

Abstract
Disulfide bond formation in periplasmic proteins is catalysed by the DsbA/DsbB system in most Gram-negative bacteria. Salmonella enterica serovar Typhimurium also encodes a paralogous pair of proteins to DsbA and DsbB, DsbL and DsbI, respectively, downstream of a periplasmic arylsulfate sulfotransferase (ASST). We show that DsbL and DsbI function as a redox pair contributing to periplasmic disulfide bond formation and, as such, affect transcription of the Salmonella pathogenicity island 1 (SPI1) type three secretion system genes and activation of the RcsCDB system, as well as ASST activity. In contrast to DsbA/DsbB, however, the DsbL/DsbI system cannot catalyse the disulfide bond formation required for flagellar assembly. Phylogenic analysis suggests that the assT dsbL dsbI genes are ancestral in the Enterobacteriaceae, but have been lost in many lineages. Deletion of assT confers no virulence defect during acute Salmonella infection of mice.
INTRODUCTION
Disulfide bond formation in periplasmic proteins is catalysed primarily by the DsbA/DsbB system (Kadokura et al., 2003). DsbA, a 21 kDa periplasmic protein with a catalytic CXXC motif, exchanges its disulfide bond with pairs of cysteines in substrate proteins (Kadokura et al., 2003). DsbA is reoxidized by DsbB, an inner-membrane protein with two periplasmic loops, each containing a pair of cysteine residues (Collet & Bardwell, 2002). DsbB oxidizes DsbA by a disulfide exchange with the cysteine pair in one periplasmic loop, and then transfers the electrons, via disulfide exchange in the second periplasmic loop, to quinones in the cytoplasmic membrane (Collet & Bardwell, 2002; Inaba & Ito, 2008).
Mutants lacking DsbA or DsbB exhibit pleiotropic phenotypes due to defects in the folding of numerous cell envelope proteins. For example, in Escherichia coli, alkaline phosphatase is inactive and unstable without disulfide bonds (Bardwell et al., 1991). These mutants also show reduced motility because of improper assembly of the flagellum (Dailey & Berg, 1993). In pathogens, mutations in dsbA can result in decreased virulence because factors such as pili, toxins and type III secretion systems (T3SSs) require disulfide bonds to function properly (Ellermeier & Slauch, 2004; Ha et al., 2003; Jackson & Plano, 1999; Miki et al., 2004; Peek & Taylor, 1992; Watarai et al., 1995).
Some Gram-negative bacteria produce DsbA and/or DsbB paralogues that either supplement or replace DsbA and DsbB activity (Grimshaw et al., 2008; Kwon & Choi, 2005; Raczko et al., 2005). For example, the Salmonella enterica virulence plasmid encodes a DsbA paralogue, SrgA, whose activity is dependent on functional DsbB (Bouwman et al., 2003). The genome sequence suggests that S. enterica serovar Typhimurium also encodes an additional oxidative system. DsbL, a DsbA paralogue, and DsbI, a DsbB paralogue, are encoded downstream of a periplasmic arylsulfate sulfotransferase (ASST), which catalyses the transfer of a sulfate group among phenolic compounds (Kim et al., 1992). Highly similar gene clusters are found throughout the genus Salmonella and in a subset of Enterobacteriaceae. ASST homologues exist in other Gram-negative proteobacteria, Gram-negative bacteroidetes, and the Gram-positive firmicutes (Grimshaw et al., 2008). DsbL and DsbI homologues, not associated with ASST enzymes, are also apparent in a variety of Gram-negative organisms (Kimball et al., 2003; Raczko et al., 2005).
Our previous work showed that dsbA in Salmonella is transcriptionally controlled by the Salmonella pathogenicity island 1 (SPI1) T3SS regulatory circuit (Ellermeier & Slauch, 2004). Loss of DsbA results in both dysfunction of the secretion apparatus and transcriptional downregulation of the SPI1 system (Lin et al., 2008). SPI1 transcriptionally responds to disulfide bond status through the flagellar apparatus and the RcsCDB system (Lin et al., 2008). Here we present evidence that DsbL and DsbI also contribute to disulfide bond formation in the Salmonella periplasm. We further prove that the Salmonella assT gene encodes an ASST, dependent on a functional disulfide bond oxidoreductase system.
METHODS
Media and reagents.
Luria–Bertani (LB), M63 glucose minimal, or low-phosphate (0.1 mM) glucose MOPS minimal media were used for growth of bacteria (Neidhardt et al., 1974; Silhavy et al., 1984). DTT was added to LB cultures to prevent free cystine from contributing to disulfide bond formation. Motility agar contained 0.3 % (w/v) agar in M63 glucose minimal medium. Bacterial strains were grown at 37 °C, except for those containing the temperature-sensitive plasmids pCP20 and pKD46 (Cherepanov & Wackernagel, 1995; Datsenko & Wanner, 2000), which were grown at 30 °C. Antibiotics were used at the following concentrations: 100 μg ampicillin ml−1, 10 μg chloramphenicol ml−1, 50 μg kanamycin ml−1, 50 μg apramycin ml−1. Enzymes were from Invitrogen and used according to the manufacturer's recommendations. Primers were from IDT.
Bacterial strains and plasmid construction.
Bacterial strains, listed in Table 1, were isogenic derivatives of S. enterica serovar Typhimurium strain 14028, E. coli K-12 strain DH5α or K-12 strain MC4100. Deletions of genes and concomitant insertion of an antibiotic resistance cassette were performed using λ Red-mediated recombination (Datsenko & Wanner, 2000; Yu et al., 2000), as described previously (Ellermeier et al., 2002), and were verified by PCR analysis. The end points of each deletion are indicated. In each case, the resulting constructs were P22-transduced into a wild-type background (Maloy et al., 1996). In some cases, antibiotic resistance cassettes were removed using plasmid pCP20 encoding FLP recombinase (Datsenko & Wanner, 2000). The wca–lacZ, dsbL–lacZ and assT–lacZ transcriptional fusions were created using FLP/FRT-mediated site-specific recombination, as described previously (Ellermeier et al., 2002). Note that these fusion constructs create null mutations in these genes. The phoA–6×His–lacZ fusion was constructed by first inserting the sequences encoding 6×His at the 3′ end of the phoA ORF followed immediately by a Kn cassette via λ Red-mediated recombination. The lacZ fusion construct was subsequently introduced as described above. The hilA–lacZ fusion, integrated at the lambda attachment site, had been constructed previously (Lin et al., 2008). The following genes were amplified using primers carrying EcoRI or BamHI sites and then cloned into vector pWKS30 (Wang & Kushner, 1991) with base-pairs indicated relative to the start site of translation of the first gene in the construct: assT (−14→+1808); assT dsbLI (−14→+3194); dsbL (−14→+687); dsbI (−14→+693); dsbLI (−14→+1379); dsbA (−30→+645); dsbB (−21→+547). All bacterial strains were rebuilt at least once and experiments were repeated with the independent constructs.
