

Abstract
The Yersinia pestis Hms+ phenotype is a manifestation of biofilm formation that causes adsorption of Congo red and haemin at 26 °C but not at 37 °C. This phenotype is required for blockage of the proventricular valve of the oriental rat flea and plays a role in transmission of bubonic plague from fleas to mammals. Genes responsible for this phenotype are located in three separate operons, hmsHFRS, hmsT and hmsP. HmsH and HmsF are outer membrane (OM) proteins, while the other four Hms proteins are located in the inner membrane. According to the Hidden Markov Method-based predictor, HmsH has a large N terminus in the periplasm, a β-barrel structure with 16 β-strands that traverse the OM, eight surface-exposed loops, and seven short turns connecting the β-strands on the periplasmic side. Here, we demonstrate that HmsH is a heat-modifiable protein, a characteristic of other β-barrel proteins, thereby supporting the bioinformatics analysis. Alanine scanning mutagenesis was used to identify conserved amino acids in the HmsH-like family that are critical for the function of HmsH in biofilm formation. Of 23 conserved amino acids mutated, four residues affected HmsH function and three likely caused protein instability. We used formaldehyde cross-linking to demonstrate that HmsH interacts with HmsF but not with HmsR, HmsS, HmsT or HmsP. Loss-of-function HmsH variants with single alanine substitutions retained their β-structure and interaction with HmsF. Finally, using a polar hmsH : : mini-kan mutant, we demonstrated that biofilm development is not important for the pathogenesis of bubonic or pneumonic plague in mice.
INTRODUCTION
Bacterial biofilms are multicellular structured communities of bacterial cells enclosed in a self-produced polymeric matrix that adheres to inert or living surfaces (Itoh et al., 2008; O'Toole et al., 1999; Stoodley et al., 2002). Biofilms provide a mode of bacterial growth that protects bacterial communities from harmful conditions in the host, such as pH changes, oxygen radicals, and attack by the immune system (Jefferson, 2004). Moreover, established biofilms can shield cells from antimicrobial agents, making biofilms extremely difficult to eradicate from living hosts (Gotz, 2002; Jefferson, 2004; O'Gara, 2007; Otto, 2008). Biofilm development by Yersinia pestis in the proventricular valve separating the oesophagus from the midgut or stomach results in blockage of fleas. Blocked fleas attempt to feed repeatedly, causing the plague bacillus to be regurgitated into the blood wound and transferring the bacteria to a mammalian host. Blocked flea transmission of plague has been a paradigm since the early observations of Bacot (Bacot & Martin, 1914; Bacot, 1915; Hinnebusch et al., 1996; Jarrett et al., 2004).
While proteins, nucleic acids, lipids/phospholipids, absorbed nutrients and metabolites are often present in biofilm matrices, exopolysaccharide (EPS) is a key component of the biofilm matrix in many biofilm-forming bacteria and may include multiple different polymers and form various types of structures (Goller & Romeo, 2008; Sutherland, 2001; Vuong et al., 2004). Poly-β-1,6-N-acetyl-d-glucosamine (poly-β-1,6-GlcNac) is a component of the biofilms of several bacterial species (Bobrov et al., 2008; Itoh et al., 2005; Izano et al., 2007; Kaplan et al., 2004; Mack et al., 1999; Wang et al., 2004). It was originally identified in Staphylococcus epidermidis and referred to as polysaccharide intercellular adhesin (PIA). Similarly, a poly-β-1,6-GlcNac EPS has been described in Staphylococcus aureus, and also appears to be made by several Gram-negative bacteria, including Escherichia coli MG1655 (termed PGA) and Y. pestis (Bobrov et al., 2008; Itoh et al., 2005; Kaplan et al., 2004; Mack et al., 1999; Wang et al., 2004). Synthesis of poly-β-1,6-GlcNAc requires the icaADBC, pgaABCD (formerly ycdSRQP) and hmsHFRS operons, respectively, in S. epidermidis, E. coli and Y. pestis (Kirillina et al., 2004; Mack et al., 1996; Wang et al., 2004).
The chromosomally borne hmsHFRS operon, located within the pgm locus, has been identified as being responsible for the adsorption of haemin and Congo red (CR) to the outer membrane (OM) of Y. pestis cells at 26 °C but not at 37 °C (Jackson & Burrows, 1956; Lillard et al., 1997; Pendrak & Perry, 1991, 1993; Perry et al., 1990; Surgalla & Beesley, 1969). CR binding is now known to be a measure of EPS production in plague biofilm development (Bobrov et al., 2008; Kirillina et al., 2004). The Y. pestis hmsHFRS gene products show high similarities to the E. coli pgaABCD gene products. HmsH, HmsF, HmsR and HmsS have amino acid sequence similarities/identities, respectively, to PgaA, PgaB, PgaC and PgaD of 58.2/41.1 %, 60.8/48.3 %, 83/66.2 % and 50/28.4 % (Itoh et al., 2005; Jones et al., 1999; Wang et al., 2004). HmsH is a proven OM protein with a predicted β-barrel structure and likely acts as a porin through which poly-β-1,6-GlcNAc is exported to the surface of the bacterium (Lillard et al., 1997; Pendrak & Perry, 1993; Perry et al., 2004). HmsF is another OM protein with a putative lipid attachment site typical of lipoproteins and a polysaccharide deacetylase domain. HmsR is a basic, 52 kDa inner membrane (IM) protein with four transmembrane domains and a central cytoplasmic loop with a glycosyltransferase (GT) domain. The GT motif (D, D, D35QXRW) is essential for CR binding in Y. pestis. Thus, HmsR is likely the key enzyme in the biosynthesis of biofilm EPS (Bobrov et al., 2008; Forman et al., 2006; Lillard et al., 1997). HmsS is a small 17.5 kDa IM protein with two transmembrane domains and cytoplasmically located N and C termini (Bobrov et al., 2008; Kirillina et al., 2004; Lillard et al., 1997). Each gene in the hmsHFRS operon is required for CR binding, and the cloned operon restores biofilm development to a Δpgm mutant (Lillard et al., 1997; Pendrak & Perry, 1991, 1993; Perry et al., 1990). In addition to hmsHFRS, two chromosomal genes outside the pgm locus, hmsT and hmsP, are involved in regulating Hms-dependent biofilm development (Jones et al., 1999; Kirillina et al., 2004; Perry et al., 2004). HmsP is an IM protein with two transmembrane domains and a cytoplasmic C terminus containing an EAL domain that is required for cyclic-di-GMP (c-di-GMP) phosphodiesterase activity (Bobrov et al., 2005, 2008; Kirillina et al., 2004). HmsT is an IM protein with four transmembrane domains and a central cytoplasmic GDDEF domain that is required for diguanylate cyclase activity (Bobrov et al., 2008; Jones et al., 1999; Perry et al., 2004). Site-directed mutagenesis has identified conserved amino acids within the GT, deacetylase, GGDEF and EAL domains of HmsR, HmsF, HmsT and HmsP, respectively, that are required for protein function (Forman et al., 2006; Kirillina et al., 2004). Genetic and biochemical analyses have shown interactions among the Hms IM proteins HmsT, HmsP, HmsR and HmsS in Y. pestis (Bobrov et al., 2008). Based on these similarities and experimental work performed in both Y. pestis and E. coli, a model of biofilm formation in Y. pestis has been developed (Fig. 1).
Fig. 1.

Y. pestis Hms-dependent biofilm model. Chains of circles represent linked monomers of the EPS component of the biofilm (filled and unfilled circles, acetylated and deacetylated monomers). Labels indicate the putative or proven enzymic activities/functions of Hms proteins. The ‘+’ sign indicates likely stimulation of HmsR enzymatic activity by c-di-GMP. At 26–34 °C, all Hms proteins are highly expressed, while at 37 °C, the levels of HmsH, HmsR and HmsT are significantly reduced to varying degrees by post-translational mechanisms. HmsT degradation reduces c-di-GMP levels. This, along with low levels of HmsR and HmsH, inhibits biofilm formation.
In this study we (1) show that OM proteins HmsH and HmsF interact with each other but not with the IM proteins HmsR, HmsS, HmsP and HmsT in Y. pestis; (2) identify seven residues important for HmsH function; (3) demonstrate that all residue changes (which cause a defective phenotype) in HmsH and HmsF do not affect their interaction; and (4) prove that biofilm formation is not important for the mammalian pathogenesis of bubonic or pneumonic plague.
METHODS
Bacterial strains and cultivation.
Strains used in this study are listed in Table 1. Y. pestis and E. coli cells were streaked on CR agar (Sigma-Aldrich) or Luria Broth (LB) agar plates, respectively, from buffered glycerol stocks stored at −80 °C (Beesley et al., 1967). Media were supplemented with ampicillin (Ap; 100 μg ml−1), chloramphenicol (Cm; 15 or 30 μg ml−1), kanamycin (Km; 50 μg ml−1) or streptomycin (Sm; 50 μg ml−1) as needed to select for retention of plasmids. For most experiments, individual colonies of Y. pestis cells from CR plates were used to inoculate Tryptose Blood Agar Base (TBA) slants and incubated at 26–30 °C for 24–48 h. Cells were washed off TBA or LB slants with an appropriate medium. OD620 was measured using a Spectronic Genesys5 spectrophotometer and was used to inoculate the medium with equivalent cell numbers. Cultures were incubated at an appropriate temperature overnight. For the crystal violet (CV)-staining assay, Y. pestis cells were grown in the defined TMH medium (Straley & Bowmer, 1986).
