 2008a), yet its absence profoundly decreases virulence in both chicken embryos and adult mice. Also, the deduced PdpA amino acid sequence shows low-level similarities to fragments of multiple proteins involved in the eukaryotic ubiquitin-proteasome pathway, and such molecular mimicry has been observed in virulence effectors of other intracellular pathogens (Angot et al., 2007). One of the motifs that we identified is a possible F-box motif in the N-terminal region of PdpA. In eukaryotic cells F-box-containing proteins are involved in protein–protein interactions, most commonly in proteins in the ubiquitin–ligase complex (Kipreos & Pagano, 2000). Inspection of the Pfam tree of F-box proteins (PF00646; http://pfam.sanger.ac.uk/family?acc=PF00646) reveals several prokaryotic F-box-containing proteins, and F-box regions have been shown to be important in two bacterial plant pathogens (Angot et al., 2006; Tzfira et al., 2004). Attempts to express domains that showed similarity to those found in ubiquitin–ligase complexes have been unsuccessful due to apparent toxicity involved in expressing PdpA protein domains, particularly those located within the amino terminus of the protein. Also, the introduction of alanine substitutions into key residues of the putative F-box failed to affect virulence of the resultant F. novicida mutants (data not shown). All efforts to detect PdpA in the host-cell cytoplasm have been unsuccessful. Although we believe PdpA is a secreted effector protein, the current tools and methodologies available have been unable to confirm this. Clearly, extensive analysis of PdpA will be required to surmise its function in virulence. The accompanying paper (Schmerck et al., 2009) reports studies on the intracellular growth phenotype of the ΔpdpA mutant and the effect of the mutation on the macrophage gene expression response to F. novicida infection.
Acknowledgments
This work was supported by grant 5R01 AI056212-02 from the National Institute of Allergy and Infectious Diseases and MOP 89812 from the Canadian Institutes of Health Research to F. E. N. The development of monoclonal antibodies was partially supported by a contract from the Biodefense and Emerging Infections Resources program at the American Type Culture Collection.
Abbreviations
FPI, Francisella pathogenicity island
T6SS, type VI secretion system
Footnotes
The GenBank/EMBL/DDBJ accession number for the sequence of the genomic regions in the pdpA deletions is EU810409.
References
- Angot, A., Peeters, N., Lechner, E., Vailleau, F., Baud, C., Gentzbittel, L., Sartorel, E., Genschik, P., Boucher, C. & Genin, S. (2006). Ralstonia solanacearum requires F-box-like domain-containing type III effectors to promote disease on several host plants. Proc Natl Acad Sci U S A 103, 14620–14625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angot, A., Vergunst, A., Genin, S. & Peeters, N. (2007). Exploitation of eukaryotic ubiquitin signaling pathways by effectors translocated by bacterial type III and type IV secretion systems. PLoS Pathog 3, e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anthony, L. D., Burke, R. D. & Nano, F. E. (1991). Growth of Francisella spp. in rodent macrophages. Infect Immun 59, 3291–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron, G. S. & Nano, F. E. (1998). MglA and MglB are required for the intramacrophage growth of Francisella novicida. Mol Microbiol 29, 247–259. [DOI] [PubMed] [Google Scholar]
- Brotcke, A. & Monack, D. M. (2008). Identification of fevR, a novel regulator of virulence gene expression in Francisella. Infect Immun 76, 3473–3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brotcke, A., Weiss, D. S., Kim, C. C., Chain, P., Malfatti, S., Garcia, E. & Monack, D. M. (2006). Identification of MglA-regulated genes reveals novel virulence factors in Francisella tularensis. Infect Immun 74, 6642–6655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charity, J. C., Costante-Hamm, M. M., Balon, E. L., Boyd, D. H., Rubin, E. J. & Dove, S. L. (2007). Twin RNA polymerase-associated proteins control virulence gene expression in Francisella tularensis. PLoS Pathog 3, e84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemens, D. L., Lee, B. Y. & Horwitz, M. A. (2004). Virulent and avirulent strains of Francisella tularensis prevent acidification and maturation of their phagosomes and escape into the cytoplasm in human macrophages. Infect Immun 72, 3204–3217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley, S. C., Gray, C. J. & Nano, F. E. (2000). Isolation and characterization of Francisella novicida mutants defective in lipopolysaccharide biosynthesis. FEMS Microbiol Lett 182, 63–67. [DOI] [PubMed] [Google Scholar]
- de Bruin, O. M., Ludu, J. S. & Nano, F. E. (2007). The Francisella pathogenicity island protein IglA localizes to the bacterial cytoplasm and is needed for intracellular growth. BMC Microbiol 7, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng, K., Blick, R. J., Liu, W. & Hansen, E. J. (2006). Identification of Francisella tularensis genes affected by iron limitation. Infect Immun 74, 4224–4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortier, A. H., Green, S. J., Polsinelli, T., Jones, T. R., Crawford, R. M., Leiby, D. A., Elkins, K. L., Meltzer, M. S. & Nacy, C. A. (1994). Life and death of an intracellular pathogen: Francisella tularensis and the macrophage. Immunol Ser 60, 349–361. [PubMed] [Google Scholar]
