

Abstract
Structures that contain a knot formed by the path of the polypeptide backbone represent some of the most complex topologies observed in proteins. How or why these topological knots arise remains unclear. By developing a method to experimentally trap and detect knots in nonnative polypeptide chains, we find that two knotted methyltransferases, YibK and YbeA, can exist in a trefoil-knot conformation even in their chemically unfolded states. The unique denatured-state topology of these molecules explains their ability to efficiently fold to their native knotted structures in vitro and offers insights into the potential role of knots in proteins. Furthermore, the high prevalence of the denatured-state knots identified here suggests that they are either difficult to untie or that threading of any untied molecules is rapid and spontaneous. The occurrence of such knotted topologies in unfolded polypeptide chains raises the possibility that they could play an important, and as yet unexplored, role in folding and misfolding processes in vivo.
Keywords: knotted protein, denatured state, protein folding, protein misfolding
Protein-folding pathways generally depend on both the intrinsic properties of the polypeptide chain and the external influence of the cellular environment. For example, whereas the shape of a protein-folding free-energy landscape is essentially determined by the amino-acid sequence, interactions with molecular chaperones are also often required to avoid energy minima that correspond to misfolded and aggregated species (1–3). The folding pathways of many small, usually monomeric, proteins have been extensively studied in an effort to understand the general rules that govern protein folding and to facilitate protein structure prediction, because both have significant implications in medicine and drug design (4, 5). However, a growing number of proteins have been identified over the last decade that challenge some long-established beliefs in the field—they possess a knot deep in their structure formed by the path of the polypeptide backbone (6–9). These molecules, which appear to be self-tying, apparently defy popular concepts that suggest that cooperativity, an increasing degree of “nativeness,” and smooth energy landscapes are required for rapid and efficient protein folding (10). Such theories imply that proteins should be knot-free. Trefoil, figure-of-eight, and penta knots with three, four, and five projected crossings of the polypeptide backbone, respectively, have been observed in proteins from all three domains of life (11–13). The functional advantages of knotted structures over their unknotted counterparts are unknown, although various computational studies have suggested that topological knots may increase protein stability or resistance to cellular translocation and degradation pathways (11, 14–16). Whereas various routes for protein knotting have been proposed, examples of which include the existence of an intermediate “slipknot” configuration or specific, attractive nonnative contacts that promote a threading event (17–19), these models have yet to be experimentally verified. For a complete understanding of the de novo folding behavior of polypeptide chains in vivo, further investigations into the mechanisms involved in protein-knot formation are necessary.
The knotted proteins YibK from Haemophilus influenzae and YbeA from Escherichia coli are single-domain, homodimeric methyltransferases (MTases), whose structures, enzymatic functions, and folding pathways have been probed using a combination of biophysical, biochemical, and computational techniques (18–27). Both proteins adopt an α/β-fold and contain a right-handed trefoil knot, where at least 40 amino acid residues have passed through a similarly sized loop (Fig. 1 A–C). They can be reversibly unfolded in vitro using chemical denaturants to a state that has no detectable structure and undergo folding reactions that involve the population of one or more monomeric intermediate states (26, 27). Experimental investigations into the folding mechanisms of engineered variants of YibK and YbeA suggest that either the polypeptide chain knots early on in the folding reaction, before the formation of secondary and tertiary structure, or that knotting may even occur in the denatured ensemble (24, 25). In addition, the identification of natural knotted MTase proteins that have large appending amino or carboxy domains, and natural tandem fusions of knotted domains within the same polypeptide chain, provides a further indication that the knotting of these proteins could involve very large loops (13). Is it possible, then, that a protein can be knotted even in its denatured state? This question has yet to be addressed due to the practical difficulties that surround the experimental determination of whether a polypeptide chain is indeed knotted at any point in time. Furthermore, the presence of a knot in a denatured polypeptide chain is somewhat unexpected due to the unfavorable entropic cost of knotting. Because of this, it is a concept that is rarely considered.
Fig. 1.

Detecting a knot in a nonnative protein chain. (A, B) The homodimeric knotted proteins YibK (160 residues, PDB ID code 1MXI) (A) and YbeA (155 residues, PDB ID code 1NS5) (B). Proteins are colored to highlight the deep trefoil knot in their structures: The knotting loop is yellow or cyan and the knotted chain is red or blue. AdoHcy molecules are displayed as ball-and-stick models to show the cofactor-binding site in the knotted region of the proteins, determined from crystallographic and modeling studies for YibK and YbeA, respectively (22, 28). (C) A topological diagram of an α/β-knot MTase to illustrate the position of the knot within the structure. The numbers refer to common secondary structure elements. (D) A strategy to trap and identify knots in protein chains using disulfide chemistry, shown here for the denatured state of a knotted protein. Only those molecules that contain a knot in their denatured states can, after circularization, refold to the correct native knotted structure.