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics* | Source or reference |
|---|---|---|
| Y. pestis strains† | ||
| KIM5(pCD1Ap)+ | Apr Pgm+ (Hms+ Ybt+) Lcr+ (pCD1Ap, ′yadA : : bla) Pla+; derived from KIM6+ | Gong et al. (2001) |
| KIM5-2008 (pCD1Ap) | Apr Hms− (hmsH2008 : : mini-kan) Lcr+ (pCD1Ap, ′yadA : : bla) Pla+; derived from KIM6-2008 | This study |
| KIM6+ | Pgm+, (Hms+) Lcr− Pla+ | Fetherston et al. (1992) |
| KIM6 | Pgm− (Δpgm; Hms−) Lcr− Pla+; derived from KIM6+ | Fetherston et al. (1992) |
| KIM6-2008 | Hms− (hmsH2008 : : mini-kan) Lcr− Pla+; derived from KIM6+ | Lillard et al. (1997); Pendrak & Perry (1991) |
| KIM6-2115 | Hms− (in-frame ΔhmsH2115) Lcr− Pla+; derived from KIM6+ | Forman et al. (2006) |
| KIM6-2116 | Hms– (non-polar ΔhmsF2116) Lcr− Pla+; derived from KIM6+ | Forman et al. (2006) |
| KIM6-2118 | Hms− (in-frame ΔhmsR2118) Lcr− Pla+; derived from KIM6+ | Forman et al. (2006) |
| KIM6-2119 | Hms− (in-frame ΔhmsS2119 : : cam) Lcr− Pla+; derived from KIM6+ | Forman et al. (2006) |
| KIM6-2051+ | Hms−, Kmr (hmsT2051 : : mini-kan) Lcr− Pla+; derived from KIM6+ | Kirillina et al. (2004) |
| KIM6-2090.2+ | Pgm+ Hms2+ (ΔhmsP2090.4) Lcr− Pla+; derived from KIM6+ | A. Bobrov and O. Kirillina, University of Kentucky |
| E. coli strains | ||
| DH5α | Cloning strain | Ausubel (1987) |
| DH5α (λ pir) | Strain for maintenance of R6K origin suicide vector | S. C. Straley, University of Kentucky |
| Plasmids | ||
| pNEB193 | 2.7 kb, Apr, cloning vector | New England Biolabs |
| pKNGΔhmsR | 9.0 kb, Smr, 2.2 kb SalI–XbaI fragment from pWSKΔhmsR ligated into SalI–XbaI sites of pKNG101 | Forman et al. (2006) |
| pNPM22 | 9.9 kb, Cmr, hmsH+hmsF′; used in construction of HmsH variant proteins | Lillard et al. (1997); Pendrak & Perry (1993) |
| pNPM22 HmsH-aa substitutions | 9.8 kb, Cmr; HmsH with various amino acid substitutions, 17 in the surface-exposed loops and seven in the predicted N-terminal periplasmic domain | This study |
*Apr, Cmr, Kmr and Smr: resistance to ampicillin, chloramphenicol, kanamycin and streptomycin, respectively. The pgm locus, type III secretion system encoded on pCD1 (Lcr), and plasminogen activator (Pla) are all required for full virulence in Y. pestis.
†Y. pestis strains with a plus sign possess an intact 102 kb pgm locus. Y. pestis Δpgm strains lack the hmsHFRS locus and the yersiniabactin iron transport system.
Plasmid construction and DNA methods.
All plasmids used in this study are listed in Table 1. Plasmid DNA was purified from bacteria cultivated overnight in Heart Infusion broth (HIB) or LB using an alkaline lysis procedure (Birnboim & Doly, 1979). Qiagen Midi- or Miniprep spin columns were used for further purification. Cloning of specific genes followed standard procedures (Maniatis et al., 1982). Amplification of DNA was performed by PCR in a MyCycler thermal cycler (Bio-Rad). Either Native Pfu (Stratagene) or Turbo Pfu (Stratagene) DNA polymerase was used for amplification. Oligonucleotide primers (listed in Supplementary Table S1) were synthesized by Integrated DNA Technologies; primers of 25 nt or longer were PAGE-purified. Transformation of E. coli was carried out by the CaCl2 method (Maniatis et al., 1982). Electroporation was used to introduce plasmids into Y. pestis, as previously described (Fetherston et al., 1995).
Construction of alanine substitutions in the predicted surface loops and N terminus of HmsH.
All amino acid substitutions were made in pNPM22 by a two-step PCR mutagenesis/cloning scheme, as described previously (Forman et al., 2006). This method uses internal primers carrying the desired alanine substitution to generate a PCR product with the desired residue change (Supplementary Table S1). The variant PCR fragments were cloned into pNEB193, then subcloned into pNPM22 or cloned directly into pNPM22 using appropriate restriction sites. Plasmid pNPM22 and its variants express HmsH and an N-terminal fragment of HmsF from their native promoter (Table 1; Forman et al., 2006; Pendrak & Perry, 1993). The construction of all mutations and PCR product amplifications were confirmed by DNA sequencing (Northwestern University Biotechnology Laboratory, IL, USA).
CV-staining assay.
CV staining was performed as previously described (Forman et al., 2006; O'Toole et al., 1999). Isolated colonies from CR plates were used to inoculate TBA slants with the appropriate antibiotics followed by incubation at 26 °C. Cells washed off the slants were used to inoculate TMH media to OD620 0.1, followed by incubation in a shaking water bath at 26 °C for 16–18 h. CV (0.01 %, w/v) was added to the tubes and incubated for 15 min with shaking. The bound CV was solubilized with 80 : 20 % (v/v) ethanol acetone mixture and the A570 was measured using a Spectronic Genesys5 spectrophotometer. Samples with a robust biofilm were diluted 1 : 3 with the ethanol/acetone mix. The A570 is proportional to the level of cell adherence and is a measure of biofilm development.
CR-binding assay.
The original CR-binding assay (Hartzell et al., 1999) was modified as previously described (Kirillina et al., 2004). From CR plates, individual colonies were grown on TBA slants at 26 °C. Cells from slants were used to inoculate HIB to OD620 0.1. Inoculated cultures were grown (∼16 h) in a shaking water bath at 26 °C. Y. pestis cells were pelleted and resuspended in HIB/CR medium (1 % HIB containing 0.2 % galactose and 30 μg CR ml−1) such that all cultures had an equivalent wet cell weight of 5 mg ml−1. Samples were incubated on a rocking platform for 3 h at room temperature (RT; ∼20 °C) or 30 °C. Cells were pelleted for 10 min. The amount of CR bound by Y. pestis cells was determined by measuring the A500 of the culture supernatants using a Spectronic Genesys5 spectrophotometer and subtracting the A500 of the supernatant from uninoculated HIB/CR medium.
SDS-PAGE and Western blot analysis.
For analysis of heat modification of HmsH, a published protocol was followed (Beher et al., 1980; Nandi et al., 2005). Y. pestis cells expressing the full-length wild-type and variant HmsH proteins were grown overnight in HIB at 30 °C. OM fractions were solubilized in 1× SDS-PAGE sample buffer containing 2 % (w/v) SDS and 5 % (v/v) β-mercaptoethanol. Half of the solubilized samples were sheared with a syringe 15 times at RT and the other half were boiled for 10 min. Samples were resolved on 8 % polyacrylamide gels.
Mini 5–10 % polyacrylamide gradient gels with a 5 % polyacrylamide stacking gel were cast using an SG100 gradient maker (Hoefer Scientific). Gels were run at 100 V and 20 mA per gel for ∼5 h and immunoblotted to PVDF membranes (Immobilon P; Millipore).
For Western blot analysis, equal amounts of protein from Y. pestis whole-cell or membrane extracts were separated by SDS-PAGE and immunoblotted to PVDF membranes. The blots were processed using a procedure modified from Towbin et al. (1979). Briefly, PVDF membranes were blocked with 5 % non-fat dry milk in 10 mM Tris/HCl (pH 7.6), 137 mM NaCl with 0.1 % Tween 20 (TBST) and then incubated with an appropriate antibody diluted in TBST. The blots were washed with TBST and incubated with horseradish peroxidase-conjugated protein A (Amersham Pharmacia Biotech). Immunoreactive proteins were detected with the ECL enhanced chemiluminescence Western blotting detection reagent (Amersham Pharmacia Biotech) and visualized on Kodak Biomax Light film. Production of polyclonal rabbit antisera against HmsH, HmsF, HmsR, HmsS, HmsT and HmsP has been described previously (Bobrov et al., 2008; Perry et al., 2004).
Isolation of membrane fractions.
Membrane fractions were isolated from Y. pestis strains grown at 30 °C overnight. Cells were harvested by centrifugation, washed once in PBS [135 mM NaCl, 2.7 mM KCl, 10 mM KH2PO4, 1.8 mM KH2PO4 (pH 7.4)] and resuspended in PBS with a cocktail of protease inhibitors (Calbiochem) to 10 OD units ml−1. The cells were disrupted by sonication using a 550 Sonic Dismembrator (Fisher Scientific). Unbroken cells were removed by centrifugation at 10 000 r.p.m. for 10 min. The membranes were pelleted by ultracentrifugation at 90 000 r.p.m. for 20 min at 4 °C in a TLA120.2 rotor using a Beckman Optima TLX ultracentrifuge. Fractions were mixed with 1× SDS sample buffer and separated by 12 % SDS-PAGE.
Formaldehyde cross-linking of Hms proteins.
Formaldehyde cross-linking (Prossnitz et al., 1988; Warren et al., 2004) was performed on strains grown in HIB at 30 °C. Cells from overnight cultures were harvested by centrifugation and resuspended to OD620 1 in 100 mM sodium phosphate buffer dibasic, pH 6.8. Samples were incubated with 1 % (v/v) formaldehyde in phosphate buffer without shaking. Following cross-linking, 1 ml aliquots of cross-linked samples were centrifuged and washed once with the same buffer. For electrophoresis, pellets were mixed with double-strength sample buffer containing 5 % β-mercaptoethanol to obtain ∼10 OD units ml−1. To reverse the cross-linking, samples were boiled for 30 min. Samples were heated for 10 min at 60 °C prior to electrophoresis through fixed 8 % or 5–10 % mini-gradient polyacrylamide gels.
Sequence analysis.
The amino acid sequences of HmsH and HmsH-like proteins were obtained from the NCBI Entrez Genome Project website (http://www.ncbi.nlm.nih.gov/). All alignments were generated in clustal w (http://align.genome.jp/) and edited by the alignments editor program geneDoc (version 2.6.02).
Virulence testing.