Table 1.
Strains and plasmids

*All Salmonella strains are isogenic derivatives of serovar Typhimurium strain 14028, unless otherwise noted.
†Numbers indicate the base pairs (inclusive) that are deleted or cloned as defined in the S. enterica serovar Typhimurium LT2 or E. coli K-12 MG1655 genome sequences (National Center for Biotechnology Information database).
‡This study unless specified otherwise.
§American Type Culture Collection.
Enzymic assays.
β-Galactosidase assays were performed using a microtitre plate assay, as described previously (Slauch & Silhavy, 1991). Strains were cultured overnight in LB medium, then subcultured in LB containing 1 % (w/v) NaCl and 0.5 mM DTT and grown statically overnight. β-Galactosidase activities were calculated as (μmol o-nitrophenol formed min−1)×106/(OD600×ml cell suspension). The values are presented as mean±sd, where n=4. Phosphatase assays were performed in an essentially identical manner except that cells were grown in MOPS medium with limited phosphate to induce phoA expression. Cells were resuspended in 1 M Tris buffer and p-nitrophenylphosphate was used as substrate.
ASST activity was quantified by monitoring the production of p-nitrophenol from the hydrolysis of p-nitrophenylsulfate (PNS), using α-naphthol as an acceptor (Kwon et al., 2001). Cultures were grown overnight at 37 °C in LB broth with 2 mM DTT and 100 μg ampicillin ml−1. A 100 μl aliquot of each culture was inoculated into 900 μl M63 glucose minimal medium with 0.1 mM PNS, 0.2 mM α-naphthol, 2 mM DTT and 100 μg ampicillin ml−1, and incubated with shaking at 37 °C for 1 h. To stop the reaction, 1 ml 1.0 M NaOH was added to each culture. The A420 was measured. ASST activities were calculated as (μmol p-nitrophenol formed min−1)×106/(OD600×ml cell suspension). The values are presented as mean±sd, where n=4.
To assay β-galactosidase activity in bacteria grown in macrophages, RAW264.7 macrophage cells (∼105 per well) grown in Dulbecco's minimal essential medium (DMEM) were infected with Salmonella opsonized in 50 % mouse serum (Equitech-bio) at an m.o.i. of 10. After 30 min, cells were rinsed with PBS three times and incubated for 20 min in DMEM containing 12.5 μg gentamicin ml−1. Macrophages were then washed with PBS and incubated in DMEM containing 12.5 μg gentamicin ml−1 for 16 h. Cells were washed with PBS three times and macrophages were scraped in PBS to remove them from the dish. Macrophage cells were disrupted with cold distilled H2O and bacteria were recovered by centrifugation and resuspended in PBS. The number of viable bacterial cells recovered was determined by diluting and plating for c.f.u. The remaining samples were used to measure β-galactosidase activity with the chemiluminescent substrate Lumigal 530 according to the manufacturer's instructions (Lumigen). The β-galactosidase activity was calculated as luminescent units per c.f.u. of bacteria in each sample. The β-galactosidase activity of bacteria grown in vitro in LB medium (16 h of growth) was measured using the same method. The values are presented as mean±sd, where n=3.
PhoA immumoblot.
Strains containing the phoA–6×His–lacZ fusions were grown overnight in MOPS medium with limiting phosphate to induce phoA expression. Each sample was assayed for β-galactosidase activity and phosphatase activity (see above). In addition, 20 μl of each culture was combined with loading buffer and proteins were separated on a 4–20 % gradient polyacrylamide gel containing SDS. After electrophoresis, proteins were electrotransferred to nitrocellulose. Mouse monoclonal anti-6-His IgG antibody (Zymed) was used as primary antibody. The secondary antibody was horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Abcam). The Western blotting and the detection of HRP-conjugated antibodies with a chemiluminescent system were done according to the instructions of the manufacturer (Amersham).
Mouse virulence assays.
Mutant and wild-type Salmonella were grown for 16 h in LB medium. Equal volumes of each culture were mixed and then diluted in sterile 0.2 M sodium phosphate buffer (pH 8.0) to 5×108 c.f.u. ml−1 (oral) or in sterile 0.15 M NaCl to 5×103 c.f.u. ml−1 [for intraperitoneal (i.p.) administration]. Six to eight-week-old female BALB/c mice (Harlan) were inoculated either orally or i.p. with 0.2 ml of a bacterial suspension. The actual c.f.u. and relative percentage of each strain were determined by direct plating of the inoculum. After 4–5 days, the mice were sacrificed by CO2 asphyxiation and the entire intestine (oral) and/or spleen (oral and i.p.) were harvested. These organs were homogenized, and serial dilutions were plated on the appropriate medium to recover bacteria. Replica plating on appropriate antibiotic medium was used to determine the percentage of each strain. Competitive index was calculated as (percentage strain A recovered/percentage strain B recovered)/(percentage strain A inoculated/percentage strain B inoculated). Student's t test was used for statistical analysis. Animal work was reviewed and approved by the University of Illinois Institutional Animal Care and Use Committee (IACUC) and performed under protocol 07070.
Phylogenetic analysis.
Sequences were aligned using clustal w (Thompson et al., 1994). Phylogeny was inferred by maximum-likelihood under the KY85 model in paup* v4b10 (Wilgenbusch & Swofford, 2003). The robustness of each alignment was determined through non-parametric bootstrap analyses consisting of 1000 replicates (Felsenstein, 1985).
