

Abstract
Studies in both humans and rodents have found that insulin+ cells appear within or near ducts of the adult pancreas, particularly following damage or disease, suggesting that these insulin+ cells arise de novo from ductal epithelium. We have found that insulin+ cells are continuous with duct cells in the epithelium that makes up the hyperplastic ducts of both chronic pancreatitis and pancreatic cancer in humans. Therefore, we tested the hypothesis that both hyperplastic ductal cells and their associated insulin+ cells arise from the same cell of origin. Using a mouse model that develops insulin+ cell-containing hyperplastic ducts in response to the growth factor TGFα, we performed genetic lineage tracing experiments to determine which cells gave rise to both hyperplastic ductal cells and duct-associated insulin+ cells. We found that hyperplastic ductal cells arose largely from acinar cells that changed their cell fate, or transdifferentiated, into ductal cells. However, insulin+ cells adjacent to acinar-derived ductal cells arose from pre-existing insulin+ cells, suggesting that islet endocrine cells can intercalate into hyperplastic ducts as they develop. We conclude that apparent pancreatic plasticity can result both from the ability of acinar cells to change fate and of endocrine cells to reorganize in association with duct structures.
Keywords: Beta cell neogenesis, Growth factor signaling, Transdifferentiation, Mouse
INTRODUCTION
Traditionally, terminally differentiated cells of mammals were thought to be immutable, that is, confined to their final differentiated state for the remainder of their lifespans. However, in cases of injury or disease, the cellular composition of organs has been noted to change. For example, in both chronic pancreatitis and pancreatic cancer, many of the enzyme-producing acinar cells in the affected region of the pancreas are absent and replaced by hyperplastic ductal epithelium (Kloppel, 1993; Kloppel and Maillet, 1998). In this and other similar cases, it has been unclear how much of the change in composition results from reprogramming of mature cells and how much from stimulation of progenitor cells residing either within the organ or in the bone marrow. Complicating the issue of origin, small clusters of endocrine cells, notably insulin-producing cells, are closely associated with the hyperplastic ducts that arise during chronic pancreatitis and pancreatic cancer (Chen et al., 1988; Esposito et al., 2007; Phillips et al., 2007). This association between duct and insulin-expressing (insulin+) cell reflects the relationship between these tissues in normal embryonic development, when insulin-producing β cells differentiate from progenitors within the duct-like epithelium of the developing pancreas. Thus, the presence of insulin+ cells in hyperplastic ducts suggests that the adult pancreas is capable of recapitulating embryonic development for the production of new insulin-producing β cells either from dedifferentiation of mature cells or from an unknown precursor. However, lineage tracing experiments that can follow the fate of different cell types have yielded conflicting results (Desai et al., 2007; Dor et al., 2004; Inada et al., 2008; Xu et al., 2008).
The need to provide new sources of insulin-producing β cells for diabetic patients has sparked considerable interest in finding ways to develop increased numbers of β cells either in vivo or in vitro. Although considerable work has indicated that β cells can replicate to increase β cell mass (Dor et al., 2004; Nir et al., 2007; Teta et al., 2007), the ability of other cells to give rise to new β cells has been controversial. Thus far, the most conclusive example of β cell neogenesis, or β cells arising from another cell type in the adult pancreas, comes from gene therapy experiments, in which three genes that regulate β cell development were introduced into pancreatic acinar cells, effectively converting many acinar cells to insulin-producing cells (Zhou et al., 2008). The enzyme-producing acinar cells are the most abundant cells of the pancreas, making them an attractive source for β cell replacement. We have shown previously that acinar cells can transdifferentiate into ductal cells in vitro in response to transforming growth factor α (TGFα) and that this is dependent upon its receptor, the epidermal growth factor receptor (EGFR) (Means et al., 2005). However, without directed transcriptional reprogramming, it remains to be determined whether acinar cells have an innate ability to transdifferentiate into insulin-producing cells. We have now addressed this issue by doing direct lineage tracing to determine the cell of origin both for hyperplastic pancreatic ducts and for the insulin+ cells that arise within them in vivo in response to TGFα. We have found that acinar cells are capable of transdifferentiating into hyperplastic ductal cells, but the insulin+ cells embedded within this ductal epithelium arise from pre-existing insulin+ cells that become intercalated into the ductal epithelium. Thus, even though one cell type is physically associated with another cell type, their origins might be completely different.
MATERIALS AND METHODS
Animals
All experiments were done with approval of the Vanderbilt Institutional Animal Care and Use Committee. Tissue-specific Cre transgenes were bred into mice carrying the R26R-EYFP [Gt(ROSA)26Sortm1(EYFP)Cos] reporter (Srinivas et al., 2001) and the MT-TGFα transgene (Sandgren et al., 1990) that contains the metallothionein promoter driving expression of TGFα. The Cre transgene and the R26R-EYFP reporter allele were never derived from the same parent as this led to embryo-wide recombination in tissues throughout the mouse. Cre transgenes included elastase500-Cre, containing 500 bp of the elastase promoter that has been shown to direct acinar-specific expression (Kruse et al., 1993) and with growth hormone 3′ untranslated sequences (Postic et al., 1999); Villin-Cre (el Marjou et al., 2004), a kind gift from Sylvie Robine; Pdx1PB-CreERT (Zhang et al., 2005), containing the 1 kilobase islet-specific enhancer region from the Pdx1 promoter and the hsp68 minimal promoter driving expression of Cre fused to a mutated estrogen receptor hormone-binding domain that renders Cre inactive until tamoxifen administration; and RIP-CreERT (Dor et al., 2004) containing the rat insulin promoter driving expression of a similar tamoxifen-inducible CreERT protein. For these mice, tamoxifen or corn oil vehicle alone was administered at 4-6 weeks of age, in 3 doses over a 5-day time period, 2 mg/dose via intraperitoneal injection. One week following the final injections, mice were placed on water containing 25 mM ZnSO4 to induce expression of the MT-TGFα promoter. No ductal hyperplasia was observed until 2-3 months of Zn2+ treatment. Following 6 months of ZnSO4 treatment, mice were euthanized and analyzed for EYFP expression.