Construction of potentially virulent strains and virulence testing were performed in a Centers for Disease Control (CDC)-approved BSL3 laboratory following Select Agent regulations. Y. pestis strains were transformed with the virulence plasmid pCD1Ap by electroporation (Forman et al., 2008; Gong et al., 2001) and plated on TBA plates containing 50 μg Ap ml−1. The plasmid profile of transformants was analysed, as well as their phenotype on CR agar (Surgalla & Beesley, 1969) and magnesium oxalate plates (Higuchi & Smith, 1961). Supernatants from cultures grown at 37 °C in the absence of CaCl2 were tested for the secretion of LcrV by Western blot analysis using polyclonal antisera against histidine-tagged LcrV (Fields et al., 1999; Forman et al., 2008). For subcutaneous infections, overnight cultures of Y. pestis cells grown in HIB at 26 °C were diluted to OD620 0.1 and incubated in HIB at 26 °C until they reached mid-exponential phase (OD620 ∼0.5). Samples were harvested and diluted in mouse isotonic PBS (149 mM NaCl, 16 mM Na2HPO4, 4 mM NaH2PO4). Groups of four 6- to 8-week-old female Swiss Webster (Hsd : : ND4) mice were injected subcutaneously with 0.1 ml of 10-fold serially diluted bacterial suspensions ranging from 100 to 105 c.f.u. ml−1. Cells used for intranasal infections were grown in HIB with 2 mM CaCl2 at 37 °C and similarly diluted in mouse isotonic PBS. Twenty microlitres of the bacterial suspension was administered to the nares of mice sedated with 100 μg ketamine and 10 mg xylazine per kg body weight. The actual administered dose was determined by plating aliquots of each dose serially diluted in mouse isotonic PBS, in duplicate, onto TBA plates containing 50 μg Ap ml−1. The colonies were counted on plates incubated at 30 °C for 2 days. Mice from two independent infections were observed daily for 2 weeks, and LD50 values were calculated according to the method of Reed & Muench (1938).
All animal care and experimental procedures were conducted in accordance with the Animal Welfare Act, Guide for the Care and Use of Laboratory Animals, PHS Policy and the US Government Principles for the Utilization of and Care for Vertebrate Animals in Teaching, Research, and Training, and approved by the University of Kentucky Institutional Animal Care and Use Committee. The University of Kentucky Animal Care Program first achieved accreditation by the Association for the Assessment and Accreditation of Laboratory Animal Care, Inc. (AAALAC) in 1966 and has maintained full accreditation continuously since that time.
RESULTS
HmsH structure
HmsH is a proven OM protein (Pendrak & Perry, 1993; Perry et al., 2004). According to the Hidden Markov Method-based predictor for β-barrel OM protein topology (PRED_TMBB; http://bioinformatics.biol.uoa.gr/PRED-TMBB), the large N terminus (first 499 aa of the processed protein) is located in the periplasm. The remaining 289 aa traverse the OM 16 times, forming a β-barrel with eight predicted surface-exposed loops and seven short turns connecting the β-strands at the periplasmic side of the protein (Bagos et al., 2004a, b) (Supplementary Fig. S1).
A general characteristic of OM β-barrel proteins is that heat treatment alters the protein conformation such that SDS-PAGE migration is modified (Beher et al., 1980; Nandi et al., 2005; Nitzan et al., 1999). Consequently, we determined whether the SDS-PAGE mobility of HmsH is modified by heat treatment. In unboiled membrane fractions, HmsH migrated with an apparent molecular mass of ∼70 kDa, while heat-treated (boiled) HmsH migrated with an apparent molecular mass of ∼100 kDa. These bands were not observed in a Δpgm strain (which lacks the hmsHFRS operon) or in a strain with a mutation in hmsH, but were present in the hmsF mutant (Fig. 2a). The difference in the migration pattern between heated and unheated HmsH suggests that HmsH is a β-barrel protein.
Fig. 2.

Effect of heat treatment of HmsH and HmsF on migration in polyacrylamide gels. Samples were suspended in sample buffer containing 2 % SDS, heated at 100 °C for 10 min or unheated (RT; ∼20 °C), then resolved by 8 % SDS-PAGE. (a) Western blot probed with HmsH antisera. Arrows identify HmsH in boiled and unboiled samples. (b) Western blot probed with HmsF antisera. The upper arrow indicates the unprocessed form of HmsF, while the lower one indicates the processed form. Strains (see also Table 1): Δpgm, KIM6; hms+, KIM6+; ΔhmsH, KIM6-2115; ΔhmsF, KIM6-2116.
As a negative control, we tested the effect of heat treatment on the migration of HmsF (an OM lipoprotein). Unlike HmsH, HmsF exhibited no change in the migration pattern between boiled and unboiled samples. Two forms of HmsF were detected with the antisera and likely correspond to unprocessed and mature forms, as demonstrated previously (Pendrak & Perry, 1993). These forms were absent in a Δpgm strain or in a strain with a mutation in hmsF, but were present in the hmsH mutant (Fig. 2b). This indicates that heat treatment of HmsF does not affect its conformation and that altered SDS-PAGE migration after heat treatment is not a general phenomenon for Y. pestis OM proteins. These results support the computational prediction that HmsH is a β-barrel protein. These studies (Fig. 2) also demonstrate that expression of HmsH is not required for normal expression/stability of HmsF and vice versa.
Alanine scanning mutagenesis in the predicted loops of HmsH
Conserved residues among homologous proteins from diverse bacteria are often critical to the function of the proteins. A blast search of microbial genomes identified several proteins related to HmsH. Fig. 3(a) shows amino acid sequence alignments of the predicted β-strands and intervening loops of HmsH with HmsH-like proteins from Pseudomonas fluorescens, Pectobacterium atrosepticum (formerly Erwinia carotovora), Xanthomonas oryzae and E. coli (Bell et al., 2004; Blattner et al., 1997; Lee et al., 2005; Paulsen et al., 2005). Of these four showing the highest similarity to Y. pestis HmsH, only E. coli PgaA has been experimentally proven to be required for biofilm development (Itoh et al., 2008; Wang et al., 2004).
Fig. 3.

Analysis of changes in conserved residues in the predicted external loops between β-strands in the C-terminal region of HmsH. (a) Alignment of these regions of HmsH and HmsH-like proteins from: Y. pestis KIM10+ (Ype); P. fluorescens Pf-5 (Pfl); Pectobacterium atrosepticum SCR11043 (Pat; formerly Erwinia carotovora); E. coli K-12 substrain MG1655 (Eco); and X. oryzae KACC10331 (Xor). The black and grey boxes indicate residue identity and similarity, while arrows show the starts (open) and ends (filled) of the eight predicted loops between β-strands. The amino acid change line shows the amino acids individually changed to alanine. (b) Quantitative CR binding after 3 h incubation at 20 °C with CR-containing HIB medium by Y. pestis cells expressing amino acid substitutions in the predicted loops of HmsH. Controls are KIM6-2115 (in-frame ΔhmsH; HmsH−), KIM6+ (Hms+) and KIM6-2115(pNPM22) (HmsH++). Plasmid pNPM22, which expresses hmsH from its native promoter, was also used to express HmsH with alanine substitutions. Thirteen mutants were not significantly different from positive controls (Table 2); only one of these, HmsH-D520A, along with the five mutants with significant decreases in CR binding, are shown. Data shown are the mean of two or more independent experiments with duplicate samples from each trial. Error bars, sd. For clarity, the secondary mutation (D520A) in HmsH-F651-D520A and HmsH-N655A-D520A is not shown in the labels.
Sixteen conserved amino acids in the eight predicted external loops of HmsH were selected for alanine scanning mutagenesis. Substitutions were not made in transmembrane regions in order to avoid disruption of the β-barrel structure (Supplementary Fig. S1). Two mutants (HmsH-N655A and HmsH-F651A) had secondary D520A mutations. Therefore, a strain bearing just the HmsH-D520A substitution was constructed. To test the effect of the alanine substitutions, Y. pestis KIM6-2115 (in-frame ΔhmsH) was transformed with pNPM22 or pNPM22 variants carrying the amino acid substitution. pNPM22 expresses hmsH from its native promoter and restores biofilm formation to KIM6-2115 (Forman et al., 2006). Strains were tested for the ability to form red colonies on CR agar after 48 h at 30 °C and to bind CR in a liquid medium over a 3 h period at RT. In addition, all strains were assayed for biofilm formation by the CV assay (Table 2, Fig. 3b). CR binding measures Y. pestis EPS levels, while CV staining quantitates cellular mass adhered to an abiotic surface (Forman et al., 2006; Kirillina et al., 2004; Simm et al., 2005). Both parameters are used to assay biofilm formation. Finally, the protein expression/stability of each HmsH variant was assessed by Western blot analysis (Table 2).
Table 2.
Effect of amino acid substitutions in HmsH on biofilm phenotypes and protein level
| Amino acid substitutions in HmsH | CR plate | CV staining (%)* | CR binding (%)* | Protein level† |
|---|---|---|---|---|
| Substitution in loop 1‡ | ||||
| HmsH-D520A | Red colonies | 80 % | 100 % | RL |
| Substitution in loop 2 | ||||
| HmsH-F549A | Red colonies | 85 % | 100 % | RL |
| Substitutions in loop 4 | ||||
| HmsH-P615A | Red colonies | 95 % | 83 % | RL |
| HmsH-R617A | Red colonies | 89 % | 100 % | RL |
| Substitutions in loop 5 | ||||
| HmsH-F651A-D520A§ | Red colonies | 105 % | 63 % | <WT |
| HmsH-D653A | Pink colonies | 83 % | 46 % | <WT |
| HmsH-N655A-D520A§ | Small pink colonies | 98 % | 60 % | <WT |
| HmsH-R657A | Red colonies | 89 % | 60 % | ≥WT <RL |
| Substitutions in loop 6 | ||||
| HmsH-P697A | Red colonies | 97 % | 90 % | ≥WT <RL |
| HmsH-D691A | Red colonies | 80 % | 100 % | RL |
| HmsH-Y694A | Red colonies | 95 % | 53 % | RL |
| Substitution in loop 7 | ||||
| HmsH-Y737A | Red colonies | 101 % | 80 % | RL |
| Substitutions in loop 8 | ||||
| HmsH-P769A | Red colonies | 93 % | 83 % | ≥WT <RL |
| HmsH-E775A | Red colonies | 97 % | 116 % | RL |
| HmsH-D771A | Red colonies | 85 % | 93 % | RL |
| HmsH-R774A | Red colonies | 85 % | 90 % | RL |
| Substitutions in the N-terminal region | ||||
| HmsH-D263A | White-to-pink, then red colonies | 58 % | 100 % | >WT |
| HmsH-E345A-D520A§ | Red colonies | 95 % | 118 % | >WT |
| HmsH-E347A | Red colonies | 103 % | 88 % | >WT |
| HmsH-D397A-D520A§ | Red colonies | 86 % | 103 % | >WT |
| HmsH-R433A-D520A§ | Red colonies | 100 % | 107 % | >WT |
| HmsH-R479A | White-to-pink, then red colonies | 46 % | 100 % | >WT |
| HmsH-R491A-D691A|| | Red colonies | 99 % | 122 % | >WT |
*CR-binding values are represented as a percentage of the hmsHFRS+ strain (100 %=0.30 A500 units). CV-staining values are represented as a percentage of the ΔhmsH(pNPM22) strain (100 %=1.72 A570 units).