Here, we demonstrate that it is possible to experimentally trap and detect knots in nonnative protein chains by the construction of mutant knotted proteins that contain terminal cysteine residues. These constructs can be circularized by formation of a disulfide bond under nonreducing conditions. This effectively tethers the termini of the protein and prevents the occurrence of any subsequent threading or unthreading events. In principle, this reaction can be carried out on any species that can be isolated during the folding process. The ability of the circularized protein to fold and form the native knotted structure can be used to verify the presence of a knot under the conditions in which the disulfide bond was first introduced. We have used this approach to investigate if the polypeptide chains of YibK and YbeA are knotted in their chemically denatured states, where there is no detectable structure (Fig. 1D). We find evidence to suggest that the denatured ensemble of both proteins contains molecules that frequently, if not predominantly, exist in the correct trefoil-knot conformation for productive folding to the native knotted structure. This result can explain the efficient folding reaction of YibK and YbeA observed in vitro (20, 26) and has important implications for general protein-folding and misfolding processes within the cellular milieu.
Results
YibK and YbeA Can Be Circularized Under Native and Strongly Denaturing Conditions.
YibK and YbeA are particularly suited to circularization due to the close proximity of their natural N and C termini (Fig. 1 A and B). The introduction of a serine-glycine spacer and a terminal cysteine residue at the amino and carboxy terminus of both proteins generated constructs that were capable of disulfide-bond formation. The terminal cysteine mutants of YibK and YbeA were purified in their linear reduced form and are here referred to as 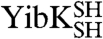 and
and 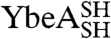 , respectively. Disulfide bonds were allowed to form in
, respectively. Disulfide bonds were allowed to form in 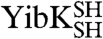 and
and 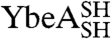 samples under both native and fully denaturing (6 M GdmCl) conditions. The oxidation reaction was monitored by quantifying the free sulfhydryl groups in solution using 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB). This resulted in both monomeric and multimeric species in an approximately equal ratio, suggesting that intra- and intermolecular disulfide bonds form at comparable rates. Monomeric disulfide-bonded species were isolated to yield the circularized proteins
samples under both native and fully denaturing (6 M GdmCl) conditions. The oxidation reaction was monitored by quantifying the free sulfhydryl groups in solution using 5,5′-dithio-bis(2-nitrobenzoic acid) (DTNB). This resulted in both monomeric and multimeric species in an approximately equal ratio, suggesting that intra- and intermolecular disulfide bonds form at comparable rates. Monomeric disulfide-bonded species were isolated to yield the circularized proteins 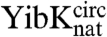 and
and 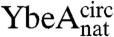 (circularized under native conditions) and
(circularized under native conditions) and 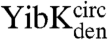 and
and 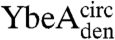 (circularized under denaturing conditions). The circularization of
(circularized under denaturing conditions). The circularization of 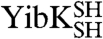 and
and 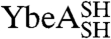 under native conditions generated reference states in which it is known that the polypeptide chain is in a knotted conformation.
under native conditions generated reference states in which it is known that the polypeptide chain is in a knotted conformation.
Circularized Forms of YibK and YbeA Can Refold to Their Correct, Native Knotted Structure.
Using our strategy, only 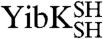 and
and 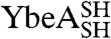 molecules with their polypeptide backbone in the correct trefoil-knot conformation when circularized can subsequently refold to a knotted, dimeric native state. This feature can be used to detect the existence of a knotted topology in the denatured state of the two proteins (Fig. 1D). A variety of structural and functional probes were employed to assess the presence of the native fold, including measures of oligomeric state, secondary structure, and ligand binding. In all cases, refolded proteins that were circularized in their chemically denatured states behaved the same as proteins that were circularized in their native states (Fig. 2). This suggests that, for both
molecules with their polypeptide backbone in the correct trefoil-knot conformation when circularized can subsequently refold to a knotted, dimeric native state. This feature can be used to detect the existence of a knotted topology in the denatured state of the two proteins (Fig. 1D). A variety of structural and functional probes were employed to assess the presence of the native fold, including measures of oligomeric state, secondary structure, and ligand binding. In all cases, refolded proteins that were circularized in their chemically denatured states behaved the same as proteins that were circularized in their native states (Fig. 2). This suggests that, for both 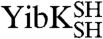 and
and 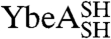 , a trefoil-knot conformation can exist in the unfolded protein. Analytical size-exclusion chromatography (SEC) confirmed that, under native conditions, the circularized constructs are able to form native-like dimers in solution, thus demonstrating that the dimer interface, located near the knotted core in YibK and YbeA, is present (Fig. 2A). A small peak corresponding to a monomer of between 10–15% was observed in the elution profiles of
, a trefoil-knot conformation can exist in the unfolded protein. Analytical size-exclusion chromatography (SEC) confirmed that, under native conditions, the circularized constructs are able to form native-like dimers in solution, thus demonstrating that the dimer interface, located near the knotted core in YibK and YbeA, is present (Fig. 2A). A small peak corresponding to a monomer of between 10–15% was observed in the elution profiles of 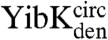 and
and 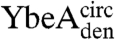 . This suggests that the dimer interface is unable to form in a minor fraction of molecules, perhaps because in these cases the polypeptide chain has become untied under the strong denaturing conditions. The far ultraviolet circular dichroism (far-UV CD) spectra of all the circularized proteins are characteristic of the MTase α/β-fold and indicate the formation of a native-like secondary structure (Fig. 2B).