Immunofluorescence
Pancreatic samples were fixed overnight at 4°C in 4% paraformaldehyde. Shorter fixation times resulted in loss of EYFP, particularly in cytoplasm. Tissues were then washed, dehydrated through a series of ethanol and Histoclear (National Diagnostics, Atlanta, GA, USA) washes and then embedded in paraffin. Five micron sections were collected, dewaxed and heated either at 60°C overnight or at high pressure in a pressure cooker (Cuisinart, East Windsor, NJ, USA) for 15 minutes in 100 mM Tris, pH 10, cooled, washed in PBS, blocked in 5% donkey serum in PBS and then incubated with indicated antibodies to GFP (Invitrogen, Carlsbad, CA or Novus Biological, Littleton, CO, USA), insulin (Linco Research, St Charles, MO, USA), glucagon (Linco Research), pancreatic polypeptide (Linco Research), somatostatin (Santa Cruz Biotechnology, Santa Cruz, CA, USA), amylase (Santa Cruz Biotechnology), cytokeratin 19 [Developmental Studies Hybridoma Bank (DSHB), Iowa City, IA, USA], Ngn3 (DSHB) and laminin (Biogenex, San Ramon, CA, USA). Antibody staining was visualized with Cy2, Cy3 or Cy5 fluorophor-conjugated anti-rabbit, anti-guinea pig, anti-mouse or anti-rat antibodies (Jackson Immunoresearch Laboratories, West Grove, PA, USA) or biotinylated anti-rabbit (Vector Laboratories, Burlingame, CA, USA) followed by Cy3-conjugated avidin (Jackson Immunoresearch Laboratories). Nuclei were stained with Toto3 (Invitrogen). Images were captured on an LSM510 confocal microscope (Carl Zeiss Microimaging, Thornwood, NY, USA) at 1 μm optic depth. Some sections were also labeled colorimetrically using the above primary antibodies and the Vectastain Elite ABC Kit (Vector Laboratories) and 3,3′-diaminobenzidine (Invitrogen) as the peroxidase substrate.
Statistical methods
Lineage tracing experiments were quantified from 2 Villin-Cre, 3 elastase500-Cre, 2 RIP-CreERT and 3 Pdx1PB-CreERT mice. Results from Villin-Cre were combined with elastase500-Cre as these lines gave comparable results. For each mouse, 500-2000 insulin+ cells within islets and 200-800 insulin+ cells in hyperplastic ducts were counted. Averages and s.e.m. were determined and compared by two-tailed t-tests.
RESULTS
Insulin+ cells arise within ductal epithelium
A number of studies have reported insulin-expressing (insulin+) cell clusters associated with pancreatic ducts, particularly with the hyperplastic ducts that arise following damage or disease (Bonner-Weir et al., 1993; Chen et al., 1988; Esposito et al., 2007; Phillips et al., 2007). In order to determine if these insulin+ cells are an integral part of the ductal epithelium, we first examined sections from human patients who had either chronic pancreatitis or pancreatic cancer. Whereas many insulin+ cells were located immediately adjacent to hyperplastic ducts (data not shown), we also found individual insulin+ cells that were continuous with the ductal epithelium (Fig. 1A,B) and thus did not reflect independent endocrine structures, but rather endocrine cells that were located within ductal epithelium. We also found that mouse models recapitulated this phenomenon. In early adenomatous lesions induced by pancreatic expression of the KrasG12D oncogene, insulin+ cells were also found within ductal epithelium (Fig. 1C). Similarly, in mice overexpressing the growth factor TGFα, a growth factor often overexpressed in human pancreatic cancer (Korc et al., 1992), insulin+ cells were found within hyperplastic ductal epithelium (Fig. 1D).
Fig. 1.

Insulin-expressing cells arise within hyperplastic ductal epithelium in humans and mice. (A,B) Pancreatic sections were immunolabeled for insulin (brown) and nuclei were counterstained (blue) in samples from human chronic pancreatitis (A) and human low-grade pancreatic intraepithelial neoplasia (PanIN) lesions associated with pancreatic ductal adenocarcinoma (B). (C) Mouse model of PanIN in which activated KrasG12D is expressed in pancreas. (D) Mouse overexpressing the growth factor TGFα. Scale bar: 50 μm.
In both human and mouse models, many more insulin+ cells were present immediately adjacent to ducts rather than being continuous with the cells of the ductal epithelium. In order to distinguish whether these closely associated cells were merely separate, juxtaposed structures or whether the insulin+ cells might have delaminated out of ductal epithelium, we determined whether the single insulin+ cells as well as clusters of insulin+ cells associated with hyperplastic ducts were each part of the ductal epithelium. We double-labeled tissue sections from TGFα-overexpressing mice for insulin and laminin. Laminin is a basement membrane protein that delineates the outer edges of islets, normal pancreatic ducts and some hyperplastic ducts, effectively marking the boundaries around these structures. As expected, when insulin+ cells were continuous with duct cells within ductal epithelium, these insulin+ cells were contained within the same basement membrane as the surrounding ductal cells (Fig. 2A,B). Clusters of insulin+ cells that appeared to be separate but closely juxtaposed to hyperplastic ducts were also contained within the basement membrane of the ducts (Fig. 2C,D). Notably, there was no laminin present separating ducts from insulin+ cells, suggesting that the insulin+ cells were an integral part of the ductal epithelium, possibly in the process of delaminating out of that epithelium as happens during embryonic development.
Fig. 2.