RESULTS
Loss of DsbL or DsbI affects transcription of hilA and wca genes
We previously showed that loss of DsbA, the primary disulfide bond oxidoreductase in the periplasm, or the cognate DsbB, decreases transcription of the SPI1 genes in serovar Typhimurium (Ellermeier & Slauch, 2004; Lin et al., 2008). During our studies, we recognized a pair of DsbA and DsbB paralogues encoded together in the Salmonella genome. DsbL (STM3193) is 27 % identical in amino acid sequence to DsbA, and has a putative CPFC active site motif. DsbI (STM3194) is 30 % identical in sequence to DsbB, and has two pairs of cysteine residues like DsbB. To test the ability of DsbLI to form disulfide bonds, we took advantage of the well-established fact that both the activity and the stability of the E. coli periplasmic alkaline phosphatase PhoA are dependent on disulfide bond formation in the protein (Bardwell et al., 1991). To monitor PhoA activity, protein levels, and expression at the same time, we tagged the C terminus of the PhoA protein with 6×His and inserted a promoterless lacZ gene immediately downstream of the phoA ORF. We then constructed isogenic strains that were dsbA+, dsbA− or dsbA− containing a low-copy-number plasmid in which the dsbL and dsbI genes were cloned downstream of a lac promoter (pDsbLI). These strains were grown in phosphate-limiting minimal medium, the resulting cells were assayed for alkaline phosphatase activity and β-galactosidase activity, and Western blot analysis was performed to monitor PhoA protein levels. As shown in Fig. 1, the dsbA+ cells had robust alkaline phosphatase and β-galactosidase activities and the PhoA protein was evident in the cell extracts. The dsbA mutant showed increased β-galactosidase activity, suggesting that transcription of the phoA gene was increased in this background. However, phosphatase activity was low and the PhoA protein was barely detectable by Western blot analysis. This is consistent with earlier results showing that the PhoA protein is unstable in the absence of disulfide bonds (Bardwell et al., 1991). Introduction of the pDsbLI plasmid into the dsbA mutant restored both phosphatase activity and PhoA protein almost to wild-type levels. These data suggest that DsbLI are able to replace DsbA and introduce disulfide bonds into the PhoA protein.
Fig. 1.
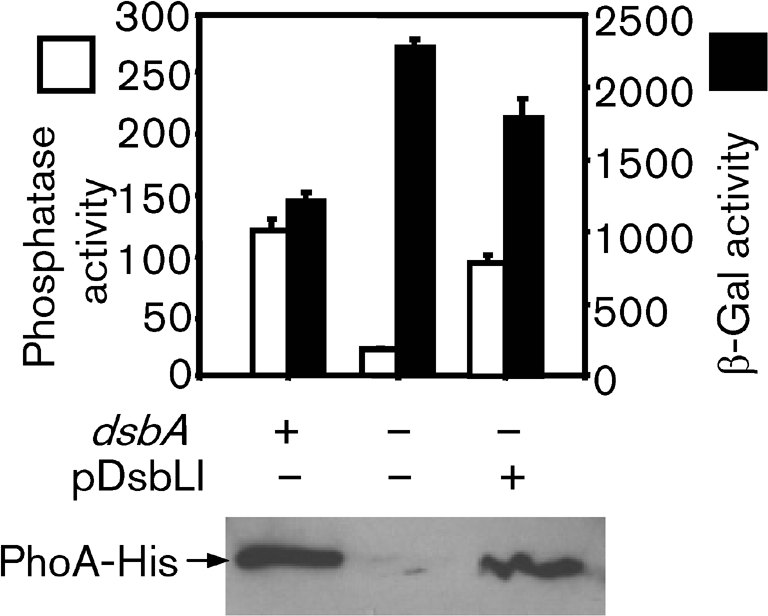
DsbLI have oxidoreductase activity. All strains contained a phoA–6×His construct followed immediately by a promoterless lacZ gene. The dsbA gene was deleted in the designated strains and the pDsbLI plasmid was introduced as indicated. Bacteria were grown overnight in low-phosphate minimal glucose MOPS medium. Cells were assayed for phosphatase activity (white bars) and β-galactosidase activity (β-Gal; black bars), and subjected to Western blot analysis using anti-6×His antibody (lower panel). Both phosphatase and β-galactosidase activities are defined as (μmol o-nitrophenol formed min−1)×106/(OD600×ml cell suspension). Data are presented as mean±sd, where n=4. The strains used were BK683, BK685 and BK685 pDsbLI.
In order to determine the function of these two proteins in Salmonella, we first tested the effect of dsbL or dsbI deletions on expression of a hilA–lacZ fusion. The hilA gene, encoding the master regulator of the SPI1 T3SS (Bajaj et al., 1995), is transcriptionally controlled by a complex regulatory circuit (Ellermeier et al., 2005). Consistent with our previous data (Lin et al., 2008), deletion of dsbA or dsbB significantly decreased the expression of hilA (Fig. 2a). SrgA is a periplasmic disulfide bond oxidoreductase encoded on the Salmonella virulence plasmid. Deletion of srgA alone had no effect on the expression of hilA, while a ΔdsbA ΔsrgA double mutation led to a greater decrease in expression than the ΔdsbA single mutation, as we have previously shown (Lin et al., 2008). Similar to srgA, deletion of dsbL or dsbI alone did not have much effect on hilA expression, but ΔdsbA ΔdsbL or ΔdsbB ΔdsbI double mutations reduced hilA expression more than either the ΔdsbA or the ΔdsbB single mutation.
Fig. 2.

Effects of oxidoreductase gene deletions on hilA and wca expression. Strains contained a hilA–lacZ fusion (a) or wca–lacZ fusion (b) with the indicated genes deleted (denoted by ‘−’). Cultures were grown statically overnight in LB [1 % (w/v) NaCl and 0.5 mM DTT] and then assayed for β-galactosidase activity, which is defined as (μmol o-nitrophenol formed min−1)×106/(OD600×ml cell suspension). Data are presented as mean±sd, where n=4. The strains used were (a) JS749, JS754, JS759, JS761 and JS859–JS863; (b) JS856 and JS864–JS871.