. This suggests that the dimer interface is unable to form in a minor fraction of molecules, perhaps because in these cases the polypeptide chain has become untied under the strong denaturing conditions. The far ultraviolet circular dichroism (far-UV CD) spectra of all the circularized proteins are characteristic of the MTase α/β-fold and indicate the formation of a native-like secondary structure (Fig. 2B).
Fig. 2.

Structural and functional characterization of linear and circularized knotted proteins. Data are shown for wild-type proteins (black), linear reduced constructs with terminal cysteine residues (cyan), proteins circularized in the native state (blue), and proteins circularized in the denatured state (red). (A) Native analytical size-exclusion chromatography. (B) Far-UV CD spectra under native (circles) and denaturing (solid lines) conditions. (C) Cofactor binding isotherms measured using ITC. Insets show the original titration traces. The continuous line represents the best fit of the data to a single-site binding model using the Origin software package (MicroCal Inc.). Data have been corrected for the heat of dilution. See also Table S1.
YibK and YbeA bind the MTase cofactor S-adenosylmethionine in a crevice that is formed by the knotted region of the protein (Fig. 1 A and B) (22, 28). The binding affinity for S-adenosyl homocysteine (AdoHcy), the product of S-adenosyl methionine after methyl-group transfer to the substrate has taken place, can be used to confirm the integrity of the cofactor binding site and therefore also the presence of the native, knotted structure (21, 24). Binding of AdoHcy was measured using isothermal titration calorimetry (ITC) for the circularized proteins; all displayed a similar affinity for AdoHcy as the equivalent linear species (Fig. 2C and Table S1). Dissociation constants were in the range of 59–74 μM and 0.6–2.4 μM for YibK and YbeA proteins, respectively. Consequently, all the structural and functional probes indicate that the circularized proteins are able to form the correct knotted, homodimeric, native structure irrespective of whether they were circularized in a native or denatured state. These results demonstrate that the polypeptide chains of 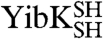 and
and 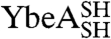 can exist in a right-handed trefoil-knot conformation in their largely unstructured, chemically denatured states.
can exist in a right-handed trefoil-knot conformation in their largely unstructured, chemically denatured states.
Knotted Denatured Polypeptide Chains as Protein-Folding Precursors for YibK and YbeA in Vitro.
We examined the stabilities and the folding pathways of the circularized knotted proteins to learn more about their folding properties. Although 6 M GdmCl was used to circularize 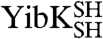 and
and 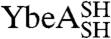 under denaturing conditions, the chemical denaturant urea was employed in these experiments to allow for comparison with previous studies on wild-type YibK and YbeA.
under denaturing conditions, the chemical denaturant urea was employed in these experiments to allow for comparison with previous studies on wild-type YibK and YbeA. 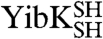 and
and 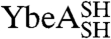 unfold at lower concentrations of urea than the parent wild-type proteins (Fig. 3). This suggests that the mutations introduced to produce proteins that are able to circularize have had a destabilizing effect. Circularization of a protein can lead to an overall change in stability depending upon the balance between the favorable entropic effect that results from a loss in conformational entropy of the denatured state and any strain caused by the introduction of a disulfide bond in the native structure (29). Equilibrium denaturation profiles measured for the circularized proteins studied here differ from those of the corresponding linear reduced forms. They display an increase in the midpoint of unfolding ([D]50%) of between 0.1–0.4 M urea and a decrease in the apparent slope of the transition, or m value (mapp), of between 0.7–1.6 kcal mol-1 M-1 (Fig. 3 and Table S2). This is consistent with circularization having a stabilizing effect and the denatured state being more compact.
unfold at lower concentrations of urea than the parent wild-type proteins (Fig. 3). This suggests that the mutations introduced to produce proteins that are able to circularize have had a destabilizing effect. Circularization of a protein can lead to an overall change in stability depending upon the balance between the favorable entropic effect that results from a loss in conformational entropy of the denatured state and any strain caused by the introduction of a disulfide bond in the native structure (29). Equilibrium denaturation profiles measured for the circularized proteins studied here differ from those of the corresponding linear reduced forms. They display an increase in the midpoint of unfolding ([D]50%) of between 0.1–0.4 M urea and a decrease in the apparent slope of the transition, or m value (mapp), of between 0.7–1.6 kcal mol-1 M-1 (Fig. 3 and Table S2). This is consistent with circularization having a stabilizing effect and the denatured state being more compact.