†WT, level of HmsH expression from the chromosome; RL, level of HmsH expression from pNPM22.
‡External loops between β-strands are numbered starting at the N terminus of HmsH.
§D520A secondary mutation identified by sequencing.
||D691A secondary mutation identified by sequencing.
There was no significant difference in the quantity of CR bound between KIM6-2115(pNPM22), where HmsH is expressed from a moderate-copy-number plasmid, and KIM6+ (Fig. 3b), suggesting that expression of HmsH from a single chromosomal locus was sufficient for maximal biofilm development and that multiple plasmid-encoded copies of hmsH did not increase biofilm formation. Most Y. pestis strains expressing an HmsH with an alanine substitution formed red colonies on CR plates by 48 h, indicating normal biofilm development (Table 2). The HmsH-D653A and HmsH-N655A-D520A mutants had an intermediate phenotype: formation of pink colonies on CR plates by 48 h. Since the HmsH-D520A mutant bound CR normally, the intermediate phenotype of HmsH-N665A-D520A was due to the N655A substitution or a combination of both changes. Short-term CR binding, as determined by a 3 h liquid CR-binding assay, revealed that cells with D653A, N655A-D520A, R657A, F651A-D520A or Y694A substitutions had an intermediate loss of CR binding when compared with KIM6-2115(pNPM22) and KIM6+ (hmsHFRS+). The modest loss of CR binding in HmsH-F651A-D520A and HmsH-N665A-D520A is likely due to the single changes alone or in combination with D520A, since HmsH-D520A retained full CR binding (Fig. 3b, Table 2). In contrast, none of the HmsH variant proteins showed a significant difference in CV staining from KIM6-2115(pNPM22), the positive control (Table 2).
Western blot analysis with antisera against HmsH was used to assess the level of variant proteins. As expected, the level of HmsH from KIM6+ (hmsHFRS+) was lower than levels obtained in KIM6-2115(pNPM22) (HmsH++) (Table 2). Since KIM6+ and KIM-2115(pNPM22) show no significant difference in CR binding or CV staining (Figs 3b and 4b, Table 2), the lower HmsH levels in KIM6+ are sufficient for normal biofilm development. Most of the variant HmsH proteins were present at a level equivalent to those of KIM6-2115(pNPM22). However, the levels of HmsH-R657A, HmsH-P697A and HmsH-P769A proteins were similar to that found in KIM6+. The amounts of HmsH-F651A-D520A, HmsH-D653A and HmsH-N655-D520A were even lower (∼85, 30 and 40 %, respectively, than the level in KIM6+) (Table 2). The lower levels of HmsH are likely due to lower protein stabilities.
Fig. 4.

Analysis of changes in conserved residues in the N terminus of HmsH. (a) Alignment of these regions of HmsH and HmsH-like proteins from Y. pestis KIM10+ (Ype); P. fluorescens Pf-5 (Pfl); Pectobacterium atrosepticum SCR11043 (Pat; formerly Erwinia carotovora); E. coli K-12 substrain MG1655 (Eco); and X. oryzae KACC10331 (Xor). The black and grey boxes indicate residue identity and similarity, respectively. Sequences start with the first residue after the SignalP predicted signal sequence. The amino acid change line shows the amino acids individually changed to alanine. (b) Quantitative CV staining by Y. pestis cells expressing amino acid substitutions in the N-terminal region of HmsH. Controls are KIM6-2115 (in-frame ΔhmsH; HmsH−), KIM6+ (Hms+) and KIM6-2115(pNPM22) (HmsH++). Data shown are the mean of two or more independent experiments with duplicate samples from each trial. Error bars, sd. For clarity, the secondary mutation (D520A) and (D691A) in HmsH-E345A-D520A, HmsH-R433-D520A, HmsH-D397A-D520A and HmsH-R491-D691A is not shown in the labels.
Alanine scanning mutagenesis in the predicted N-terminal periplasmic domain of HmsH
The N-terminal region of HmsH, which is predicted to be in the periplasm, was aligned with HmsH-like proteins from other organisms (Fig. 4a). Seven conserved residues among these proteins were selected for alanine substitutions. The HmsH protein levels of these variants were all greater than that for KIM6+ (hmsHFRS+) (Table 2). Sequencing revealed that four mutants had secondary mutations. Thus, HmsH-E345, HmsH-D397A and HmsH-433A had an additional mutation in loop 1 (D520A). HmsH-R491A also contained a D691A substitution in loop 6 (Table 2). None of these mutations had a significant effect on biofilm development as measured by the three assays (Table 2, Fig. 4b). However, Y. pestis expressing HmsH-D263A or HmsH-R479A proteins were defective in adherence as measured by CV-staining assay (Fig. 4b). Both these mutants formed white-to-pink colonies after 48 h incubation at 30 °C. Oddly, the 3 h liquid CR-binding assay failed to demonstrate a defect in EPS production.
In addition to the length of time cells are exposed to CR, the two CR-binding assays differ in incubation temperature. Since the liquid CR-binding assay is performed at RT (∼20 °C), we incubated CR plates streaked with controls and the HmsH-D263A and HmsH-R479A mutants at this temperature. At the lower temperature both mutants formed colonies that were distinctly redder than the same mutants incubated at 30 °C (data not shown). To quantify this apparent temperature-dependent phenotype, we performed the liquid CR-binding assays at 20 and 30 °C. The HmsH-D263A and HmsH-R479A variants complemented the hmsH mutation at the lower temperature but not at 30 °C. In contrast, wild-type HmsH complemented the hmsH mutant at both temperatures, while HmsH-Y694A failed to complement at either temperature (Figs 5 and 3b; data not shown). Thus, a temperature-dependent phenotype is caused by the D263A and R479A substitutions in HmsH.
Fig. 5.
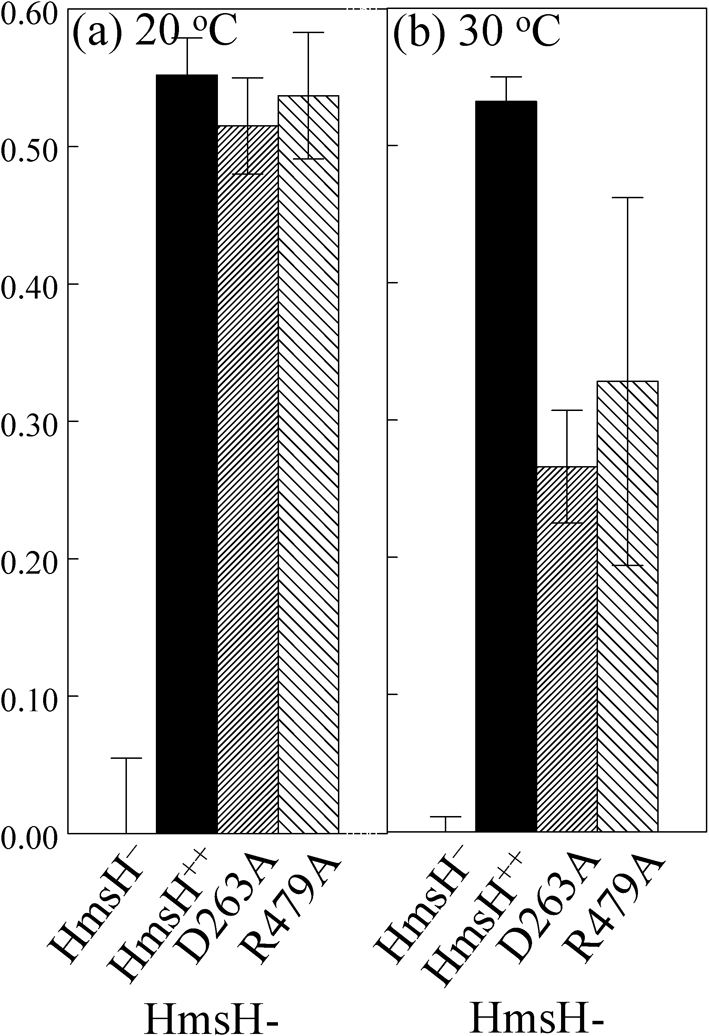
CR binding by HmsH-D263A and HmsH-R479A variants is temperature-dependent. Quantitative CR binding is shown for Y. pestis KIM6-2115 (in-frame ΔhmsH; HmsH−), KIM6-2115(pNPM22) (HmsH++) and the two variants after 3 h incubation at 20 °C (a) or 30 °C (b) with CR-containing HIB medium. Data shown are the mean of two or more independent experiments with duplicate samples from each trial. Error bars, sd.
The HmsH-Y694A and HmsH-T657A variants exhibited the opposite phenotype, forming red colonies on CR plates but showing reduced CR binding in the liquid assay (Table 2). Consequently, we hypothesize that these two mutants could also have a temperature-dependent phenotype with the higher temperature being required for these HmsH variants to be functional. However, liquid CR-binding assays performed at 20 and 30 °C showed no difference in these two variants.