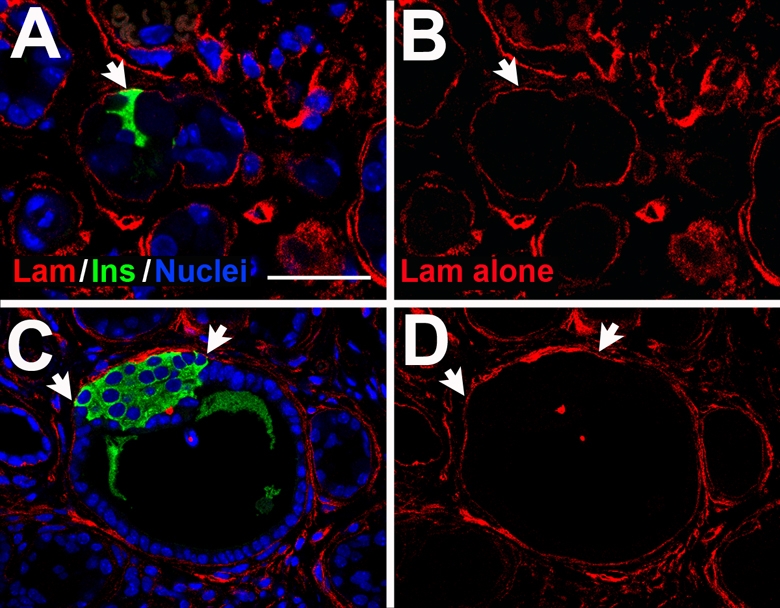
Insulin+ cell clusters are contained within ductal basement membrane. Sections from TGFα-overexpressing pancreas were immunolabeled for insulin (green) and laminin (red) as a marker of the basement membrane, and counterstained to show nuclei (blue). (A) Several insulin+ cells (arrow) that are continuous with the ductal epithelium are contained within a laminin-positive basement membrane. (B) Laminin staining alone from A. (C) A larger cluster of insulin+ cells (arrows) is contained within a ductal basement membrane. (D) Laminin alone from C. Scale bar: 50 μm.
For comparison, we also examined the formation of basement membrane as endocrine cells normally delaminated from ductal epithelium during late embryonic development. At embryonic day 17.5, laminin marked basement membrane around forming ducts and islets (see Fig. S1 in the supplementary material). However, at discreet points, no laminin was detected between the ducts and forming islets. At these points, laminin formed a continuous layer from the duct, extending out around the forming islet. This continuity of basement membrane from duct to delaminating endocrine cells is similar between embryonic tissue and TGFα-induced hyperplastic tissue. However, in the TGFα-overexpressing mice, areas of contact between duct and islet clusters that were devoid of laminin were much broader than in embryonic pancreas. For example, approximately 12 endocrine cells were juxtaposed to ductal epithelium in Fig. 2C, whereas only 1-3 endocrine cells were immediately juxtaposed to duct cells in embryonic pancreas (see Fig. S1 in the supplementary material). As larger ductal endocrine clusters were observed in TGFα-overexpressing mice, they tended to wrap around the duct, maintaining a close association rather than separating from the ductal epithelium. This morphology suggests that if endocrine cells are indeed delaminating from ductal epithelium, they remain within the original basement membrane structure for some period of time.
Acinar cells transdifferentiate into duct cells but not endocrine cells
Although the juxtaposition of single insulin+ cells and insulin+ cell clusters to ductal epithelium suggested that these endocrine cells might have arisen from the same progenitors as the hyperplastic duct cells, we used genetic lineage tracing to define the precise cell of origin for both the hyperplastic ducts and the insulin+ cells associated with them. In order to label large numbers of specific cell types in vivo, we made use of the R26R-EYFP allele, which is transcriptionally silent until Cre recombinase removes a transcriptional stop cassette allowing expression of the EYFP protein (Srinivas et al., 2001). Once recombined, EYFP is expressed from the ubiquitously active Rosa26 locus, even if Cre is subsequently lost from the cell. Because expression of EYFP results from genomic recombination of the R26R-EYFP allele, it represents a heritable genetic trait that will be durably expressed throughout the life of the cell and also passed on to any progeny cells. Thus, tissue-specific activation of the R26R-EYFP allele provides an indelible marker of both Cre-expressing cells and of any cells that arise from those cells. We employed two Cre transgenes, Villin-Cre (el Marjou et al., 2004) and elastase500-Cre to determine the fate of acinar cells in response to TGFα. We previously showed that Villin-Cre activity is specific to acinar cells in the pancreas, although it is also expressed in other cells of other organs (Means et al., 2005). We also used 500 basepairs of the elastase promoter (Kruse et al., 1993) to drive acinar expression of Cre in the elastase500-Cre transgene. However, as mice aged, we observed a small degree of Cre activity in endocrine cells, with approx. 5% of endocrine cells being labeled in addition to virtually all acinar cells in both Villin-Cre and elastase500-Cre mice. Endocrine recombination was lower in young mice (Means et al., 2005) (see Fig. S2 in the supplementary material) and probably resulted from rare misexpression of the transgenes during the 8-month timecourse of our experiments. However, we cannot rule out the possibility that there is a low rate of transdifferentiation of acinar cells into islet cells in normal pancreas.
The Villin-Cre or elastase500-Cre transgenes along with the R26R-EYFP reporter were bred into mice expressing the MT-TGFα transgene. Mice were placed on water containing 25 mM ZnSO4 to induce expression from this metallothionein promoter and were examined 6 months later to determine if acinar cells had given rise to the hyperplastic ductal cells and/or duct-associated insulin+ cells. Confirming previous work showing that acinar cells can transdifferentiate into ducts in vitro (Means et al., 2005), hyperplastic ductal cells carried the EYFP lineage label, indicating that acinar cells gave rise to the majority of hyperplastic ducts that developed in response to TGFα expression (Fig. 3). However, with both acinar cell-specific Cre transgenes, there was no EYFP label present in the majority of insulin-producing cells found either within the hyperplastic ducts or in normal islet structures (Fig. 4). As discussed above and quantified below, both elastase500-Cre and Villin-Cre labeled 5.4±1.9% of normal islet β cells and a similar 5.3±1.1% of duct-associated insulin+ cells. When single insulin+ cells were observed in hyperplastic ducts, they were continuous with EYFP+ ductal cells. Thus, acinar cells were not the cells of origin for ductal insulin+ cells even though they gave rise to duct cells immediately adjacent to these insulin+ cells. These data indicate that although acinar cells can transdifferentiate into ductal cells, they do not seem to be capable of giving rise to insulin-producing cells in response to TGFα.
Fig. 3.