Disulfide bond status is one of several signals that can activate the RcsCDB phosphorelay system, composed of the sensor RcsC, the phosphotransfer protein RcsD (YojN), and the response regulator RcsB (Lin et al., 2008; Majdalani & Gottesman, 2005). Activation of the capsule synthesis operon, wcaA–F is controlled by RcsCDB (Majdalani & Gottesman, 2005), resulting in a mucoid phenotype. We noticed that the ΔdsbA ΔdsbL or ΔdsbB ΔdsbI double mutants are mucoid, just like a ΔdsbA ΔsrgA double mutant. In contrast, serovar Typhimurium dsbA or dsbB single mutants are not obviously mucoid. Therefore, expression of the wca genes can be used to monitor disulfide bond status in the periplasm. As shown in Fig. 2(b), wca expression was induced ∼37-fold in the dsbA mutant, while deletion of both dsbA and srgA led to more than a 100-fold induction of the wca fusion. Consistent with the results shown in Fig. 2(a), deletion of dsbL or dsbI alone had no effect on wca expression. However, a ΔdsbA ΔdsbL or ΔdsbB ΔdsbI double mutation activated wca expression to a higher level than either the ΔdsbA or the ΔdsbB single mutation. Furthermore, deletion of dsbL in the dsbA srgA background led to a further induction of the wca fusion. Taken together, these data suggest that DsbL and DsbI contribute to disulfide bond formation in the periplasm.
DsbL and DsbI function as a redox pair for disulfide bond formation
To further understand the role of DsbL and DsbI in Salmonella, we investigated the ability of DsbL or DsbI to complement dsbA or dsbB mutations. First, we tested whether overproduction of DsbL or DsbI was able to suppress the activation of the wca genes. We introduced plasmids pDsbA, pDsbB, pDsbL, pDsbI or pDsbLI (expressing both proteins), or the vector control pWKS30 into a wca–lacZ fusion strain in wild-type, dsbA, dsbA srgA, dsbB or dsbA dsbB deletion backgrounds. These plasmids conferred very minor effects on expression of wca genes in an otherwise wild-type background (Fig. 3a, b). As expected, production of DsbA from a plasmid significantly suppressed the induction of wca in dsbA or dsbA srgA deletion backgrounds (Fig. 3a). In contrast, overproduction of DsbL only slightly suppressed wca activation in these backgrounds. However, the co-expression from the plasmid of both DsbL and DsbI decreased expression of wca similarly to overexpression of DsbA. The DsbB-expressing plasmid was also able to fully overcome the activation of wca expression in the ΔdsbB mutant (Fig. 3b). Overproduction of DsbI alone also fully suppressed wca induction in the ΔdsbB mutation. The plasmid pDsbLI completely suppressed the induction in both the ΔdsbB and the ΔdsbA ΔdsbB mutant backgrounds. These results suggest that DsbI is able to reoxidize DsbA in the absence of DsbB, but that DsbL is not efficiently reoxidized by DsbB. However, DsbL does efficiently contribute to disulfide bond formation in the presence of DsbI, suggesting that these proteins function best together as a redox pair in Salmonella.
Fig. 3.
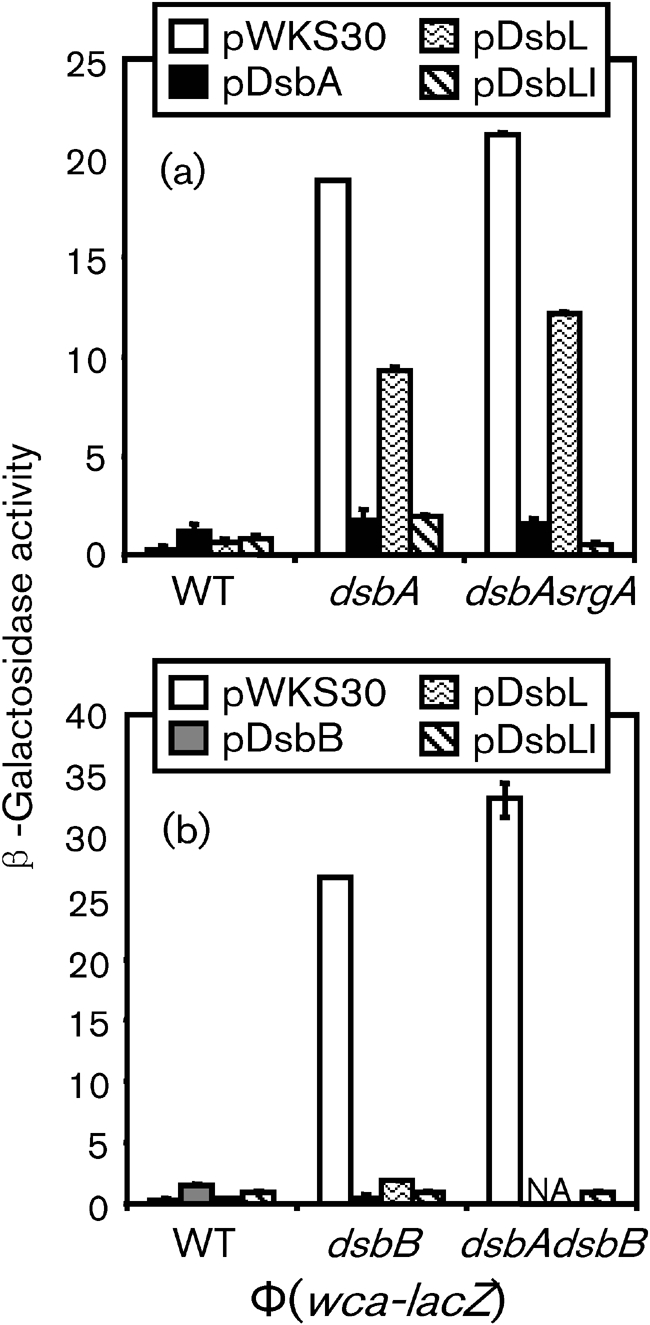
Complementation of dsbA and/or dsbB mutations by dsbL and dsbI. Strains contained the wca–lac fusion with the deletions of genes designated below each graph and the indicated plasmids. Cultures were grown statically overnight in LB [1 % (w/v) NaCl and 0.5 mM DTT] and then assayed for β-galactosidase activity, which is defined as (μmol o-nitrophenol formed min−1)×106/(OD600×ml cell suspension). Data are presented as mean±sd, where n=4. The strains used were plasmid-containing derivatives of (a) JS856, JS864 and JS868; (b) JS856, JS869 and JS872. WT, wild-type.