HmsH and HmsF interact to form an Hms OM complex
We used formaldehyde cross-linking to study Hms OM protein interactions since (a) cross-linking with formaldehyde occurs over a short distance (2 Å); (b) formaldehyde permeates the cell membrane and is rather non-specific; and (c) formaldehyde cross-linkages are reversible (Prossnitz et al., 1988; Sutherland et al., 2008; Vasilescu et al., 2004). Y. pestis KIM6+ (hmsHFRS+) cells were cross-linked with 1 % formaldehyde, a concentration that has been shown to be optimal for detecting protein–protein interactions in other bacteria (Prossnitz et al., 1988; Skare et al., 1993; Warren et al., 2004). Initial time-courses were performed to determine the optimal incubation time with 1 % formaldehyde. Cross-linked samples were separated by SDS-PAGE and blots probed with HmsF antisera. By 15 min, two bands of ∼142 and 134 kDa as well as the HmsF monomer were detected (Fig. 6).
Fig. 6.

Western blot of formaldehyde-cross-linked Y. pestis strains probed with HmsF antisera. (a) Y. pestis KIM6+ (Hms+), KIM6-2116 (ΔhmsF) and KIM6-2116(pBADhmsF) (hmsF++). Samples were cross-linked (X), not cross-linked (UX), or cross-linked and then reversed (XR). The grey arrow points to the HmsF monomer band. (b) Y. pestis KIM6+ (Hms+), KIM6-2115 (ΔhmsH) and KIM6-2115(pNPM22) (hmsH++). Except for lane 1, all samples were cross-linked. The HmsF monomer is not shown in (b). Black arrows in both panels point to the cross-linked complexes containing HmsF. Molecular masses of protein standards are shown within the range of 250–64 kDa. For (b), only the location of the 148 kDa standard is shown.
These bands were absent from the ΔhmsF mutant and present in the complemented ΔhmsF strain (Fig. 6a). After reversal of the cross-linking by boiling, the two cross-linked complexes were not detected, while an ∼72 kDa band corresponding to the HmsF monomer remained (Fig. 6a). The two cross-linked bands were not detected in ΔhmsH samples but were present in the sample from the complemented ΔhmsH strain (Fig. 6b). To demonstrate the presence of HmsH in these cross-linked complexes directly, Y. pestis samples were probed with antisera against HmsH. The two bands corresponding to the HmsH–HmsF complex were detected in cells expressing HmsH and HmsF but not in mutants lacking either HmsH or HmsF (Fig. 7). To determine whether additional Hms proteins are part of the HmsH–HmsF complexes, we analysed the effect of mutations in other hms genes on complex formation. None of the mutations in genes for the Hms IM proteins (in hmsR, hmsS, hmsT or hmsP) caused any apparent alterations in the two OM complex bands containing HmsH and HmsF (Fig. 8). These data suggest that HmsF and HmsH are not interacting with HmsR, HmsS, HmsT and HmsP at least at a level that can be detected by cross-linking.
Fig. 7.
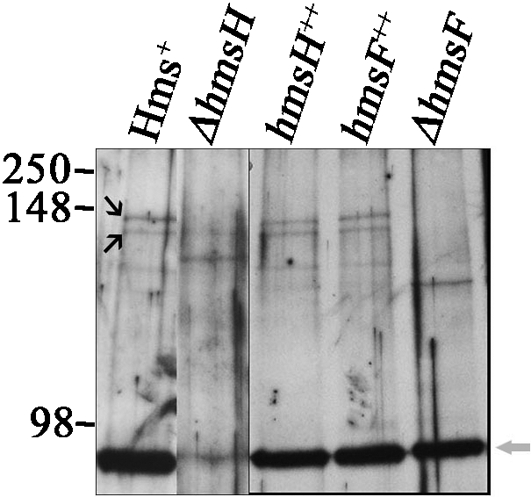
Western blot of formaldehyde-cross-linked Y. pestis strains probed with HmsH antisera. Strains: Y. pestis KIM6+ (Hms+), KIM6-2115 (ΔhmsH), KIM6-2115(pNPM22) (hmsH++), KIM6-2116(pBADhmsF) (hmsF++) and KIM6-2116 (ΔhmsF). Black arrows identify the two HmsH–HmsF cross-linked complexes, while the grey arrow shows the HmsH monomer. Molecular masses of protein standards are shown within the range of 250–98 kDa.
Fig. 8.
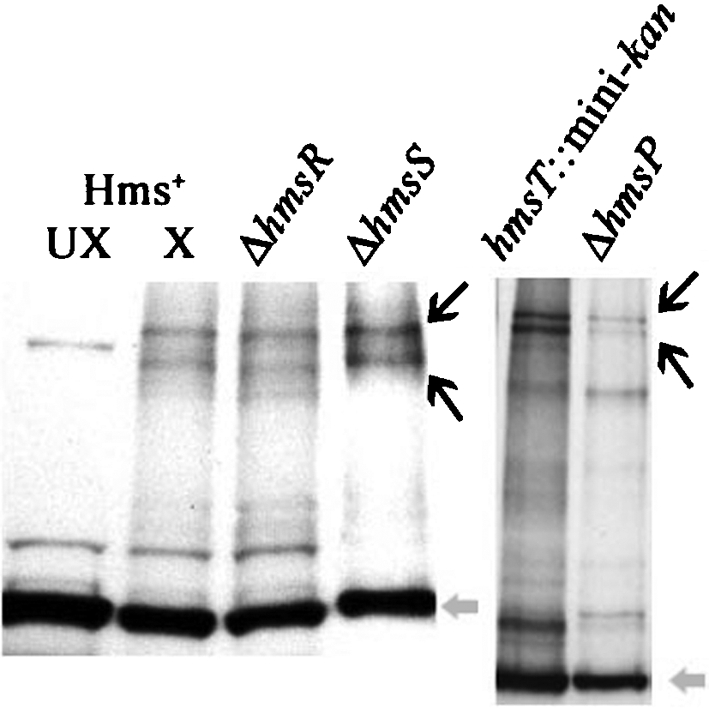
Western blot of formaldehyde-cross-linked Y. pestis strains KIM6+ (Hms+), KIM6-2118 (ΔhmsR), KIM6-2119 (ΔhmsS), KIM6-2051 (hmsT : : mini-kan) and KIM6-2090.2+ (ΔhmsP) probed with HmsF antisera. Except for one Hms+ control (Hms+ UX), all samples were cross-linked (X) with formaldehyde. Black arrows identify the cross-linked HmsH–HmsF complex, while the grey arrow identifies the HmsF monomer.
Residue substitutions in HmsH and HmsF do not alter protein structure or interactions
Our mutagenesis studies identified some amino acid substitutions that affected HmsH protein level and/or function. All of the mutant proteins retained their apparent β-barrel structure, as heat treatment did not alter their SDS-PAGE migration (data not shown).
Some of the alanine substitutions within the putative N- and C-terminal domains of HmsH gave a biofilm-negative or -reduced phenotype. One possibility for this phenotype is that these mutant proteins are defective for interaction with HmsF. However, all of the Y. pestis strains expressing an HmsH variant protein with a reduced or biofilm-negative phenotype still exhibited two cross-linked complexes (data not shown). Thus, there is no clear defect in HmsF–HmsH interactions due to these residue substitutions in HmsH.
Previously, single amino acid changes within the putative deacetylase domain of HmsF (D114, D115 and H184) all exhibited loss of biofilm formation (Forman et al., 2006). It is possible that alteration of these residues affects interaction of HmsF with the OM complexes. Examination of cross-linked Y. pestis cells expressing HmsF or one of the three variants (HmsF-D114A, HmsF-D115A and HmsF-H184A) revealed the presence of the two OM complexes (data not shown). Consequently, these residue changes in HmsF do not appear to affect interaction with HmsH and any other proteins in this complex.
Biofilm development is not critical to the pathogenesis of bubonic or pneumonic plague in mice
Previously, a ΔhmsR mutant (6 bp deletion) in a slightly attenuated background (yopJ psa) was shown to be as virulent as its hmsR+ parent in a mouse model of bubonic plague (Lillard et al., 1999). While the ΔhmsR mutation causes a complete loss of biofilm formation in vitro, the HmsR deletion protein and all other Hms proteins are fully expressed. Here, we used an hmsH2008 : : mini-kan mutant (which does not express HmsH, HmsF, HmsR or HmsS) to examine the role of biofilm formation in mammalian disease.
The hmsH2008 : : mini-kan mutant was fully virulent in mice via a subcutaneous route of infection mimicking bubonic plague. The hmsH2008 : : mini-kan mutant had an LD50 of 89 cells (±22) for the mutant compared with 23 cells (±14) for the wild-type parent. The inability to form a biofilm also failed to affect pathogenesis in a lung infection of mice (pneumonic plague). The hmsH2008 : : mini-kan mutant had an LD50 of 330 cells (±212), while the wild-type strain had an LD50 of 329 cells (±105). Thus, biofilm development is not critical for plague pathogenesis in mice.
DISCUSSION
Biofilm formation is an important aspect of many bacterial diseases, including osteomyelitis, dental caries, middle ear infections, ocular implant infections, and chronic lung infections in cystic fibrosis patients (Jefferson, 2004; Mack et al., 2007; O'Gara, 2007). Biofilms are also involved in prostatitis, biliary tract infection, and urinary catheter cystitis caused by E. coli, as well as systemic and device-related infections by S. aureus and/or S. epidermidis (Kropec et al., 2005; Van Houdt & Michiels, 2005; Vuong et al., 2004; Wang et al., 2004). Our results indicate that Hms-dependent biofilm formation by Y. pestis does not play a significant role in mammalian disease (bubonic plague or pneumonic plague). However, it has been shown that the Hms system is required for blockage of the flea (Hinnebusch et al., 1996).