Acinar cells contribute to hyperplastic ducts. In TGFα-overexpressing mice, acini were labeled for expression of the EYFP protein via the Villin-Cre transgene and R26R-EYFP reporter. Mice were placed on Zn2+-containing water at 6 weeks of age and hyperplastic ducts were allowed to develop for 6 months. (A) Co-immunostaining demonstrated that EYFP (green) was expressed in amylase-positive acinar cells (red) as expected, but not all EYFP-positive cells co-expressed amylase, indicating that some previously labeled acinar cells were no longer acinar cells. (B) Amylase staining alone from A. (C) EYFP staining alone from A. (D) EYFP was also expressed in cytokeratin-19-postive (red) hyperplastic ducts, but not in cytokeratin-19-positive ducts of normal morphology (arrowheads), indicating that hyperplastic ducts, but not normal ducts, arose from acinar-to-ductal transdifferentiation. (E) Cytokeratin 19 staining alone from D. (F) EYFP staining alone from D. Arrows, co-labeled cells; arrowheads, normal pancreatic duct; asterisks, non-specific staining of luminal duct contents. Scale bar: 50 μm.
Fig. 4.
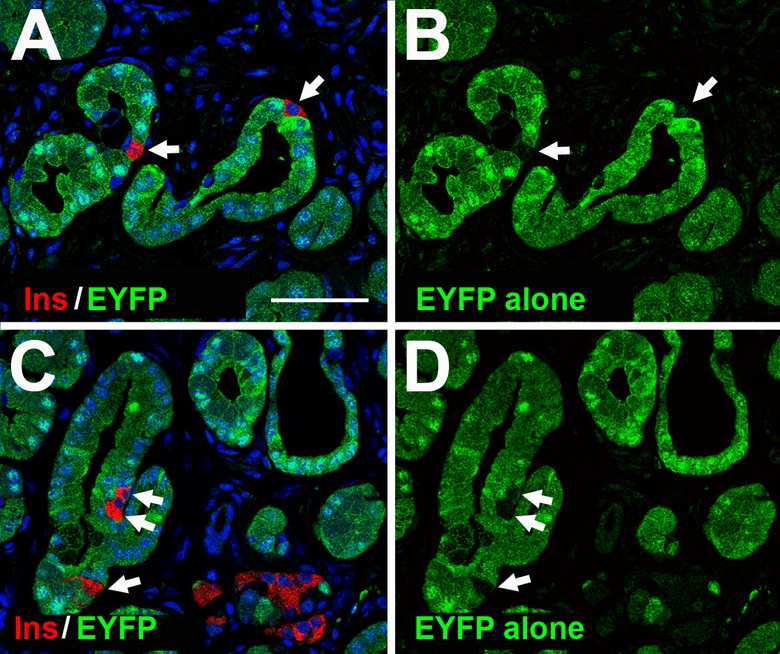
Acinar cells do not give rise to duct-associated insulin+ cells. Acinar cells were labeled using the Villin-Cre transgene to activate EYFP expression. Following induction of hyperplastic ducts, sections were co-immunostained for EYFP (green) and insulin (red) and nuclei were counterstained in blue. (A,C) Although ductal cells immediately surrounding insulin+ cells were lineage-labeled with EYFP expression, ductal insulin+ cells (arrows) were not labeled. (B,D) EYFP only staining of A and C, respectively. Scale bar: 50 μm.
Duct-associated insulin+ cells arise from pre-existing endocrine cells
Although it was possible that either duct cells or an unknown progenitor cell might have given rise to duct-associated insulin+ cells, we first sought to rule out the possibility that pre-existing islet cells could have intercalated into the epithelium of the hyperplastic ducts. The Pdx1PB-CreERT transgene is expressed specifically in islets, principally in β cells but also in 4-5% of glucagon-producing α cells and somatostatin-producing δ cells (Zhang et al., 2005). However, Pdx1PB-CreERT is only active following treatment with tamoxifen, thus any endocrine cells that arise after tamoxifen has been cleared from the bloodstream will not be recombined or labeled with EYFP. Thus, we could investigate whether insulin-producing cells within ducts were derived from islet cells that were present before the onset of ductal hyperplasia. We also used a second islet-specific Cre transgene, RIP-CreERT (Dor et al., 2004) but found that, over the 8-month timecourse of these experiments, this transgene mediated recombination in more than half of all insulin+ cells, even in the absence of tamoxifen (data not shown). Additionally, RIP-CreERT labeled a relatively large percentage of acinar cells. We examined two RIP-CreERT; R26R-EYFP mice, not overexpressing TGFα, at 7-8 months of age (the average age of MT-TGFα mice examined), and found that 3-5% of all acinar cells were labeled with EYFP (see Fig. S3 in the supplementary material). Therefore, we did not use the RIP-CreERT transgene for further lineage tracing experiments. Pdx1PB-CreERT, conversely, did not show any labeling of acinar cells in 7- to 8-month-old mice (see Fig. S3 in the supplementary material) and labeled only 0.31±0.09% of endocrine cells with no tamoxifen over that same timecourse (n=3 mice, 2000-5000 cells counted for each).
We interbred mice carrying the islet-specific Pdx1PB-CreERT transgene to mice carrying the R26R-EYFP reporter and the MT-TGFα transgene. These mice were injected with three low doses of tamoxifen at 1 month of age to yield EYFP labeling in approximately 60-65% of islet β cells. At this stage, MT-TGFα mice have a morphologically normal pancreas, and require at least 2-3 months of zinc induction before hyperplastic ducts appear. This slow development precluded the inadvertent labeling of insulin+ cells as they arose in hyperplastic ducts, so that only pre-existing insulin-producing β cells would be labeled. As reported previously (Zhang et al., 2005), no labeling was seen in non-endocrine cells. One week after the final tamoxifen injection, mice were placed on water containing zinc to induce TGFα expression. After 6 months of zinc induction, mice were analyzed for EYFP expression in different pancreatic cell types (Fig. 5). As discussed above, EYFP labeling was found only in islets in control mice not carrying the TGFα transgene. However, in TGFα-overexpressing mice, insulin+ cells found either within or closely associated with the ductal epithelium also carried the EYFP label, indicating that these cells arose from the endocrine cells that were present at the time of tamoxifen injection. The number of ductal insulin+ cells expressing EYFP reflected the percentage labeled in islets. That is, 64.2±1.2% of islet β cells were labeled, whereas 63.1±3.4% of duct-associated insulin+ cells were labeled. Supporting a β cell origin for ductal insulin+ cells, we did not detect any Ngn3 expression in MT-TGFα mice nor did ductal insulin+ cells co-express ductal markers such as cytokeratin 19 (Fig. 5) or bind DBA (data not shown). We also determined whether β cells could act as multipotent progenitor cells and give rise to other cell types using Pdx1PB-CreERT to label β cells. No EYFP-labeling was found in duct cells, acinar cells or the majority of glucagon-expressing (glucagon+) cells, both in islets and in hyperplastic ducts (Fig. 5; data not shown). A very small number of glucagon+ cells containing the EYFP lineage label were seen in both islets and in ductal structures, reflecting the small number of glucagon+ cells normally labeled by the Pdx1PB-CreERT transgene. Thus, pre-existing β cells gave rise to ductal insulin+ cells but did not give rise to other duct-associated cell types.