Fig. 3.

Equilibrium denaturation profiles of linear and circularized knotted proteins. (A, B) Normalized urea denaturation profiles for YibK (A) and YbeA (B) variants to demonstrate the slope and midpoint of the unfolding transition. Data were measured at 1-μM protein and are colored as in Fig. 2. The solid lines represent the fit of the data to a two-state dimer denaturation model. See also Table S2.
Information on the rate and mechanism of folding of the circularized knotted proteins was obtained from analysis of kinetic folding and unfolding data. Wild-type YibK folds from a urea-denatured state with a complex kinetic mechanism that involves four reversible folding phases. These correspond to the formation of two different intermediates on parallel pathways that fold via a third sequential monomeric intermediate to form a native dimer in a slow rate-limiting dimerization reaction (Fig. 4A) (27). In contrast, wild-type YbeA exhibits two reversible kinetic phases that relate to native dimer formation from a monomeric intermediate state (Fig. 4B) (26). The folding kinetics of 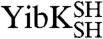 and
and 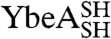 and their associated circularized forms were measured over a range of urea concentrations, and a global analysis of the kinetic data was performed similar to that used to determine the folding pathways of the wild-type proteins (25, 26). The mutant proteins were all found to fold with the same number of reversible, [urea]-dependent phases as the parent wild-type proteins (Figs. S1 and S2), and the kinetic data were used to calculate chevron plots (Fig. 5 and Table S3). The sum of the kinetic m values (mkin), which are the slopes of the chevron plots, for all the folding and unfolding phases observed for
and their associated circularized forms were measured over a range of urea concentrations, and a global analysis of the kinetic data was performed similar to that used to determine the folding pathways of the wild-type proteins (25, 26). The mutant proteins were all found to fold with the same number of reversible, [urea]-dependent phases as the parent wild-type proteins (Figs. S1 and S2), and the kinetic data were used to calculate chevron plots (Fig. 5 and Table S3). The sum of the kinetic m values (mkin), which are the slopes of the chevron plots, for all the folding and unfolding phases observed for 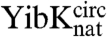 and
and 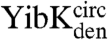 is some 0.7–1.3 kcal mol-1 M-1 lower than for
is some 0.7–1.3 kcal mol-1 M-1 lower than for 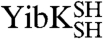 . Conversely, the kinetic stability of
. Conversely, the kinetic stability of 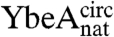 and
and 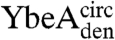 is at least 1.5 kcal-1 mol-1 greater than for
is at least 1.5 kcal-1 mol-1 greater than for 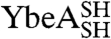 (Table S3). Both these observations are consistent with the proteins being circularized. Overall, however, the kinetic folding data suggest that the gross features of the folding free-energy landscape of the knotted proteins have not been altered upon circularization; circularized and linear variants appear to fold through essentially the same pathway as the parent wild-type proteins and populate similar intermediate species. Moreover, the refolding rate constants measured for the circularized proteins are comparable to those calculated for the parent wild-type protein (Fig. 5 and Table S3). This indicates that the trapping of the knot has had little impact on the folding speed or the rate-determining step. Consequently, a productive folding trajectory in vitro for wild-type YibK and YbeA most likely begins from a knotted denatured state.
(Table S3). Both these observations are consistent with the proteins being circularized. Overall, however, the kinetic folding data suggest that the gross features of the folding free-energy landscape of the knotted proteins have not been altered upon circularization; circularized and linear variants appear to fold through essentially the same pathway as the parent wild-type proteins and populate similar intermediate species. Moreover, the refolding rate constants measured for the circularized proteins are comparable to those calculated for the parent wild-type protein (Fig. 5 and Table S3). This indicates that the trapping of the knot has had little impact on the folding speed or the rate-determining step. Consequently, a productive folding trajectory in vitro for wild-type YibK and YbeA most likely begins from a knotted denatured state.
Fig. 4.

The folding pathways of YibK and YbeA. The folding mechanisms proposed for wild-type dimeric YibK (A) and YbeA (B), based on previous kinetic analysis of folding and unfolding reactions (26, 27).
Fig. 5.

Characterization of the folding free-energy landscapes of linear and circularized knotted proteins. (A–D) Chevron plots calculated from the global analysis of kinetic transients measured in urea for variants of YibK (A, B) and YbeA (C, D). Data were measured at 1-μM protein and are colored as in Fig. 2. Rate constants for each phase are denoted by open diamonds (very fast), filled diamonds (fast), open circles (slow), and filled circles (very slow dimerization). Data were obtained from single-jump unfolding or refolding experiments, except for the very fast, fast, and slow unfolding arms of the YibK chevron plots that were calculated from interrupted refolding experiments. See also Figs. S1 and S2 and Table S3.