The DsbLI system is unable to substitute for DsbAB in motility
Loss of DsbA or DsbB prevents proper assembly of the flagellar apparatus, causing a severe motility defect (Dailey & Berg, 1993). In order to determine the ability of DsbL and DsbI to facilitate flagellar assembly, we monitored motility in various mutant backgrounds while overproducing DsbL or DsbI. As expected, deletion of dsbL or dsbI, like srgA, conferred no obvious motility phenotype (Fig. 4a). Loss of DsbA led to a severe motility defect (Fig. 4b), a phenotype exacerbated by also deleting srgA (Fig. 4c). Interestingly, while overproduction of DsbA could fully complement the motility defect in the dsbA or dsbA srgA deletion strains, overproduction of DsbL had no apparent effect (Fig. 4b, c). Moreover, even overproducing DsbL and DsbI together had no significant effect on the motility defect in the dsbA or dsbA srgA mutant backgrounds.
Fig. 4.

DsbLI are unable to substitute for DsbAB in motility. Single colonies of the indicated strains were stabbed onto motility agar plates and incubated overnight. The genotype of the background strain is marked for each panel unless otherwise noted in the individual labels. The strains used were: (a) 14028, JS741, JS850 and JS851; plasmid-containing derivatives of (b) JS326, (c) JS748, (d) JS849, (e) JS326, JS849 and JS873, except for WT=14028.
Loss of DsbB also conferred a significant motility defect (Fig. 4d). Overproduction of DsbI in this background partially suppressed this phenotype (Fig. 4d). Overproduction of DsbL and DsbI together further suppressed the motility defect in the dsbB mutant, but not to wild-type levels. These results are consistent with those described above, suggesting that although DsbI can work with DsbA, it functions better with DsbL. However, these data also indicate that the DsbLI system cannot fully overcome the motility deficiency caused by loss of the DsbAB system. To further confirm these results, we also introduced plasmid pDsbLI into a ΔdsbA ΔdsbB double mutant background. Consistent with our previous observations, the double mutant was mucoid and non-motile. Introduction of the plasmid expressing both DsbL and DsbI apparently relieved the mucoid phenotype, but had little effect on motility (Fig. 4e). Thus, although the DsbLI redox system does contribute to disulfide bond formation, as evidenced by the change in the mucoid phenotype, it cannot substitute for the DsbAB system in flagellar assembly.
DsbL and DsbI contribute to ASST activity
DsbL and DsbI are encoded downstream of a putative ASST encoded by the assT gene. It has been reported that in Enterobacter amnigenus and uropathogenic E. coli (UPEC) disulfide bond formation is required for efficient production of functional ASST in the periplasm (Grimshaw et al., 2008; Kwon & Choi, 2005). Therefore, a physiological role for the DsbLI system is likely to be the activation of ASST in Salmonella. In the serovar Typhimurium LT2 genome sequence, assT is annotated as two ORFs, STM3191 and STM3192 (McClelland et al., 2001). We cloned and sequenced this region from serovar Typhimurium strain 14028. Sequence analysis showed a single ORF encoding a putative protein homologous over its entire length to known ASST enzymes. Analysis suggested that the LT2 genome has acquired a frame-shift mutation that inactivates assT (see below).
E. coli K-12 does not have the assT dsbLI or any other apparent redox system besides DsbAB, so we first tested for ASST activity in wild-type, dsbA or dsbB deletion backgrounds in E. coli K-12 strain DH5α. We cloned the putative assT gene with or without dsbLI. As shown in Fig. 5(a), the introduction of pAssT or pAssT DsbLI into the wild-type strain conferred high-level ASST activity. The deletion of dsbA or dsbB reduced ASST activity 4.6- and threefold, respectively, in strains making only AssT. In contrast, co-expression of DsbL and DsbI resulted in significant ASST activity even in dsbA or dsbB mutant backgrounds.
Fig. 5.

DsbL and DsbI contribute to ASST activity in E. coli (a) and Salmonella (b). The strains contained pWKS30 (vector), pAssT or pAssT DsbLI in the mutant backgrounds as indicated below the graphs. Strains were grown overnight at 37 °C in LB broth with 2 mM DTT and 100 μg ampicillin ml−1, and assessed for ASST activity as described in Methods. ASST activity=(μmol p-nitrophenol formed min−1)×106/(OD600×ml cell suspension). The data are presented as mean±sd, where n=4. Strains used were plasmid-containing derivatives of (a) DH5αλpir, DX1616 and DX1617; (b) JS853 and JS874–JS877.
We then introduced these plasmids into various Salmonella strains in which the chromosomal assT dsbLI genes were deleted. Fig. 5(b) shows that the plasmid producing AssT conferred robust ASST activity. This activity was reduced 3.7-fold in the dsbA mutant and further reduced in a ΔdsbA ΔsrgA double mutant background. Deletion of srgA alone had little effect. As expected, a dsbB deletion had a similar effect to that of the dsbA srgA double mutations. In contrast, when we overproduced DsbL and DsbI from the plasmid, the ASST activity was independent of DsbA, SrgA and DsbB, suggesting that DsbL and DsbI efficiently introduce disulfide bounds into the ASST protein in Salmonella.