While blocked flea transmission of plague has been the paradigm since the early observations of Bacot, blockage also causes flea mortality, thus reducing the transmission period (Bacot & Martin, 1914; Bacot, 1915; Darby et al., 2002; Hinnebusch et al., 1996; Hinnebusch & Erickson, 2008; Jarrett et al., 2004; Kirillina et al., 2004; Perry & Fetherston, 1997). Recent studies have shown that early-phase transmission without the macrophenomenon of blockage is an important transmission mechanism that is likely responsible for the rapid spread of Y. pestis during outbreaks in rodent populations and human bubonic plague epidemics (Eisen et al., 2006, 2007a, b; Wilder et al., 2008). Nevertheless, flea blockage extends the transmission period beyond the early-phase period of 3 days. Blockage also appears to reduce clearance of Y. pestis by fleas, which may prolong the infection or increase the infection rate in fleas. These factors and the increased feeding attempts may be required to maintain low levels of infected fleas in reservoirs between epidemic outbreaks (Bazanova et al., 1991; Burroughs, 1947; Gage & Kosoy, 2005; Hinnebusch et al., 1996; Kartman et al., 1958; Kutyrev et al., 1992).
Flea blockage requires the expression of the Hms proteins. HmsT is a diguanylate cyclase, while HmsP is a phosphodiesterase; these two proteins control biofilm formation by affecting c-di-GMP levels. HmsR, likely in concert with HmsS, is a putative GT that is probably responsible for the production of biofilm EPS, which is likely modified by HmsF. The Hidden Markov Method-based predictor (Bagos et al., 2004a, b) indicates that HmsH is a β-barrel protein with the N terminus in the periplasm (first 499 residues of the processed protein) and C-terminal amphipathic β-sheets that traverse the OM 16 times with eight predicted surface-exposed loops and seven short turns connecting the β-strands (Supplementary Fig. S1). Heat treatment alters the electrophoretic mobility of HmsH (Fig. 2), a general characteristic of β-barrel proteins that is caused by a conformational change to a more α-helical structure (Beher et al., 1980; Nakamura & Mizushima, 1976; Nandi et al., 2005; Nitzan et al., 1999). This supports the bioinformatics prediction of a β-barrel structure for this OM protein. Itoh et al. (2008) recently demonstrated that the E. coli PgaA (an HmsH-like protein) is required for export of the deacetylated polymer of β-1,6-GlcNAc. Given the bioinformatics structural analysis, similarity to PgaA, and our experimental data, it is almost certain that HmsH plays a similar role in biofilm development in Y. pestis.
We used structural analysis and the alignment of HmsH-like proteins from other bacteria with Y. pestis HmsH to select conserved residues for mutagenesis. To avoid disruption of the β-barrel structure of HmsH, we targeted conserved residues in the predicted N terminus and surface-exposed loops between β-strands in the C terminus of HmsH for mutagenesis. This approach resulted in four categories of mutants.
The first category (16 mutants) retained full HmsH function as determined by CR-binding and CV-staining assays. Although HmsH-P697A and HmsH-P769 were fully functional, they did exhibit protein levels lower than wild-type HmsH expressed from a recombinant plasmid but higher than levels expressed from a single chromosomal gene. The reason for the reduced protein levels in these two mutants is undetermined but may be due to lower protein stability.
The number of conserved residues that could be mutated without affecting HmsH function was surprising given the extent of identity among HmsH orthologues from relatively diverse organisms (e.g. Pseudomonas, Xanthomonas and Pectobacterium vs Yersinia and Escherichia). Horizontal transfer could account for the presence of highly conserved residues that are not required for protein function; however, there is no evidence that the hmsHFRS locus was part of a mobile element. In all five organisms, the GC content of the hms and hms-like loci is similar to that of the whole genome of the relevant organisms (Table 3). Thus, the reason why so many conserved residues in HmsH/HmsH-like proteins appear to be non-critical for protein function remains obscure.
Table 3.
Comparison of the GC content of the whole genome with that of hms/pga loci in five bacterial species
| Species | Whole genome GC (mol%) | hms/pga loci GC (mol%) | Difference in GC (mol%) |
|---|---|---|---|
| Y. pestis KIM10+ | 47 % | 49 % | 3 % |
| E. coli MG1655 | 50 % (51 %)* | 46 % (44 %)* | 4 % (7 %)* |
| P. fluorescens Pf-5 | 63 % | 67 % | 4 % |
| X. oryzae KACC10331 | 63 % | 63.4 % | 0.4 % |
| Pectobacterium atrosepticum SCR11043 | 51 % | 49 % | 2 % |
*GC (mol%) in parentheses are values calculated and published by Wang et al. (2004).
The second category of mutants contains two variants, HmsH-Y694A in loop 6 and HmsH-R657A in loop 5 (Table 2). It is clear that these residue changes did not disrupt the β-barrel structure of HmsH, nor did they prevent formation of HmsH–HmsF-containing complexes (data not shown). Both variants formed red colonies on CR plates but had reduced levels of CR binding over a 3 h period (Fig. 3). They also exhibited normal CV staining (data not shown). This suggests that both mutations reduced the function of HmsH sufficiently to show a phenotype in a short-term liquid assay (quantitative CR binding) but not in a longer-term assay (>48 h on CR plates, overnight for CV staining). One possible interpretation of this result is that these two mutations result in an EPS that has lower affinity for CR, perhaps by altering interactions with HmsF to affect the degree of EPS deacetylation, which might affect CR binding. A second possibility is that the altered residues reduce attachment of the EPS to the cell surface. In the CR plate assay, any EPS that was released would not diffuse away from the colony, resulting in an apparently normal phenotype. While HmsH-Y694A yielded a protein level similar to HmsH expressed from pNPM22, HmsH-R657A had a lower level similar to that of wild-type HmsH expressed from the chromosome. The level of HmsH-R657A should be sufficient to restore normal biofilm development if the residue change affected only the level of the variant protein. Expression of HmsH from a moderate-copy-number plasmid did not increase biofilm formation (Figs 3b and 4b), indicating that HmsH function is not a rate-limiting step in biofilm development. Thus, residues Y694 and R657 play an important but not essential role in HmsH function and the R657A substitution also has a modest effect on HmsH protein levels.
The third category had variant protein levels lower than those observed for wild-type HmsH expressed from the chromosome. Thus, the reduced ability of HmsH-D653A, HmsH-N655A-D520A and HmsH-F651A-D520A to bind CR might simply reflect these lower protein levels. On the longer-term CR plate assay, the first two variants formed pink colonies, while the slightly higher level of HmsH-F651A-D520A correlated with the ability of this strain to form red colonies on CR plates. Again, CV-staining levels with all the strains expressing these three variant HmsH proteins were similar to those observed with the Hms+ strain. The reason for the lower levels of variant proteins is undetermined but may be due to protein instability.
The final category contains HmsH-D263A and HmsH-R479A (both in the N terminus). These strains had variant protein levels similar to those observed for wild-type HmsH expressed from the chromosome, and formed white-to-pink colonies on CR plates after 48 h at 30 °C that turned red after storage at RT or 4 °C. In addition, CV staining was ∼50 % of wild-type. Both of these assays would correlate with a lower level of EPS production. However, there was no defect in CR binding in the quantitative liquid assay at RT. Further studies demonstrated that the apparent differences in CR binding were temperature-dependent. When assays were performed at RT (∼20 °C), these variants exhibited a wild-type phenotype but were defective at 30 °C (Fig. 5). This is the first description of a temperature-dependent phenotype for HmsH.
Previously, our laboratory (Bobrov et al., 2008) has demonstrated interactions between the Hms IM proteins using the BACTH system and biochemical analyses. That study identified homotypic interactions with HmsP, HmsT and HmsS as well as heterotypic interactions of HmsP with HmsR and HmsR with HmsS. Since the BACTH system cannot be used to study OM proteins, we used formaldehyde cross-linking to study the interaction of Y. pestis HmsH and HmsF.
This study provides the first biochemical evidence for interaction of the Hms OM proteins HmsH and HmsF. HmsH forms a complex with HmsF, exhibiting two cross-linked bands, both of which were disrupted by either ΔhmsF or ΔhmsH mutations (Figs 6 and 7). This suggests that both proteins are required for these interactions (i.e. neither cross-linked band is due to homodimers or homopolymers). Interaction of HmsH and HmsF could be necessary for modification and export of partially deacetylated polysaccharide through the OM. Cross-linking studies with Y. pestis strains expressing HmsH-variant proteins with a phenotype and selected HmsF-variant proteins (alteration in the deacetylase domain causing loss of biofilm formation) yielded the same two cross-linked bands observed in the Hms+ strain. Thus, at this level of analysis, none of the mutations tested affected protein–protein interactions between HmsH and HmsF, indicating that this is not the cause for the defect in biofilm development. The domains involved in the interaction between these two proteins are unknown. The N-terminal domain of HmsH might serve as a plug for the porin but may also be required for interaction with HmsF. In E. coli, the PgaA N terminus shows similarity to the tetratricopeptide repeat domains of human nucleoporin O-linked GlcNAc transferase, which forms an elongated superhelical structure that mediates protein–protein interactions (Itoh et al., 2008). It is possible that the N-terminal domain of HmsH is required for such protein–protein interactions.
The composition and size of the two OM complexes containing HmsH and HmsF remain to be determined. Estimation of the molecular masses of the complexes based on the electrophoretic mobility of cross-linked proteins is unreliable. While it is possible that proteins in addition to HmsH and HmsF are important components of these complexes, similar cross-linking studies with ΔhmsR, ΔhmsS, ΔhmsP and ΔhmsT mutants (Fig. 8) indicate that these mutations do not significantly affect the formation of the two OM cross-linked complexes.
Acknowledgments
This study was supported by Public Health Service grant AI25098 from the US National Institutes of Health. We thank A. Bobrov and O. Kirillina for providing Y. pestis strain KIM6-2090.2+.
Abbreviations
c-di-GMP, cyclic-di-GMP
CR, Congo red
CV, crystal violet
EPS, exopolysaccharide
GT, glycosyltransferase
IM, inner membrane
OM, outer membrane
poly-β-1,6-GlcNac, poly-β-1,6-N-acetyl-d-glucosamine
RT, room temperature
Footnotes
A supplementary table, showing the primers used in this study, and a supplementary figure, showing a graphical representation of the predicted positions of the transmembrane strands of HmsH with respect to the lipid bilayer, are available with the online version of this paper.