Knotting Characteristics of the Denatured Polypeptide Chains of YibK and YbeA.
Our data suggest that the large majority of denatured 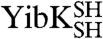 and
and 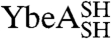 molecules have the correct trefoil-knot conformation such that after circularization they can still fold to the native state (Fig. 2). This implies that, during disulfide-bond formation under denaturing conditions, either the polypeptide chains rarely (or never) become untied or knotted and unknotted states exist in equilibrium and circularization of the knotted species is favored. We investigated the effect of denaturing the wild-type proteins in 6 M GdmCl at several temperatures for various lengths of time. The refolding efficiencies of both YibK and YbeA, as determined by their ability to dimerize and regain the characteristic far-UV CD and fluorescence signals of the native proteins, remain close to 100%, even after molecules have been left to denature for up to four days at 37 °C (Fig. S3). Successful refolding to a dimeric structure was observed after denatured proteins were incubated at temperatures ranging from 50 to 90 °C; however, mass spectrometry, SEC, and SDS-PAGE indicated that a significant proportion of the polypeptide chains decompose into smaller fragments under these conditions. This suggests that, at physiological temperatures, the denatured-state knots in YibK and YbeA are relatively hard to untie and remain present even after long periods of time, or that any untied molecules can reknot rapidly and spontaneously before folding. Taken together, these data demonstrate that occurrences of chains in knotted conformations are frequent, if not prevalent, in the denatured ensemble of these proteins.
molecules have the correct trefoil-knot conformation such that after circularization they can still fold to the native state (Fig. 2). This implies that, during disulfide-bond formation under denaturing conditions, either the polypeptide chains rarely (or never) become untied or knotted and unknotted states exist in equilibrium and circularization of the knotted species is favored. We investigated the effect of denaturing the wild-type proteins in 6 M GdmCl at several temperatures for various lengths of time. The refolding efficiencies of both YibK and YbeA, as determined by their ability to dimerize and regain the characteristic far-UV CD and fluorescence signals of the native proteins, remain close to 100%, even after molecules have been left to denature for up to four days at 37 °C (Fig. S3). Successful refolding to a dimeric structure was observed after denatured proteins were incubated at temperatures ranging from 50 to 90 °C; however, mass spectrometry, SEC, and SDS-PAGE indicated that a significant proportion of the polypeptide chains decompose into smaller fragments under these conditions. This suggests that, at physiological temperatures, the denatured-state knots in YibK and YbeA are relatively hard to untie and remain present even after long periods of time, or that any untied molecules can reknot rapidly and spontaneously before folding. Taken together, these data demonstrate that occurrences of chains in knotted conformations are frequent, if not prevalent, in the denatured ensemble of these proteins.
Discussion
The existence of a knot in the denatured states of YibK and YbeA, while perhaps unexpected, is consistent with our earlier work that examined the folding pathways of these proteins and can explain the relative ease with which they can form their native states during in vitro folding experiments (24, 26, 27). This includes the surprising result that the fusion of a large, stable structured domain at both the N and C termini of YibK and YbeA has little effect on the folding kinetics (24, 26, 27). It is also in agreement with previous mutational studies on YibK, which suggest that the threading of the polypeptide chain and the formation of protein structure occur independently, as successive events (25). Moreover, the mechanisms by which the knotted denatured states of YibK and YbeA fold to their native structures in vitro may be more similar than we originally thought to those already proposed for the folding of unknotted proteins, which involve cocooperativity and smooth folding energy landscapes to channel the protein toward the native state.
The denatured knots identified here may arise from conformational preferences in the unfolded polypeptide chains, or they may represent kinetically trapped states. In either case, the ability of a polypeptide chain to adopt a knot in its denatured state is somewhat surprising due to the unfavorable entropic cost of knotting. Whether the denatured knots are loosely or tightly tied, are stabilized by either native or nonnative contacts, or are in a dynamic equilibrium with unknotted chains are areas for future research. In fact, it was postulated over a decade ago that knots in native proteins might originate from knotted denatured states (30). Our results are consistent with computational and experimental studies that have demonstrated that long flexible strings and homopolymers, similar to denatured polypeptide chains, have a high chance of becoming entangled of their own accord (6, 31–34).