Fig. 5.

Ductal insulin+ cells arise from pre-existing β cells. β cells were labeled via the Pdx1PB-CreERT transgene with activity induced by tamoxifen injection. One week following the final tamoxifen injections, mice were given Zn2+ to induce TGFα expression and analyzed 6 months later for expression of EYFP in different cell types. (A-D) EYFP (green) was found in single insulin+ cells (red; arrow) continuous with ductal epithelium (cytokeratin 19, blue), indicating that the insulin+ cell arose from a pre-existing β cell. Note that a second insulin+ cell was not labeled by EYFP, reflecting the ~65% labeling efficiency for the Pdx1PB-CreERT transgene. A is the merge of panels B-D. (E-H) A cluster of insulin+ cells that is in contact with the ductal lumen was also labeled with EYFP, indicating that those cells arose from pre-existing β cells. E is the merge of panels F-G. Arrows indicate locations that can be compared across rows of panels. Scale bar: 10 μm.
When the EYFP-insulin double labeling efficiency is compared between mice labeled with acinar-specific Cres and mice labeled with an islet-specific Cre, it is clear that pre-existing endocrine cells, not acinar cells, gave rise to duct-associated insulin+ cells (Fig. 6). Examining acinar-specific Cre activity alone with its low-efficiency labeling of endocrine cells leaves open the possibility that a small percentage of endocrine cells, in islets as well as in ducts, arose via acinar cell transdifferentiation. However, the nearly identical labeling of endocrine cells in islets and in ducts by Pdx1PB-CreERT without any acinar labeling indicates that any acinar cell transdifferentation is a minor component at best. By combining analyses of different lineages, we conclude that acinar cells transdifferentiated into hyperplastic ductal epithelium and that pre-existing endocrine cells became intercalated into these structures. However, we could not rule out that insulin+ cells were present normally in exocrine structures and maintained their exocrine association as ductal hyperplasia occurred. Insulin+ cells have been reported in normal ducts of rats, with a frequency that decreases with age (Madden and Sarras, 1989). Therefore, we examined whether insulin+ cells are present in normal pancreatic ducts as mice age. Examining random cross-sections of pancreas from 2-month-old normal mice, no insulin+ cells were found within either ductal or acinar structures (n=3 mice, 3-4 cross-sections each). Because the main pancreatic duct has been proposed as a site of β cell neogenesis (Sharma et al., 1999), we also specifically examined sections through the main pancreatic duct. Two-month-old mice did indeed contain 1-2 clusters each of insulin+ cells continuous with ductal epithelium per approximately 1000 ductal cells of the main pancreatic duct or closely associated interlobular duct branches. Each cluster comprised 1-3 insulin+ cells for an average of 0.32±0.13% of main duct cells (see Fig. S4 in the supplementary material; data not shown). However, in four 6-month-old mice examined, we found no insulin+ cells within the main pancreatic duct, indicating that less than 0.1% of normal main duct cells are insulin+, which is comparable with the results from rat pancreas (Madden and Sarras, 1989). Thus, it appears that mice lose insulin+ cells within their larger ducts as they age. Because we began Zn2+ induction of TGFα expression at approximately 6 weeks of age, it was possible that TGFα merely expanded the population present in these large ducts of the pancreas. Therefore, we examined the occurrence and distribution of duct-associated insulin+ cells in MT-TGFα mice. We found that every cross-section of the pancreas, not just in main duct regions, contained insulin+ cells within hyperplastic ducts, with an average of 12±1.3 ducts per cross-section containing insulin+ cells (n=5 MT-TGFα mice, 2-3 sections counted per mouse). Thus, it is unlikely that TGFα-overexpression expanded the number of insulin+ cells normally present in ductal epithelium.
Fig. 6.
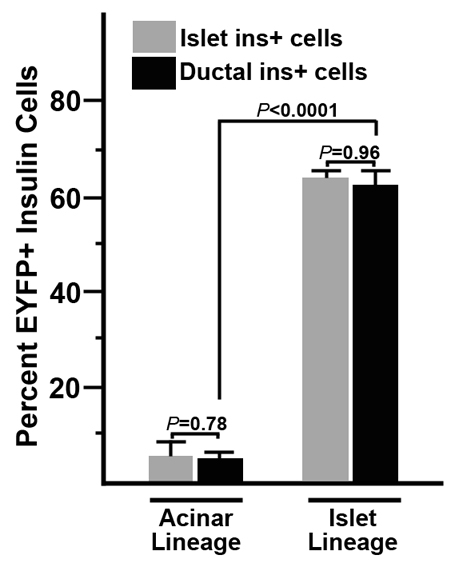
Pre-existing β cells, not acinar cells, give rise to ductal insulin+ cells in response to TGFα signaling. Quantification of the results from Figs 4 and 5 compare the ability of acinar cells and islet cells to give rise to duct-associated insulin+ cells. For acinar lineage tracing (left two bars), results from 3 elastase500-Cre and 2 Villin-Cre mice were averaged. Note that although these transgenes labeled virtually all acinar cells, they also labeled ~5% of islet (gray bar) β cells by the time mice were 8 months old. Similarly, 5% of duct-associated (black bar) insulin+ cells were labeled in these mice. For islet lineage tracing (right two bars), 3 Pdx1PB-CreERT mice were used to label islet cells with tamoxifen injected one week before MT-TGFα transgene induction and ~2 months before the appearance of hyperplastic ducts. At the tamoxifen dosage used, 64.2±1.2% of islet (gray bar) β cells were labeled with the EYFP lineage reporter. Similarly, 63.1±3.4% of duct-associated (black bar) insulin+ cells were labeled. Together, these data indicate that acinar cells do not contribute to ductal insulin+ cells and that the majority of duct-associated insulin+ cells arise from pre-existing β cells. Percentages were determined from 3 elastase500-Cre, 2 Villin-Cre and 3 Pdx1PB-CreERT mice, with 500-2000 islet insulin+ cells and 200-800 ductal insulin+ cells counted per mouse.