References
- Ausubel, F. M. (1987). Current protocols in Molecular Biology. New York: Greene Publishing & Wiley-Interscience.
- Bacot, A. W. (1915). LXXXI. Further notes on the mechanism of the transmission of plague by fleas. J Hyg (Lond) 14 (Plague Suppl. 4), 744–776. [PMC free article] [PubMed] [Google Scholar]
- Bacot, A. W. & Martin, C. J. (1914). LXVII. Observations on the mechanism of the transmission of plague by fleas. J Hyg (Lond) 13 (Plague Suppl. 3), 423–439. [PMC free article] [PubMed] [Google Scholar]
- Bagos, P. G., Liakopoulos, T. D., Spyropoulos, I. C. & Hamodrakas, S. J. (2004a). A Hidden Markov Model method, capable of predicting and discriminating beta-barrel outer membrane proteins. BMC Bioinformatics 5, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagos, P. G., Liakopoulos, T. D., Spyropoulos, I. C. & Hamodrakas, S. J. (2004b). PRED-TMBB: a web server for predicting the topology of beta-barrel outer membrane proteins. Nucleic Acids Res 32, W400–W404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bazanova, L. P., Zhovtyi, I., Maevskii, M. P., Klimov, V. Y. & Popkov, A. F. (1991). The seasonal dynamics of blocking in the flea Citellophorus tesquorum altaicus from the Tuva natural plague focus. Med Parazitol (Mosk) Jan-Feb. 24–26. [PubMed]
- Beesley, E. D., Brubaker, R. R., Janssen, W. A. & Surgalla, M. J. (1967). Pesticins. III. Expression of coagulase and mechanism of fibrinolysis. J Bacteriol 94, 19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beher, M. G., Schnaitman, C. A. & Pugsley, A. P. (1980). Major heat-modifiable outer membrane protein in Gram-negative bacteria: comparison with the OmpA protein of Escherichia coli. J Bacteriol 143, 906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell, K. S., Sebaihia, M., Pritchard, L., Holden, M. T., Hyman, L. J., Holeva, M. C., Thomson, N. R., Bentley, S. D., Churcher, L. J. & other authors (2004). Genome sequence of the enterobacterial phytopathogen Erwinia carotovora subsp. atroseptica and characterization of virulence factors. Proc Natl Acad Sci U S A 101, 11105–11110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birnboim, H. C. & Doly, J. (1979). A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res 7, 1513–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blattner, F. R., Plunkett, G., III, Bloch, C. A., Perna, N. T., Burland, V., Riley, M., Collado-Vides, J., Glasner, J. D., Rode, C. K. & other authors (1997). The complete genome sequence of Escherichia coli K-12. Science 277, 1453–1474. [DOI] [PubMed] [Google Scholar]
- Bobrov, A. G., Kirillina, O. & Perry, R. D. (2005). The phosphodiesterase activity of the HmsP EAL domain is required for negative regulation of biofilm formation in Yersinia pestis. FEMS Microbiol Lett 247, 123–130. [DOI] [PubMed] [Google Scholar]
- Bobrov, A. G., Kirillina, O., Forman, S., Mack, D. & Perry, R. D. (2008). Insights into Yersinia pestis biofilm development: topology and co-interaction of Hms inner membrane proteins involved in exopolysaccharide production. Environ Microbiol 10, 1419–1432. [DOI] [PubMed] [Google Scholar]
- Burroughs, A. L. (1947). Sylvatic plague studies. The vector efficiency of nine species of fleas compared with Xenopsylla cheopis. J Hyg (Lond) 45, 371–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darby, C., Hsu, J. W., Ghori, N. & Falkow, S. (2002). Caenorhabditis elegans: plague bacteria biofilm blocks food intake. Nature 417, 243–244. [DOI] [PubMed] [Google Scholar]
- Eisen, R. J., Bearden, S. W., Wilder, A. P., Montenieri, J. A., Antolin, M. F. & Gage, K. L. (2006). Early-phase transmission of Yersinia pestis by unblocked fleas as a mechanism explaining rapidly spreading plague epizootics. Proc Natl Acad Sci U S A 103, 15380–15385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisen, R. J., Lowell, J. L., Montenieri, J. A., Bearden, S. W. & Gage, K. L. (2007a). Temporal dynamics of early-phase transmission of Yersinia pestis by unblocked fleas: secondary infectious feeds prolong efficient transmission by Oropsylla montana (Siphonaptera: Ceratophyllidae). J Med Entomol 44, 672–677. [DOI] [PubMed] [Google Scholar]
- Eisen, R. J., Wilder, A. P., Bearden, S. W., Montenieri, J. A. & Gage, K. L. (2007b). Early-phase transmission of Yersinia pestis by unblocked Xenopsylla cheopis (Siphonaptera: Pulicidae) is as efficient as transmission by blocked fleas. J Med Entomol 44, 678–682. [DOI] [PubMed] [Google Scholar]
- Fetherston, J. D., Schuetze, P. & Perry, R. D. (1992). Loss of the pigmentation phenotype in Yersinia pestis is due to the spontaneous deletion of 102 kb of chromosomal DNA which is flanked by a repetitive element. Mol Microbiol 6, 2693–2704. [DOI] [PubMed] [Google Scholar]
- Fetherston, J. D., Lillard, J. W., Jr & Perry, R. D. (1995). Analysis of the pesticin receptor from Yersinia pestis: role in iron-deficient growth and possible regulation by its siderophore. J Bacteriol 177, 1824–1833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields, K. A., Nilles, M. L., Cowan, C. & Straley, S. C. (1999). Virulence role of V antigen of Yersinia pestis at the bacterial surface. Infect Immun 67, 5395–5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forman, S., Bobrov, A. G., Kirillina, O., Craig, S. K., Abney, J., Fetherston, J. D. & Perry, R. D. (2006). Identification of critical amino acid residues in the plague biofilm Hms proteins. Microbiology 152, 3399–3410. [DOI] [PubMed] [Google Scholar]
- Forman, S., Wulff, C. R., Myers-Morales, T., Cowan, C., Perry, R. D. & Straley, S. C. (2008). yadBC of Yersinia pestis, a new virulence determinant for bubonic plague. Infect Immun 76, 578–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gage, K. L. & Kosoy, M. Y. (2005). Natural history of plague: perspectives from more than a century of research. Annu Rev Entomol 50, 505–528. [DOI] [PubMed] [Google Scholar]
- Goller, C. C. & Romeo, T. (2008). Environmental influences on biofilm development. Curr Top Microbiol Immunol 322, 37–66. [DOI] [PubMed] [Google Scholar]
- Gong, S., Bearden, S. W., Geoffroy, V. A., Fetherston, J. D. & Perry, R. D. (2001). Characterization of the Yersinia pestis Yfu ABC inorganic iron transport system. Infect Immun 69, 2829–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gotz, F. (2002). Staphylococcus and biofilms. Mol Microbiol 43, 1367–1378. [DOI] [PubMed] [Google Scholar]
- Hartzell, P. L., Millstein, J. & LaPaglia, C. (1999). Biofilm formation in hyperthermophilic Archaea. Methods Enzymol 310, 335–349. [DOI] [PubMed] [Google Scholar]
- Higuchi, K. & Smith, J. L. (1961). Studies on the nutrition and physiology of Pasteurella pestis. VI. A differential plating medium for the estimation of the mutation rate to avirulence. J Bacteriol 81, 605–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch, B. J. & Erickson, D. L. (2008). Yersinia pestis biofilm in the flea vector and its role in the transmission of plague. Curr Top Microbiol Immunol 322, 229–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinnebusch, B. J., Perry, R. D. & Schwan, T. G. (1996). Role of the Yersinia pestis hemin storage (hms) locus in the transmission of plague by fleas. Science 273, 367–370. [DOI] [PubMed] [Google Scholar]
- Itoh, Y., Wang, X., Hinnebusch, B. J., Preston, J. F., III & Romeo, T. (2005). Depolymerization of β-1,6-N-acetyl-d-glucosamine disrupts the integrity of diverse bacterial biofilms. J Bacteriol 187, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh, Y., Rice, J. D., Goller, C., Pannuri, A., Taylor, J., Meisner, J., Beveridge, T. J., Preston, J. F., III & Romeo, T. (2008). Roles of pgaABCD genes in synthesis, modification, and export of the Escherichia coli biofilm adhesin poly-beta-1,6-N-acetyl-d-glucosamine. J Bacteriol 190, 3670–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izano, E. A., Sadovskaya, I., Vinogradov, E., Mulks, M. H., Velliyagounder, K., Ragunath, C., Kher, W. B., Ramasubbu, N., Jabbouri, S. & other authors (2007). Poly-N-acetylglucosamine mediates biofilm formation and antibiotic resistance in Actinobacillus pleuropneumoniae. Microb Pathog 43, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, S. & Burrows, T. W. (1956). The pigmentation of Pasteurella pestis on a defined medium containing haemin. Br J Exp Pathol 37, 570–576. [PMC free article] [PubMed] [Google Scholar]
- Jarrett, C. O., Deak, E., Isherwood, K. E., Oyston, P. C., Fischer, E. R., Whitney, A. R., Kobayashi, S. D., DeLeo, F. R. & Hinnebusch, B. J. (2004). Transmission of Yersinia pestis from an infectious biofilm in the flea vector. J Infect Dis 190, 783–792. [DOI] [PubMed] [Google Scholar]
- Jefferson, K. K. (2004). What drives bacteria to produce a biofilm? FEMS Microbiol Lett 236, 163–173. [DOI] [PubMed] [Google Scholar]
- Jones, H. A., Lillard, J. W., Jr & Perry, R. D. (1999). HmsT, a protein essential for expression of the haemin storage (Hms+) phenotype of Yersinia pestis. Microbiology 145, 2117–2128. [DOI] [PubMed] [Google Scholar]