The results presented here can be related to events in the cellular environment. Our data allow us to propose that denatured knotted polypeptide chains are precursors to the folding pathways of YibK and YbeA in vitro. It is possible that species similar to the denatured knots detected in this study are also populated in vivo during the posttranslational folding of YibK and YbeA, after they are synthesized on the ribosome and knotting for the first time. Evidence that large loops are required during the de novo folding of these proteins comes from the successful biosynthesis of recombinant knotted fusion proteins of both YibK and YbeA with a sizable (91-residue) domain attached at both termini (24). A large number of natural knotted fusion proteins with substantial domains at either the amino or carboxy terminus have also been identified, including several that involve two tandem knotted protein domains in the same polypeptide chain (13). It has been proposed that the knotting of these proteins either must involve very large loops, perhaps like the denatured-state knots identified here, through which the whole domains are threaded, or that knot formation may occur from both directions by the threading of either the amino or carboxy terminal regions (13). How the knot forms in the first place after protein synthesis is unknown. It is possible that the polypeptide chain is initially guided into the correct knotted conformation by a slipknot intermediate, or by specific nonnative contacts, as suggested in previous simulation studies on YibK and YbeA (18, 19).
We can consider the effect of a knotted denatured state on the overall folding free-energy landscape of a knotted protein and how this might be useful during the folding and unfolding events that occur frequently within the cell. A chain that exists in a knotted topology will have fewer accessible conformations and therefore a lower entropy compared to an unknotted chain of equal length. This will restrict the free-energy landscape and facilitate the approach of the polypeptide chain to the native state structure, thus reducing the probability of misfolding and aggregation events (Fig. 6). In this way, the presence of a knot in the denatured state might be considered an intrinsic “chaperone-like” property of the protein to promote efficient folding. Consequently, our results allow us to speculate that the origin of the function of a knot in a protein may not lie in its effects on the native structure, but instead in the unique properties of the denatured state. To exist in a knotted conformation when unfolded in the crowded environment of the cell may be an advantage if it leads to an increased ability to avoid misfolding and aggregation. A knotted denatured state may have reduced exposed hydrophobic surface and volume, both of which are important properties when considering folding vs. misfolding in the cellular environment. Further evidence to suggest that a knotted precursor state encourages efficient protein folding comes from the observed “robustness” of the folding pathways of variants of YibK and YbeA in vitro. Formation of the native knotted structure is fully reversible and is not significantly affected by circularization (in either the native or the denatured state) or the attachment of domains at the amino, carboxy, or both termini (20, 24, 26). This can be compared to some initial views on knotted protein structures that considered such topologies likely to hinder rather than aid folding (35).
Fig. 6.

Knotted denatured states could promote efficient protein folding. Schematic representations of hypothetical folding free-energy landscapes for knotted and unknotted proteins of equal length to demonstrate the potential effect of a knot in the denatured state on the folding mechanism of a protein. See also Fig. S3.
We have demonstrated that denatured polypeptide chains of natively knotted proteins can exist in knotted conformations, even under conditions where spectroscopic probes indicate little structure. In some ways, this is similar to existing ideas that residual structure in the denatured states of nonknotted proteins can be important for folding (36–38). Previous studies have shown that native-like topology can persist in the denatured ensembles of monomeric proteins, either by use of simulations to examine the mean structure of the denatured state (36) or through analysis of residual dipolar coupling constants measured experimentally (37). In addition, global residual structure, including oligomer formation, has been observed in a multimeric protein under unfolding conditions (38). Our results suggest that the polypeptide knots identified here form with ease, or prevail, in the denatured state. Consequently, denatured-state topologies such as this may have a significant, and as yet unexplored, role in general protein-folding and misfolding processes. For example, do knotted conformations exist in the denatured states of unknotted proteins? Can knotted species similar to those identified here lead to kinetic traps or misfolding, and, if so, how do unknotted proteins avoid or escape these knots during folding? Is there a role for molecular chaperones in promoting or preventing knotting in nonnative polypeptide chains? Is it possible to predict if a denatured polypeptide chain has a propensity to knot, and is this an important consideration for the de novo folding of nascent polypeptide chains in vivo? The approach we have used here to determine the presence of knots in polypeptide chains is general and can be used in the future to address many of these questions.
Materials and Methods
Expression and Purification of Proteins.
The genetic sequences for YibK and YbeA were extended by polymerase chain reaction to encode for a serine-glycine linker and a terminal cysteine residue at each terminus and inserted into a pET-17b (Novagen) vector. The cysteine residues Cys22 and Cys83 for YibK and Cys122 for YbeA were mutated to alanine to prevent any ambiguity in the position of any disulfide bond. The resultant proteins were expressed and purified under reducing conditions (10 mM DTT) as described for the parent wild-type protein (20, 26). DNA sequencing, SDS-PAGE, mass spectrometry, and amino acid analysis confirmed the identity and purity of each protein. All protein concentrations are in monomer units.
Creation of Circularized Species.