- Golovliov, I., Baranov, V., Krocova, Z., Kovarova, H. & Sjostedt, A. (2003a). An attenuated strain of the facultative intracellular bacterium Francisella tularensis can escape the phagosome of monocytic cells. Infect Immun 71, 5940–5950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovliov, I., Sjostedt, A., Mokrievich, A. & Pavlov, V. (2003b). A method for allelic replacement in Francisella tularensis. FEMS Microbiol Lett 222, 273–280. [DOI] [PubMed] [Google Scholar]
- Gray, C. G., Cowley, S. C., Cheung, K. K. & Nano, F. E. (2002). The identification of five genetic loci of Francisella novicida associated with intracellular growth. FEMS Microbiol Lett 215, 53–56. [DOI] [PubMed] [Google Scholar]
- Kipreos, E. T. & Pagano, M. (2000). The F-box protein family. Genome Biol 1, REVIEWS3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, X. H., Golovliov, I. & Sjostedt, A. (2004). Expression of IglC is necessary for intracellular growth and induction of apoptosis in murine macrophages by Francisella tularensis. Microb Pathog 37, 225–230. [DOI] [PubMed] [Google Scholar]
- Lauriano, C. M., Barker, J. R., Nano, F. E., Arulanandam, B. P. & Klose, K. E. (2003). Allelic exchange in Francisella tularensis using PCR products. FEMS Microbiol Lett 229, 195–202. [DOI] [PubMed] [Google Scholar]
- Lauriano, C. M., Barker, J. R., Yoon, S.-S., Nano, F. E., Arulanandam, B. P., Hassett, D. J. & Klose, K. E. (2004). MglA regulates transcription of virulence factors necessary for Francisella tularensis intraamoebae and intramacrophage survival. Proc Natl Acad Sci U S A 101, 4246–4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenco, J., Hubalek, M., Larsson, P., Fucikova, A., Brychta, M., Macela, A. & Stulik, J. (2007). Proteomics analysis of the Francisella tularensis LVS response to iron restriction: induction of the F. tularensis pathogenicity island proteins IglABC. FEMS Microbiol Lett 269, 11–21. [DOI] [PubMed] [Google Scholar]
- Ludu, J. S., de Bruin, O. M., Duplantis, B. N., Schmerk, C. L., Chou, A. Y., Elkins, K. L. & Nano, F. E. (2008a). The Francisella pathogenicity island protein PdpD is required for full virulence and associates with homologues of the type VI secretion system. J Bacteriol 190, 4584–4595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludu, J. S., Nix, E. B., Duplantis, B. N., de Bruin, O. M., Gallagher, L. A., Hawley, L. M. & Nano, F. E. (2008b). Genetic elements for selection, deletion mutagenesis and complementation in Francisella spp. FEMS Microbiol Lett 278, 86–93. [DOI] [PubMed] [Google Scholar]
- Mohapatra, N. P., Soni, S., Bell, B. L., Warren, R., Ernst, R. K., Muszynski, A., Carlson, R. W. & Gunn, J. S. (2007). Identification of an orphan response regulator required for the virulence of Francisella spp. and transcription of pathogenicity island genes. Infect Immun 75, 3305–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nano, F. E. & Schmerk, C. (2007). The Francisella pathogenicity island. Ann N Y Acad Sci 1105, 122–137. [DOI] [PubMed] [Google Scholar]
- Nano, F. E., Zhang, N., Cowley, S. C., Klose, K. E., Cheung, K. K., Roberts, M. J., Ludu, J. S., Letendre, G. W., Meierovics, A. I. & other authors (2004). A Francisella tularensis pathogenicity island required for intramacrophage growth. J Bacteriol 186, 6430–6436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nix, E. B., Cheung, K. K., Wang, D., Zhang, N., Burke, R. D. & Nano, F. E. (2006). Virulence of Francisella spp. in chicken embryos. Infect Immun 74, 4809–4816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osborn, M. J., Gander, J. E. & Parisi, E. (1972). Mechanism of assembly of the outer membrane of Salmonella typhimurium. Site of synthesis of lipopolysaccharide. J Biol Chem 247, 3973–3986. [PubMed] [Google Scholar]
- Proctor, R. A., White, J. D., Ayala, E. & Canonico, P. G. (1975). Phagocytosis of Francisella tularensis by Rhesus monkey peripheral leukocytes. Infect Immun 11, 146–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santic, M., Molmeret, M., Klose, K. E., Jones, S. & Kwaik, Y. A. (2005). The Francisella tularensis pathogenicity island protein IglC and its regulator MglA are essential for modulating phagosome biogenesis and subsequent bacterial escape into the cytoplasm. Cell Microbiol 7, 969–979. [DOI] [PubMed] [Google Scholar]
- Schmerk, C. L., Duplantis, B. N., Howard, P. L. & Nano, F. E. (2009). A Francisella novicida pdpA mutant exhibits limited intracellular replication and remains associated with the lysosomal marker LAMP-1. Microbiology 155, 1498–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tempel, R., Lai, X. H., Crosa, L., Kozlowicz, B. & Heffron, F. (2006). Attenuated Francisella novicida transposon mutants protect mice against wild-type challenge. Infect Immun 74, 5095–5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzfira, T., Vaidya, M. & Citovsky, V. (2004). Involvement of targeted proteolysis in plant genetic transformation by Agrobacterium. Nature 431, 87–92. [DOI] [PubMed] [Google Scholar]
- Wang, R. F. & Kushner, S. R. (1991). Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100, 195–199. [PubMed] [Google Scholar]
- Weiss, D. S., Brotcke, A., Henry, T., Margolis, J. J., Chan, K. & Monack, D. M. (2007). In vivo negative selection screen identifies genes required for Francisella virulence. Proc Natl Acad Sci U S A 104, 6037–6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, H. Y., Chung, P. C., Shih, H. W., Wen, S. R. & Lai, E. M. (2008). Secretome analysis uncovers an Hcp-family protein secreted via a type VI secretion system in Agrobacterium tumefaciens. J Bacteriol 190, 2841–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]