- Kaplan, J. B., Velliyagounder, K., Ragunath, C., Rohde, H., Mack, D., Knobloch, J. K. & Ramasubbu, N. (2004). Genes involved in the synthesis and degradation of matrix polysaccharide in Actinobacillus actinomycetemcomitans and Actinobacillus pleuropneumoniae biofilms. J Bacteriol 186, 8213–8220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartman, L., Prince, F. M., Quan, S. F. & Stark, H. E. (1958). New knowledge on the ecology of sylvatic plague. Ann N Y Acad Sci 70, 668–711. [DOI] [PubMed] [Google Scholar]
- Kirillina, O., Fetherston, J. D., Bobrov, A. G., Abney, J. & Perry, R. D. (2004). HmsP, a putative phosphodiesterase, and HmsT, a putative diguanylate cyclase, control Hms-dependent biofilm formation in Yersinia pestis. Mol Microbiol 54, 75–88. [DOI] [PubMed] [Google Scholar]
- Kropec, A., Maira-Litran, T., Jefferson, K. K., Grout, M., Cramton, S. E., Gotz, F., Goldmann, D. A. & Pier, G. B. (2005). Poly-N-acetylglucosamine production in Staphylococcus aureus is essential for virulence in murine models of systemic infection. Infect Immun 73, 6868–6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kutyrev, V. V., Filippov, A. A., Oparina, O. S. & Protsenko, O. A. (1992). Analysis of Yersinia pestis chromosomal determinants Pgm+ and Psts associated with virulence. Microb Pathog 12, 177–186. [DOI] [PubMed] [Google Scholar]
- Lee, B. M., Park, Y. J., Park, D. S., Kang, H. W., Kim, J. G., Song, E. S., Park, I. C., Yoon, U. H., Hahn, J. H. & other authors (2005). The genome sequence of Xanthomonas oryzae pathovar oryzae KACC10331, the bacterial blight pathogen of rice. Nucleic Acids Res 33, 577–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lillard, J. W., Jr, Fetherston, J. D., Pedersen, L., Pendrak, M. L. & Perry, R. D. (1997). Sequence and genetic analysis of the hemin storage (hms) system of Yersinia pestis. Gene 193, 13–21. [DOI] [PubMed] [Google Scholar]
- Lillard, J. W., Jr, Bearden, S. W., Fetherston, J. D. & Perry, R. D. (1999). The haemin storage (Hms+) phenotype of Yersinia pestis is not essential for the pathogenesis of bubonic plague in mammals. Microbiology 145, 197–209. [DOI] [PubMed] [Google Scholar]
- Mack, D., Fischer, W., Krokotsch, A., Leopold, K., Hartmann, R., Egge, H. & Laufs, R. (1996). The intercellular adhesin involved in biofilm accumulation of Staphylococcus epidermidis is a linear β-1,6-linked glucosaminoglycan: purification and structural analysis. J Bacteriol 178, 175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack, D., Riedewald, J., Rohde, H., Magnus, T., Feucht, H. H., Elsner, H. A., Laufs, R. & Rupp, M. E. (1999). Essential functional role of the polysaccharide intercellular adhesin of Staphylococcus epidermidis in hemagglutination. Infect Immun 67, 1004–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack, D., Davies, A. P., Harris, L. G., Rohde, H., Horstkotte, M. A. & Knobloch, J. K. (2007). Microbial interactions in Staphylococcus epidermidis biofilms. Anal Bioanal Chem 387, 399–408. [DOI] [PubMed] [Google Scholar]
- Maniatis, T., Fritsch, E. F. & Sambrook, J. (1982). Molecular Cloning: a Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
- Nakamura, K. & Mizushima, S. (1976). Effects of heating in dodecyl sulfate solution on the conformation and electrophoretic mobility of isolated major outer membrane proteins from Escherichia coli K-12. J Biochem 80, 1411–1422. [DOI] [PubMed] [Google Scholar]
- Nandi, B., Nandy, R. K., Sarkar, A. & Ghose, A. C. (2005). Structural features, properties and regulation of the outer-membrane protein W (OmpW) of Vibrio cholerae. Microbiology 151, 2975–2986. [DOI] [PubMed] [Google Scholar]
- Nitzan, Y., Pechatnikov, I., Bar-El, D. & Wexler, H. (1999). Isolation and characterization of heat-modifiable proteins from the outer membrane of Porphyromonas asaccharolytica and Acinetobacter baumannii. Anaerobe 5, 43–50. [DOI] [PubMed] [Google Scholar]
- O'Gara, J. P. (2007). ica and beyond: biofilm mechanisms and regulation in Staphylococcus epidermidis and Staphylococcus aureus. FEMS Microbiol Lett 270, 179–188. [DOI] [PubMed] [Google Scholar]
- O'Toole, G. A., Pratt, L. A., Watnick, P. I., Newman, D. K., Weaver, V. B. & Kolter, R. (1999). Genetic approaches to study of biofilms. Methods Enzymol 310, 91–109. [DOI] [PubMed] [Google Scholar]
- Otto, M. (2008). Staphylococcal biofilms. Curr Top Microbiol Immunol 322, 207–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulsen, I. T., Press, C. M., Ravel, J., Kobayashi, D. Y., Myers, G. S., Mavrodi, D. V., DeBoy, R. T., Seshadri, R., Ren, Q. & other authors (2005). Complete genome sequence of the plant commensal Pseudomonas fluorescens Pf-5. Nat Biotechnol 23, 873–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendrak, M. L. & Perry, R. D. (1991). Characterization of a hemin-storage locus of Yersinia pestis. Biol Met 4, 41–47. [DOI] [PubMed] [Google Scholar]
- Pendrak, M. L. & Perry, R. D. (1993). Proteins essential for expression of the Hms+ phenotype of Yersinia pestis. Mol Microbiol 8, 857–864. [DOI] [PubMed] [Google Scholar]
- Perry, R. D. & Fetherston, J. D. (1997). Yersinia pestis – etiologic agent of plague. Clin Microbiol Rev 10, 35–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, R. D., Pendrak, M. L. & Schuetze, P. (1990). Identification and cloning of a hemin storage locus involved in the pigmentation phenotype of Yersinia pestis. J Bacteriol 172, 5929–5937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry, R. D., Bobrov, A. G., Kirillina, O., Jones, H. A., Pedersen, L. L., Abney, J. & Fetherston, J. D. (2004). Temperature regulation of the hemin storage (Hms+) phenotype of Yersinia pestis is posttranscriptional. J Bacteriol 186, 1638–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prossnitz, E., Nikaido, K., Ulbrich, S. J. & Ames, G. F. (1988). Formaldehyde and photoactivatable cross-linking of the periplasmic binding protein to a membrane component of the histidine transport system of Salmonella typhimurium. J Biol Chem 263, 17917–17920. [PubMed] [Google Scholar]
- Reed, L. J. & Muench, H. (1938). A simple method of estimating fifty percent endpoints. Am J Hyg 27, 493–497. [Google Scholar]
- Simm, R., Fetherston, J. D., Kader, A., Romling, U. & Perry, R. D. (2005). Phenotypic convergence mediated by GGDEF-domain-containing proteins. J Bacteriol 187, 6816–6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skare, J. T., Ahmer, B. M., Seachord, C. L., Darveau, R. P. & Postle, K. (1993). Energy transduction between membranes. TonB, a cytoplasmic membrane protein, can be chemically cross-linked in vivo to the outer membrane receptor FepA. J Biol Chem 268, 16302–16308. [PubMed] [Google Scholar]
- Stoodley, P., Sauer, K., Davies, D. G. & Costerton, J. W. (2002). Biofilms as complex differentiated communities. Annu Rev Microbiol 56, 187–209. [DOI] [PubMed] [Google Scholar]
- Straley, S. C. & Bowmer, W. S. (1986). Virulence genes regulated at the transcriptional level by Ca2+ in Yersinia pestis include structural genes for outer membrane proteins. Infect Immun 51, 445–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surgalla, M. J. & Beesley, E. D. (1969). Congo red-agar plating medium for detecting pigmentation in Pasteurella pestis. Appl Microbiol 18, 834–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland, I. (2001). Biofilm exopolysaccharides: a strong and sticky framework. Microbiology 147, 3–9. [DOI] [PubMed] [Google Scholar]
- Sutherland, B. W., Toews, J. & Kast, J. (2008). Utility of formaldehyde cross-linking and mass spectrometry in the study of protein-protein interactions. J Mass Spectrom 43, 699–715. [DOI] [PubMed] [Google Scholar]
- Towbin, H., Staehelin, T. & Gordon, J. (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci U S A 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Houdt, R. & Michiels, C. W. (2005). Role of bacterial cell surface structures in Escherichia coli biofilm formation. Res Microbiol 156, 626–633. [DOI] [PubMed] [Google Scholar]
- Vasilescu, J., Guo, X. & Kast, J. (2004). Identification of protein-protein interactions using in vivo cross-linking and mass spectrometry. Proteomics 4, 3845–3854. [DOI] [PubMed] [Google Scholar]
- Vuong, C., Kocianova, S., Voyich, J. M., Yao, Y., Fischer, E. R., DeLeo, F. R. & Otto, M. (2004). A crucial role for exopolysaccharide modification in bacterial biofilm formation, immune evasion, and virulence. J Biol Chem 279, 54881–54886. [DOI] [PubMed] [Google Scholar]
- Wang, X., Preston, J. F., III & Romeo, T. (2004). The pgaABCD locus of Escherichia coli promotes the synthesis of a polysaccharide adhesin required for biofilm formation. J Bacteriol 186, 2724–2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren, M. A., Kucharski, L. M., Veenstra, A., Shi, L., Grulich, P. F. & Maguire, M. E. (2004). The CorA Mg2+ transporter is a homotetramer. J Bacteriol 186, 4605–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilder, A. P., Eisen, R. J., Bearden, S. W., Montenieri, J. A., Gage, K. L. & Antolin, M. F. (2008). Oropsylla hirsuta (Siphonaptera: Ceratophyllidae) can support plague epizootics in black-tailed prairie dogs (Cynomys ludovicianus) by early-phase transmission of Yersinia pestis. Vector Borne Zoonotic Dis 8, 359–367. [DOI] [PubMed] [Google Scholar]