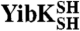 and
and  were fully reduced [5 mM tris(2-carboxyethyl) phosphine] in either native [50 mM Tris-HCl (pH 7.5), 200 mM KCl, 10% (vol/vol) glycerol] or denaturing [0.1 M Tris-HCl (pH 8.0), 6 M GdmCl] conditions. The reducing agent was then removed by buffer exchange. The subsequent oxidation reaction was monitored by quantifying the free sulfhydryl groups in solution using 5,5′-dithio-bis(2-nitrobenzoic acid) and resulted in both monomeric and multimeric species. The circularized monomeric proteins were isolated using denaturing gel-filtration chromatography, and the presence of a disulfide bond in these molecules was verified by mass spectrometry and an absence of free thiol groups (see SI Materials and Methods).
were fully reduced [5 mM tris(2-carboxyethyl) phosphine] in either native [50 mM Tris-HCl (pH 7.5), 200 mM KCl, 10% (vol/vol) glycerol] or denaturing [0.1 M Tris-HCl (pH 8.0), 6 M GdmCl] conditions. The reducing agent was then removed by buffer exchange. The subsequent oxidation reaction was monitored by quantifying the free sulfhydryl groups in solution using 5,5′-dithio-bis(2-nitrobenzoic acid) and resulted in both monomeric and multimeric species. The circularized monomeric proteins were isolated using denaturing gel-filtration chromatography, and the presence of a disulfide bond in these molecules was verified by mass spectrometry and an absence of free thiol groups (see SI Materials and Methods).
Protein Characterization.
Analytical SEC, ITC, far-UV CD, fluorescence, and thermodynamic and kinetic folding experiments were carried out as previously described (20, 21, 24, 25, 27). Additional information on protein characterization can be found in the SI Materials and Methods.
Supplementary Material
Acknowledgments.
We thank J. Clarke, E. Werrell, and S. Humphrey for critical readings of the manuscript. This research was supported by a fellowship and a grant from St. John’s College, University of Cambridge (to A.L.M.).
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0912161107/DCSupplemental.
References
- 1.Clark PL. Protein folding in the cell: Reshaping the folding funnel. Trends Biochem Sci. 2004;29:527–534. doi: 10.1016/j.tibs.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 2.Dobson CM. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 3.Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol: From nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408. [DOI] [PubMed] [Google Scholar]
- 4.Hartl FU, Hayer-Hartl M. Converging concepts of protein folding in vitro and in vivo. Nat Struct Mol Biol. 2009;16:574–581. doi: 10.1038/nsmb.1591. [DOI] [PubMed] [Google Scholar]
- 5.Service RF. Problem solved* (*sort of) Science. 2008;321:784–786. doi: 10.1126/science.321.5890.784. [DOI] [PubMed] [Google Scholar]
- 6.Mallam AL. How does a knotted protein fold? FEBS J. 2009;276:365–375. doi: 10.1111/j.1742-4658.2008.06801.x. [DOI] [PubMed] [Google Scholar]
- 7.Taylor WR, Lin K. Protein knots: A tangled problem. Nature. 2003;421:25. doi: 10.1038/421025a. [DOI] [PubMed] [Google Scholar]
- 8.Taylor WR. A deeply knotted protein structure and how it might fold. Nature. 2000;406:916–919. doi: 10.1038/35022623. [DOI] [PubMed] [Google Scholar]
- 9.Yeates TO, Norcross TS, King NP. Knotted and topologically complex proteins as models for studying folding and stability. Curr Opin Chem Biol. 2007;11:595–603. doi: 10.1016/j.cbpa.2007.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Onuchic JN, Wolynes PG. Theory of protein folding. Curr Opin Struc Biol. 2004;14:70–75. doi: 10.1016/j.sbi.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 11.Virnau P, Mirny LA, Kardar M. Intricate knots in proteins: Function and evolution. PLOS Comput Biol. 2006;2:1074–1079. doi: 10.1371/journal.pcbi.0020122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Taylor WR. Protein knots and fold complexity: Some new twists. Comput Biol Chem. 2007;31:151–162. doi: 10.1016/j.compbiolchem.2007.03.002. [DOI] [PubMed] [Google Scholar]
- 13.Tkaczuk KL, Dunin-Horkawicz S, Purta E, Bujnicki JM. Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases. BMC Bioinformatics. 2007;8:73. doi: 10.1186/1471-2105-8-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dzubiella J. Sequence-specific size, structure, and stability of tight protein knots. Biophys J. 2009;96:831–839. doi: 10.1016/j.bpj.2008.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang L, Makarov DE. Translocation of a knotted polypeptide through a pore. J Chem Phys. 2008;129:121107. doi: 10.1063/1.2968554. [DOI] [PubMed] [Google Scholar]