DISCUSSION
We have found that pancreatic acinar cells have a limited ability to give rise to other cell types within the adult mouse pancreas in vivo. Using rigorous genetic lineage tracing, we have demonstrated that pancreatic acinar cells have the ability to transdifferentiate into the hyperplastic ducts that develop upon overexpression of TGFα. Although numerous insulin-expressing cells were part of this hyperplastic ductal epithelium, we have shown that these ductal insulin+ cells originated from pre-existing β cells and were not the result of β cell neogenesis. Although acinar cells are capable of giving rise to ductal cells in response to TGFα signaling both in vitro and in vivo, they were not the cell of origin for endocrine cells found associated with such ducts in vivo.
Phylogenetic differences in β cell location
Although the majority of β cells are clustered in pancreatic islets in mammals, other species show a variety of localizations of insulin-producing cells (for a review, see Gannon and Wright, 1999). Tunicates and cephalochordates such as amphioxus have individual insulin+ cells located within gut epithelium, providing an evolutionary precedent for insulin+ cells within epithelial sheets. However, vertebrates as distant as agnathans organize β cells into distinct islet clusters. Thus, the ability of β cells to emigrate out of epithelial sheets wherein they arise and cluster evolved even before development of a distinct exocrine pancreas.
Acinar cells transdifferentiate into ductal cells
In addition to demonstrating that ductal insulin+ cells do not arise from acinar cells, this study demonstrates that β cells do not give rise to hyperplastic ducts in response to TGFα-overexpression. Rather, TGFα induces transdifferentiation of acinar cells into hyperplastic ducts. In contrast to our studies, Strobel et al. (Strobel et al., 2007) found that acinar cells gave rise to only a small percentage of the hyperplastic ducts that arose in response to cerulein, a cholecystokinin analog that induces pancreatitis in mice. This difference might result from specific effects of TGFα on acinar cells and/or because cerulein treatment results in widespread apoptosis of acinar cells. It is probable that both acinar cells and duct cells can give rise to hyperplastic ducts in the pancreas, with acinar cells predominating in our study owing to their overwhelming abundance.
Gidekel Friedlander et al. (Gidekel Friedlander et al., 2009) performed lineage studies to demonstrate that activation of the KrasG12D oncogene via the insulin promoter could give rise to ductal adenomas, suggesting that islets could be a cell of origin for pancreatic ductal adenocarcinoma. However, this group used the RIP-CreERT transgene to activate KrasG12D expression. The authors described little or no labeling of acinar cells with this CreERT transgene. We have shown here that RIP-CreERT recombines 3-6 percent of acinar cells. The discrepancy in our studies might arise from the reporter gene used to assess recombination. Gidekel Friedlander et al. (Gidekel Friedlander et al., 2009) used the R26R reporter that expresses β-galactosidase (βgal) upon Cre-mediated recombination (Soriano, 1999) rather than the EYFP reporter that we used (Srinivas et al., 2001). We have found that it is very difficult to preserve βgal activity in adult acinar cells. In our hands, reliable βgal detection in acinar cells requires transcardial perfusion of the fixative, rapid freezing and re-fixation of tissue sections on the slide. Even EYFP is less well-preserved in acinar cytoplasm than in islet cytoplasm (see Fig. S3 in the supplementary material) but its small size allows EYFP to diffuse into the nucleus where it is more readily detected. Thus, the ability of KrasG12D to induce islet cells to give rise to ductal adenomas will have to await experiments with a more specific islet CreERT, such as the Pdx1PB-CreERT used here.
Origin of β cells in the adult pancreas
Various cell types have been postulated as possible sources for β cell renewal in vivo. Many researchers have suggested that there are facultative progenitor cells residing within normal ductal epithelium that lead to islet cell regeneration. These studies have primarily relied on morphological analysis after the stimulation of pancreas-wide damage by either partial pancreatectomy (Ackermann Misfeldt et al., 2008; Bonner-Weir et al., 1993) or ductal ligation (Wang et al., 1995) that results in the presence of insulin+ cells closely associated with ducts. The appearance of these insulin+ cells has been interpreted as new islets budding from the ductal epithelium (Esposito et al., 2007; Gu and Sarvetnick, 1993; Phillips et al., 2007; Song et al., 1999). However, a recent report from Desai et al. (Desai et al., 2007) demonstrated that insulin+ cells in islets observed following surgical removal of part of the pancreas did not result from acinar cell transdifferentiation, a result similar to ours using growth factor induction of ductal hyperplasia. Desai et al. (Desai et al., 2007) did not determine what the cell of origin was in their partial pancreatectomy model, nor did they examine duct-associated insulin+ cells. In order to determine if duct cells could transdifferentiate into insulin-producing cells, Solar et al. (Solar et al., 2009) used the HNF1β promoter to drive CreERT in pancreatic ducts. They found that duct cells in adult mice did not contribute to insulin-producing cells even after pancreatic duct ligation, but they appeared to examine only endocrine cells in islet structures, not within ducts, and they did not identify what the origin was for these endocrine cells. Inada et al. (Inada et al., 2008) used the carbonic anhydrase II (CAII) promoter to drive CreERT expression in pancreatic ducts. After partial pancreatectomy, both insulin+ and ductal cells were labeled with the Cre reporter, suggesting that ducts could give rise to new β cells. However, this CAII-CreERT transgene was not characterized for its expression pattern or labeling efficiency, thus putting any conclusions on hold until those are determined.