The assT and dsbLI genes are independently transcribed
Previous studies in UPEC and Enterobacter amnigenus imply that assT and dsbLI are encoded in one operon (Grimshaw et al., 2008; Kwon & Choi, 2005). To determine whether assT and dsbLI are in one operon in Salmonella, we inserted a chloramphenicol resistance cassette into assT with transcription of the cassette directed antisense to assT transcription. Then we measured the effect of this insertion on the expression of a lacZ transcriptional fusion in the downstream gene dsbL. The results indicated that there was little effect of the ΔassT : : Cm mutation on dsbL expression (16.58±0.96 β-galactosidase units, assT+; 14.01±0.04, assT : : Cm). This suggests that assT and dsbL are transcribed independently in Salmonella. Further analysis showed that deletion of dsbA and srgA or dsbB had no effect on transcription of dsbL or dsbI (data not shown). It is not clear if these genes are regulated in response to any particular stimuli.
Although earlier microarray data show that the expression of assT in serovar Typhimurium is induced 3.28-fold after infection of macrophages (Eriksson et al., 2003), our data suggested that expression of assT was not induced in macrophages (155.6±0.35 units c.f.u.−1 in LB; 128.8±5.67 units c.f.u.−1 in macrophages). In contrast to assT, expression of dsbL was slightly induced (467.15±1.06 units c.f.u.−1 in LB; 855.8±90.9 units c.f.u.−1 in macrophages), consistent with the hypothesis that assT and dsbL are transcribed independently.
ASST has no apparent function during acute infection
To explicitly test for a role of AssT in virulence, we performed competition assays after either i.p. or oral infection. Deletion of assT conferred no virulence defect compared with an isogenic wild-type strain after i.p. infection (Table 2). STM4098 encodes an additional putative ASST in the Salmonella chromosome. Deletion of stm4098 along with deletion of assT still conferred no virulence phenotype in bacteria recovered from the spleen after i.p. infection or in the intestines or spleens of mice infected orally (Table 2). However, insertions in assT have been identified in a screen for mutations affecting long-term persistence in mice (Lawley et al., 2006). Thus, although loss of AssT activity does not affect acute infection, this enzyme might have some more subtle effect that can only be observed during a longer infection period.
Table 2.
Effects of ΔassT in mouse virulence assays
| Strain* | Route of infection† | Tissue | Median CI‡ | Number of mice | P§ |
|---|---|---|---|---|---|
| ΔassT | i.p. | Spleen | 1.23 | 4 | 0.39 |
| ΔassT Δstm4098 | i.p. | Spleen | 3.05 | 3 | 0.22 |
| ΔassT Δstm4098 | Oral | Spleen | 1.69 | 5 | 0.07 |
| ΔassT Δstm4098 | Oral | Intestine | 3.77 | 5 | 0.02 |
*All inocula contained a 1 : 1 ratio of the designated mutant and isogenic wild-type strain. Strains used: i.p., JS852, JS878 and 14028; oral, JS879 and JS107.
†Doses: 103 c.f.u. i.p., 108 c.f.u. oral.
‡Competitive index=output(mutant/wild-type)/input(mutant/wild-type).
§Student's t test comparing output versus inoculum.
DISCUSSION
The DsbA/DsbB catalytic system in E. coli K-12 has been widely studied and serves as the paradigm for disulfide bond oxidoreductases (Kadokura et al., 2003). In Salmonella, the virulence plasmid-encoded DsbA paralogue SrgA has also been characterized (Bouwman et al., 2003). Here we show that Salmonella encodes an additional pair of DsbA/DsbB paralogues, DsbL and DsbI, in the chromosome associated with an ASST. DsbL/DsbI contribute to overall periplasmic disulfide bond formation, as evidenced by effects on SPI1 expression and activation of the RcsCDB system. However, DsbL/DsbI are unable to compensate for DsbA/DsbB in motility.
Grimshaw et al. (2008) have characterized the orthologous system from the UPEC strain CFT073. Although DsbL and DsbI are 88 % and 87 % identical, respectively, between the two bacteria, there are notable phenotypic differences. Consistent with our data, Grimshaw and colleagues concluded that UPEC DsbL/DsbI could not complement dsbA/dsbB mutants for motility, but could form disulfide bonds in ASST. They suggested that, whereas DsbL is inherently more oxidizing than DsbA, charged amino acids around the active site might limit potential substrates. Indeed, they concluded that DsbL/DsbI are specific for ASST. While it is apparent that neither DsbL orthologue is capable of oxidizing a key flagellar protein(s), motility and ASST activity were the only in vivo functional assays performed in their study and it is not known if DsbLI facilitate disulfide bond formation in additional proteins in the UPEC strain. These authors also noted, as did we, that DsbL and DsbI function best together as a redox pair. However, their data suggest that the UPEC DsbL can partially function with DsbB, while DsbA cannot function with UPEC DsbI. In striking contrast, we noted that Salmonella DsbL could function only weakly with DsbB, whereas DsbA provided robust activity in conjunction with Salmonella DsbI (Figs 3 and 4).
The assT gene was originally identified in various uropathogenic strains of E. coli (Lloyd et al., 2007). Mobley and colleagues had shown that assT transcription is increased in UPEC cells recovered from the urine of infected mice (Snyder et al., 2004). This result and the fact that the assT (and dsbLI) genes are missing in several strains of E. coli, including K12 strains, led these authors to conclude that assT was ‘uropathogen-specific’, with the implication that this gene is somehow involved in virulence, an assumption that has been propagated in subsequent papers (Grimshaw et al., 2008; Malojcic et al., 2008). However, analysis shows that the assT dsbLI genes are present in the same genomic context in a variety of Enterobacteriaceae, including: all the S. enterica subsp. enterica serovars, as well as S. enterica subsp. arizonae and Salmonella bongori; numerous E. coli strains, including non-UPEC isolates, but not K12 or O157 isolates; other Escherichia species; and Citrobacter. Phylogenetic analysis of the assT DNA sequence from these various organisms shows a relationship that reflects the accepted phylogeny of the overall genomes (Fig. 6; Vernikos et al., 2007). The simplest interpretation of these data is that the assT dsbLI genes were present in the common ancestor of these species, but that these genes were lost during the evolution of certain lineages. In both S. enterica serovar Typhimurium strain LT2 (McClelland et al., 2001) and Salmonella bongori strain 12149 (Sanger Institute), the assT genes apparently contain independent frameshift mutations. As shown here, the assT gene encodes a functional enzyme in serovar Typhimurium strain 14028.
Fig. 6.