- 16.Sulkowska JI, Sulkowski P, Szymczak P, Cieplak M. Stabilizing effect of knots on proteins. Proc Natl Acad Sci USA. 2008;105:19714–19719. doi: 10.1073/pnas.0805468105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.King NP, Yeates EO, Yeates TO. Identification of rare slipknots in proteins and their implications for stability and folding. J Mol Biol. 2007;373:153–166. doi: 10.1016/j.jmb.2007.07.042. [DOI] [PubMed] [Google Scholar]
- 18.Wallin S, Zeldovich KB, Shakhnovich EI. The folding mechanics of a knotted protein. J Mol Biol. 2007;368:884–893. doi: 10.1016/j.jmb.2007.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sulkowska JI, Sulkowski P, Onuchic J. Dodging the crisis of folding proteins with knots. Proc Natl Acad Sci USA. 2009;106:3119–3124. doi: 10.1073/pnas.0811147106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mallam AL, Jackson SE. Folding studies on a knotted protein. J Mol Biol. 2005;346:1409–1421. doi: 10.1016/j.jmb.2004.12.055. [DOI] [PubMed] [Google Scholar]
- 21.Mallam AL, Jackson SE. The dimerization of an alpha/beta-knotted protein is essential for structure and function. Structure. 2007;15:111–122. doi: 10.1016/j.str.2006.11.007. [DOI] [PubMed] [Google Scholar]
- 22.Purta E, Kaminska KH, Kasprzak JM, Bujnicki JM, Douthwaite S. YbeA is the m3Psi methyltransferase RlmH that targets nucleotide 1915 in 23S rRNA. RNA. 2008;14:2234–2244. doi: 10.1261/rna.1198108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ero R, Peil L, Liiv A, Remme J. Identification of pseudouridine methyltransferase in Escherichia coli. RNA. 2008;14:2223–2233. doi: 10.1261/rna.1186608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mallam AL, Onuoha SC, Grossmann JG, Jackson SE. Knotted fusion proteins reveal unexpected possibilities in protein folding. Mol Cell. 2008;30:642–648. doi: 10.1016/j.molcel.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 25.Mallam AL, Morris ER, Jackson SE. Exploring knotting mechanisms in protein folding. Proc Natl Acad Sci USA. 2008;105:18740–18745. doi: 10.1073/pnas.0806697105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mallam AL, Jackson SE. A comparison of the folding of two knotted proteins: YbeA and YibK. J Mol Biol. 2007;366:650–665. doi: 10.1016/j.jmb.2006.11.014. [DOI] [PubMed] [Google Scholar]
- 27.Mallam AL, Jackson SE. Probing nature’s knots: The folding pathway of a knotted homodimeric protein. J Mol Biol. 2006;359:1420–1436. doi: 10.1016/j.jmb.2006.04.032. [DOI] [PubMed] [Google Scholar]
- 28.Lim K, et al. Structure of the YibK methyltransferase from Haemophilus influenzae (HI0766): A cofactor bound at a site formed by a knot. Proteins Struct Funct Genet. 2003;51:56–67. doi: 10.1002/prot.10323. [DOI] [PubMed] [Google Scholar]
- 29.Creighton TE. Disulphide bonds and protein stability. Bioessays. 1988;8:57–63. doi: 10.1002/bies.950080204. [DOI] [PubMed] [Google Scholar]
- 30.Mansfield ML. Fit to be tied. Nat Struct Biol. 1997;4:166–167. doi: 10.1038/nsb0397-166. [DOI] [PubMed] [Google Scholar]
- 31.Virnau P, Kantor Y, Kardar M. Knots in globule and coil phases of a model polyethylene. J Am Chem Soc. 2005;127:15102–15106. doi: 10.1021/ja052438a. [DOI] [PubMed] [Google Scholar]
- 32.Raymer DM, Smith DE. Spontaneous knotting of an agitated string. Proc Natl Acad Sci USA. 2007;104:16432–16437. doi: 10.1073/pnas.0611320104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mansfield ML. Efficient knot group identification as a tool for studying entanglements of polymers. J Chem Phys. 2007;127:244901. doi: 10.1063/1.2806928. [DOI] [PubMed] [Google Scholar]
- 34.Mansfield ML. Development of knotting during the collapse transition of polymers. J Chem Phys. 2007;127:244902. doi: 10.1063/1.2806929. [DOI] [PubMed] [Google Scholar]
- 35.Mansfield ML. Are there knots in proteins? Nat Struct Biol. 1994;1:213–214. doi: 10.1038/nsb0494-213. [DOI] [PubMed] [Google Scholar]
- 36.Zagrovic B, Snow CD, Khaliq S, Shirts MR, Pande VS. Native-like mean structure in the unfolded ensemble of small proteins. J Mol Biol. 2002;323:153–164. doi: 10.1016/s0022-2836(02)00888-4. [DOI] [PubMed] [Google Scholar]
- 37.Shortle D, Ackerman MS. Persistence of native-like topology in a denatured protein in 8 M urea. Science. 2001;293:487–489. doi: 10.1126/science.1060438. [DOI] [PubMed] [Google Scholar]
- 38.Perham M, Chen M, Ma J, Wittung-Stafshede P. Unfolding of heptameric co-chaperonin protein follows “fly casting” mechanism: Observation of transient nonnative heptamer. J Am Chem Soc. 2005;127:16402–16403. doi: 10.1021/ja055574o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.