Other studies have also shown that duct-associated insulin+ cells can arise in mouse pancreas, for example, in response to adenoviral-mediated overexpression of two EGFR ligands, HB-EGF and betacellulin (Kozawa et al., 2005; Tokui et al., 2006). The ability of both these ligands to activate EGFR, as does TGFα used in the studies presented here, suggests that the increase in β cells might have resulted from relocalization and/or expansion of pre-existing β cells.
In addition to insulin+ cells appearing in ducts, neurogenin 3 (Ngn3) has also been detected in pancreatic ducts in response to damage. Partial pancreatectomy that retains the main pancreatic duct in the tail of the pancreas while removing most other exocrine and endocrine tissue resulted in the appearance of Ngn3+ cells in ducts (Ackermann Misfeldt et al., 2008), whereas partial pancreatectomy completely removing the distal main duct did not result in Ngn3 expression (Lee et al., 2006). Xu et al. (Xu et al., 2008) demonstrated that partial pancreatic duct ligation, a method of constricting flow through the main pancreatic duct resulting in damage distal to the site of ligation, also resulted in the appearance of Ngn3-expressing cells. Ngn3 is a regulator of β cell neogenesis during embryonic development, so its expression in adult pancreas might suggest that β cell neogenesis could be occurring. In either of the above studies it is not known which cells gave rise to the Ngn3+ cells. No Ngn3+ cells were detected in MT-TGFα mice. However, cellular changes in response to TGFα occur slowly over many months so transient Ngn3 expression could have been missed. We chose this model because we could be sure that tamoxifen would be cleared from the bloodstream long before ductal insulin+ cells were observed. However, we have also examined mice that overexpress a particular isoform of a related family member, soluble HB-EGF (sHB-EGF) (Ray et al., 2009). In these mice, ductal hyperplasia occurs beginning around 1-2 months of age and many ductal insulin+ cells are observed by 2 months of age (data not shown). Even in these mice, no Ngn3 was detected. The discrepancies in Ngn3 detection throughout the literature might be owing to different inductive cues and/or differences in antibodies used for detection. In none of the studies were adult Ngn3+ cells lineage mapped to determine their cell of origin. Wang et al. (Wang et al., 2009) have demonstrated a low level of Ngn3 expression in adult islets and have demonstrated that its loss in adult β cells affected islet function. Therefore, Ngn3 is not only a marker of endocrine precursors but might also be a marker of some adult β cells.
In the studies by Xu et al., adult Ngn3-expressing cells were not shown to give rise to β cells in vivo, but could be induced to differentiate into insulin-producing cells ex vivo, suggesting that adult pancreatic cells are capable of generating new β cells but the adult environment prohibits completion of this pathway. In support of this hypothesis, others have reported ex vivo expansion of insulin+ cells from pancreatic cultures consisting primarily of acinar cells (Baeyens et al., 2005; Minami et al., 2005). Thus, it might be possible to derive ex vivo methods for increasing β cell mass from adult pancreas for subsequent islet transplantation into diabetic patients.
We have shown previously that acinar cells (Means et al., 2005) isolated from mature mouse pancreas could transdifferentiate into ductal cells ex vivo under the influence of exogenous TGFα and that this process involved a dedifferentiation step. Here we show that this is recapitulated in vivo with acinar cells giving rise to hyperplastic ducts. The use of transgenes, allowing for both acinar cell- and islet cell-specific Cre recombinase expression coupled with the R26R-EYFP reporter, enabled us to determine the level of plasticity of adult pancreatic acinar cells in response to TGFα in vivo. Our findings that pre-existing β cells gave rise to duct-associated insulin+ cells are in agreement with the recent results of others, that only β cells can give rise to new β cells in vivo. Dor et al. (Dor et al., 2004) used an elegant insulin-promoter-based approach to show that existing β cells are the sole source of new β cells in the adult mouse under normal conditions. Along the same lines, an innovative DNA analog-based lineage tracing method has been utilized to demonstrate that β cell generation is the result of self duplication during both normal growth conditions and in cases of acute β cell expansion, and does not involve specialized progenitors (Teta et al., 2007).
Interestingly, multiple studies that demonstrated an increase in insulin-producing cells both in vivo and in vitro used EGFR ligands to stimulate this β cell expansion (Baeyens et al., 2005; Minami et al., 2005; Rooman et al., 2000; Suarez-Pinzon et al., 2005a; Suarez-Pinzon et al., 2005b). Although our findings indicate that EGFR signaling does not lead to islet neogenesis in vivo, EGFR signaling may allow alterations in the β cell environment, such as ductal localization, that allow increased proliferation or survival. It will be interesting to determine in future studies whether the ductal environment promotes the proliferation of functional β cells. Such studies could aid our ability to increase endocrine function in diabetic patients that retained some β cell mass.