Maximum-likelihood phylogenetic tree of assT DNA sequences. Sequences are from representative genomes in which the assT dsbLI genes are in the same overall genomic context. Those branches with <70 % bootstrap support from 1000 replicates are marked with an asterisk.
The presence of assT in UPEC strains and its apparent induction during growth in the bladder led to the suggestion that the gene product has a role in UPEC virulence, although no direct assay was performed. Although a number of interesting molecules such as the catecholamine epinephrine (Kobashi et al., 1987), known to affect virulence gene expression in intestinal pathogens (Walters & Sperandio, 2006), and various phenolic antibiotics (Kim et al., 1992) have been shown in vitro to be modified by ASST enzymes, we have shown that loss of assT confers no phenotype during acute Salmonella infection in mice. Mutations in assT do confer a defect in long-term infection of mice (Lawley et al., 2006). Thus, any role in virulence is subtle at best and the physiological role of ASST in the Enterobacteriaceae is yet to be determined. DsbI has been shown to affect the ability of Helicobacter pylori to colonize mouse stomach, but this result is not surprising given that DsbI (not associated with assT or dsbL genes) appears to be the only DsbB-like protein in this organism (Godlewska et al., 2006).
Acknowledgments
This work was supported by Public Health Service grant AI063230 from the National Institute of Allergy and Infectious Diseases. We thank Jim Bardwell and Rex Gaskins for helpful discussions and Rachel Whittaker for help with the phylogenetic analyses.
Abbreviations
ASST, arylsulfate sulfotransferase
i.p., intraperitoneal(ly)
SPI1, Salmonella pathogenicity island 1
T3SS, type III secretion system
UPEC, uropathogenic E. coli
References
- Bajaj, V., Hwang, C. & Lee, C. A. (1995). HilA is a novel OmpR/ToxR family member that activates the expression of Salmonella typhimurium invasion genes. Mol Microbiol 18, 715–727. [DOI] [PubMed] [Google Scholar]
- Bardwell, J. C., McGovern, K. & Beckwith, J. (1991). Identification of a protein required for disulfide bond formation in vivo. Cell 67, 581–589. [DOI] [PubMed] [Google Scholar]
- Bouwman, C. W., Kohli, M., Killoran, A., Touchie, G. A., Kadner, R. J. & Martin, N. L. (2003). Characterization of SrgA, a Salmonella enterica serovar Typhimurium virulence plasmid-encoded paralogue of the disulfide oxidoreductase DsbA, essential for biogenesis of plasmid-encoded fimbriae. J Bacteriol 185, 991–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherepanov, P. P. & Wackernagel, W. (1995). Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene 158, 9–14. [DOI] [PubMed] [Google Scholar]
- Collet, J. F. & Bardwell, J. C. (2002). Oxidative protein folding in bacteria. Mol Microbiol 44, 1–8. [DOI] [PubMed] [Google Scholar]
- Dailey, F. E. & Berg, H. C. (1993). Mutants in disulfide bond formation that disrupt flagellar assembly in Escherichia coli. Proc Natl Acad Sci U S A 90, 1043–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko, K. A. & Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97, 6640–6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellermeier, C. D. & Slauch, J. M. (2004). RtsA coordinately regulates DsbA and the Salmonella pathogenicity island 1 type III secretion system. J Bacteriol 186, 68–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellermeier, C. D., Janakiraman, A. & Slauch, J. M. (2002). Construction of targeted single copy lac fusions using lambda Red and FLP-mediated site-specific recombination in bacteria. Gene 290, 153–161. [DOI] [PubMed] [Google Scholar]
- Ellermeier, C. D., Ellermeier, J. R. & Slauch, J. M. (2005). HilD, HilC and RtsA constitute a feed forward loop that controls expression of the SPI1 type three secretion system regulator hilA in Salmonella enterica serovar Typhimurium. Mol Microbiol 57, 691–705. [DOI] [PubMed] [Google Scholar]
- Eriksson, S., Lucchini, S., Thompson, A., Rhen, M. & Hinton, J. C. (2003). Unravelling the biology of macrophage infection by gene expression profiling of intracellular Salmonella enterica. Mol Microbiol 47, 103–118. [DOI] [PubMed] [Google Scholar]
- Felsenstein, J. (1985). Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791. [DOI] [PubMed] [Google Scholar]
- Godlewska, R., Dzwonek, A., Mikula, M., Ostrowski, J., Pawlowski, M., Bujnicki, J. M. & Jagusztyn-Krynicka, E. K. (2006). Helicobacter pylori protein oxidation influences the colonization process. Int J Med Microbiol 296, 321–324. [DOI] [PubMed] [Google Scholar]
- Grimshaw, J. P., Stirnimann, C. U., Brozzo, M. S., Malojcic, G., Grutter, M. G., Capitani, G. & Glockshuber, R. (2008). DsbL and DsbI form a specific dithiol oxidase system for periplasmic arylsulfate sulfotransferase in uropathogenic Escherichia coli. J Mol Biol 380, 667–680. [DOI] [PubMed] [Google Scholar]
- Ha, U. H., Wang, Y. & Jin, S. (2003). DsbA of Pseudomonas aeruginosa is essential for multiple virulence factors. Infect Immun 71, 1590–1595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haldimann, A. & Wanner, B. L. (2001). Conditional-replication, integration, excision, and retrieval plasmid–host systems for gene structure–function studies of bacteria. J Bacteriol 183, 6384–6393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inaba, K. & Ito, K. (2008). Structure and mechanisms of the DsbB–DsbA disulfide bond generation machine. Biochim Biophys Acta 1783, 520–529. [DOI] [PubMed] [Google Scholar]
- Jackson, M. W. & Plano, G. V. (1999). DsbA is required for stable expression of outer membrane protein YscC and for efficient Yop secretion in Yersinia pestis. J Bacteriol 181, 5126–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadokura, H., Katzen, F. & Beckwith, J. (2003). Protein disulfide bond formation in prokaryotes. Annu Rev Biochem 72, 111–135. [DOI] [PubMed] [Google Scholar]
- Kim, D. H., Yoon, H. K., Koizumi, M. & Kobashi, K. (1992). Sulfation of phenolic antibiotics by sulfotransferase obtained from a human intestinal bacterium. Chem Pharm Bull (Tokyo) 40, 1056–1057. [DOI] [PubMed] [Google Scholar]
- Kimball, R. A., Martin, L. & Saier, M. H., Jr (2003). Reversing transmembrane electron flow: the DsbD and DsbB protein families. J Mol Microbiol Biotechnol 5, 133–149. [DOI] [PubMed] [Google Scholar]
- Kobashi, K., Kim, D. H. & Morikawa, T. (1987). A novel type of arylsulfotransferase. J Protein Chem 6, 237–244. [Google Scholar]
- Kwon, A. R. & Choi, E. C. (2005). Role of disulfide bond of arylsulfate sulfotransferase in the catalytic activity. Arch Pharm Res 28, 561–565. [DOI] [PubMed] [Google Scholar]
- Kwon, A. R., Yun, H. J. & Choi, E. C. (2001). Kinetic mechanism and identification of the active site tyrosine residue in Enterobacter amnigenus arylsulfate sulfotransferase. Biochem Biophys Res Commun 285, 526–529. [DOI] [PubMed] [Google Scholar]
- Lawley, T. D., Chan, K., Thompson, L. J., Kim, C. C., Govoni, G. R. & Monack, D. M. (2006). Genome-wide screen for Salmonella genes required for long-term systemic infection of the mouse. PLoS Pathog 2, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, D., Rao, C. V. & Slauch, J. M. (2008). The Salmonella SPI1 type three secretion system responds to periplasmic disulfide bond status via the flagellar apparatus and the RcsCDB system. J Bacteriol 190, 87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd, A. L., Rasko, D. A. & Mobley, H. L. (2007). Defining genomic islands and uropathogen-specific genes in uropathogenic Escherichia coli. J Bacteriol 189, 3532–3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majdalani, N. & Gottesman, S. (2005). The Rcs phosphorelay: a complex signal transduction system. Annu Rev Microbiol 59, 379–405. [DOI] [PubMed] [Google Scholar]
- Malojcić, G., Owen, R. L., Grimshaw, J. P., Brozzo, M. S., Dreher-Teo, H. & Glockshuber, R. (2008). A structural and biochemical basis for PAPS-independent sulfuryl transfer by aryl sulfotransferase from uropathogenic Escherichia coli. Proc Natl Acad Sci U S A 105, 19217–19222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloy, S. R., Stewart, V. J. & Taylor, R. K. (1996). Genetic Analysis of Pathogenic Bacteria: a Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Mann, B. A. & Slauch, J. M. (1997). Transduction of low-copy number plasmids by bacteriophage P22. Genetics 146, 447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClelland, M., Sanderson, K. E., Spieth, J., Clifton, S. W., Latreille, P., Courtney, L., Porwollik, S., Ali, J., Dante, M. & other authors (2001). Complete genome sequence of Salmonella enterica serovar Typhimurium LT2. Nature 413, 852–856. [DOI] [PubMed] [Google Scholar]
- Miki, T., Okada, N. & Danbara, H. (2004). Two periplasmic disulfide oxidoreductases, DsbA and SrgA, target outer membrane protein SpiA, a component of the Salmonella pathogenicity island 2 type III secretion system. J Biol Chem 279, 34631–34642. [DOI] [PubMed] [Google Scholar]
- Neidhardt, F. C., Bloch, P. L. & Smith, D. F. (1974). Culture medium for enterobacteria. J Bacteriol 119, 736–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peek, J. A. & Taylor, R. K. (1992). Characterization of a periplasmic thiol : disulfide interchange protein required for the functional maturation of secreted virulence factors of Vibrio cholerae. Proc Natl Acad Sci U S A 89, 6210–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raczko, A. M., Bujnicki, J. M., Pawlowski, M., Godlewska, R., Lewandowska, M. & Jagusztyn-Krynicka, E. K. (2005). Characterization of new DsbB-like thiol-oxidoreductases of Campylobacter jejuni and Helicobacter pylori and classification of the DsbB family based on phylogenomic, structural and functional criteria. Microbiology 151, 219–231. [DOI] [PubMed] [Google Scholar]
- Silhavy, T. J., Berman, M. L. & Enquist, L. W. (1984). Experiments with Gene Fusions. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Slauch, J. M. & Silhavy, T. J. (1991). cis-acting ompF mutations that result in OmpR-dependent constitutive expression. J Bacteriol 173, 4039–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder, J. A., Haugen, B. J., Buckles, E. L., Lockatell, C. V., Johnson, D. E., Donnenberg, M. S., Welch, R. A. & Mobley, H. L. (2004). Transcriptome of uropathogenic Escherichia coli during urinary tract infection. Infect Immun 72, 6373–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson, J. D., Higgins, D. G. & Gibson, T. J. (1994). clustal w: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22, 4673–4680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vernikos, G. S., Thomson, N. R. & Parkhill, J. (2007). Genetic flux over time in the Salmonella lineage. Genome Biol 8, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walters, M. & Sperandio, V. (2006). Quorum sensing in Escherichia coli and Salmonella. Int J Med Microbiol 296, 125–131. [DOI] [PubMed] [Google Scholar]
- Wang, R. F. & Kushner, S. R. (1991). Construction of versatile low-copy-number vectors for cloning, sequencing and gene expression in Escherichia coli. Gene 100, 195–199. [PubMed] [Google Scholar]
- Watarai, M., Tobe, T., Yoshikawa, M. & Sasakawa, C. (1995). Disulfide oxidoreductase activity of Shigella flexneri is required for release of Ipa proteins and invasion of epithelial cells. Proc Natl Acad Sci U S A 92, 4927–4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilgenbusch, J. C. & Swofford, D. (2003). Inferring evolutionary trees with PAUP*. Curr Protoc Bioinformatics Chapter 6, Unit 6.4. [DOI] [PubMed] [Google Scholar]
- Yu, D., Ellis, H. M., Lee, E. C., Jenkins, N. A., Copeland, N. G. & Court, D. L. (2000). An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci U S A 97, 5978–5983. [DOI] [PMC free article] [PubMed] [Google Scholar]