Supplementary Material
Acknowledgements
We thank Steven Leach for collaborating to generate the elastase500-Cre mouse and for critically reading this manuscript. This work was supported by JDRF research grant 1-2006-759 (A.L.M.), and through the Vanderbilt Digestive Diseases Research Center (NIH P30DK058404, M.K.W.) and the use of the Vanderbilt Cell Imaging Shared Resource (NIH CA68485, DK20593, DK58404, HD15052, DK59637 and EY08126). Deposited in PMC for release after 12 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.048421/-/DC1
References
- Ackermann Misfeldt A., Costa R. H., Gannon M. (2008). Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes 57, 3069-3077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeyens L., De Breuck S., Lardon J., Mfopou J. K., Rooman I., Bouwens L. (2005). In vitro generation of insulin-producing beta cells from adult exocrine pancreatic cells. Diabetologia 48, 49-57 [DOI] [PubMed] [Google Scholar]
- Bonner-Weir S., Baxter L. A., Schuppin G. T., Smith F. E. (1993). A second pathway for regeneration of adult exocrine and endocrine pancreas. A possible recapitulation of embryonic development. Diabetes 42, 1715-1720 [DOI] [PubMed] [Google Scholar]
- Chen J., Baithun S. I., Pollock D. J., Berry C. L. (1988). Argyrophilic and hormone immunoreactive cells in normal and hyperplastic pancreatic ducts and exocrine pancreatic carcinoma. Virchows Arch. A Pathol. Anat. Histopathol. 413, 399-405 [DOI] [PubMed] [Google Scholar]
- Desai B. M., Oliver-Krasinski J., De Leon D. D., Farzad C., Hong N., Leach S. D., Stoffers D. A. (2007). Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J. Clin. Invest. 117, 971-977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y., Brown J., Martinez O. I., Melton D. A. (2004). Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 429, 41-46 [DOI] [PubMed] [Google Scholar]
- el Marjou F., Janssen K. P., Chang B. H., Li M., Hindie V., Chan L., Louvard D., Chambon P., Metzger D., Robine S. (2004). Tissue-specific and inducible Cre-mediated recombination in the gut epithelium. Genesis 39, 186-193 [DOI] [PubMed] [Google Scholar]
- Esposito I., Seiler C., Bergmann F., Kleeff J., Friess H., Schirmacher P. (2007). Hypothetical progression model of pancreatic cancer with origin in the centroacinar-acinar compartment. Pancreas 35, 212-217 [DOI] [PubMed] [Google Scholar]
- Gannon M., Wright C. V. (1999). Endodermal patterning and organogenesis. In Cell Lineage and Fate Determination (ed. Moody S. A.), pp. 583-616 San Diego, CA: Academic Press; [Google Scholar]
- Gidekel Friedlander S. Y., Chu G. C., Snyder E. L., Girnius N., Dibelius G., Crowley D., Vasile E., DePinho R. A., Jacks T. (2009). Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 16, 379-389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu D., Sarvetnick N. (1993). Epithelial cell proliferation and islet neogenesis in IFN-g transgenic mice. Development 118, 33-46 [DOI] [PubMed] [Google Scholar]
- Inada A., Nienaber C., Katsuta H., Fujitani Y., Levine J., Morita R., Sharma A., Bonner-Weir S. (2008). Carbonic anhydrase II-positive pancreatic cells are progenitors for both endocrine and exocrine pancreas after birth. Proc. Natl. Acad. Sci. USA 105, 19915-19919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloppel G. (1993). Pathology of nonendocrine pancreatic tumors. In The Pancreas: Biology, Pathobiology, and Disease (ed. Go V. L. W.), pp. 871-897 New York: Raven Press; [Google Scholar]
- Kloppel G., Maillet B. (1998). Pathology of chronic pancreatitis. In The Pancreas, vol. 1 (ed. Beger H. G., Warshaw A. L., Buchler M. W., Carr-Locke D. L., Neoptolemos J. P., Russell C., Sarr M. G.), pp. 720-723 London: Blackwell Science; [Google Scholar]
- Korc M., Chandrasekar B., Yamanaka Y., Friess H., Buchier M., Beger H. G. (1992). Overexpression of the epidermal growth factor receptor in human pancreatic cancer is associated with concomitant increases in the levels of epidermal growth factor and transforming growth factor alpha. J. Clin. Invest. 90, 1352-1360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozawa J., Tokui Y., Moriwaki M., Li M., Ohmoto H., Yuan M., Zhang J., Iwahashi H., Imagawa A., Yamagata K., et al. (2005). Regenerative and therapeutic effects of heparin-binding epidermal growth factor-like growth factor on diabetes by gene transduction through retrograde pancreatic duct injection of adenovirus vector. Pancreas 31, 32-42 [DOI] [PubMed] [Google Scholar]
- Kruse F., Rose S. D., Swift G. H., Hammer R. E., MacDonald R. J. (1993). An endocrine-specific element is an integral component of an exocrine-specific pancreatic enhancer. Genes Dev. 7, 774-786 [DOI] [PubMed] [Google Scholar]
- Lee C. S., De Leon D. D., Kaestner K. H., Stoffers D. A. (2006). Regeneration of pancreatic islets after partial pancreatectomy in mice does not involve the reactivation of neurogenin-3. Diabetes 55, 269-272 [PubMed] [Google Scholar]
- Madden M. E., Sarras M. P., Jr (1989). The pancreatic ductal system of the rat: cell diversity, ultrastructure, and innervation. Pancreas 4, 472-485 [DOI] [PubMed] [Google Scholar]
- Means A. L., Meszoely I. M., Suzuki K., Miyamoto Y., Rustgi A. K., Coffey R. J., Jr, Wright C. V., Stoffers D. A., Leach S. D. (2005). Pancreatic epithelial plasticity mediated by acinar cell transdifferentiation and generation of nestin-positive intermediates. Development 132, 3767-3776 [DOI] [PubMed] [Google Scholar]
- Minami K., Okuno M., Miyawaki K., Okumachi A., Ishizaki K., Oyama K., Kawaguchi M., Ishizuka N., Iwanaga T., Seino S. (2005). Lineage tracing and characterization of insulin-secreting cells generated from adult pancreatic acinar cells. Proc. Natl. Acad. Sci. USA 102, 15116-15121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nir T., Melton D. A., Dor Y. (2007). Recovery from diabetes in mice by beta cell regeneration. J. Clin. Invest. 117, 2553-2561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips J. M., O'Reilly L., Bland C., Foulis A. K., Cooke A. (2007). Patients with chronic pancreatitis have islet progenitor cells in their ducts, but reversal of overt diabetes in NOD mice by anti-CD3 shows no evidence for islet regeneration. Diabetes 56, 634-640 [DOI] [PubMed] [Google Scholar]
- Postic C., Shiota M., Niswender K. D., Jetton T. L., Chen Y., Moates J. M., Shelton K. D., Lindner J., Cherrington A. D., Magnuson M. A. (1999). Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 274, 305-315 [DOI] [PubMed] [Google Scholar]
- Ray K. C., Blaine S. A., Washington M. K., Braun A. H., Singh A. B., Harris R. C., Harding P. A., Coffey R. J., Means A. L. (2009). Transmembrane and soluble isoforms of heparin-binding epidermal growth factor-like growth factor regulate distinct processes in the pancreas. Gastroenterology 137, 1785-1794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooman I., Heremans Y., Heimberg H., Bouwens L. (2000). Modulation of rat pancreatic acinoductal transdifferentiation and expression of PDX-1 in vitro. Diabetologia 43, 907-914 [DOI] [PubMed] [Google Scholar]
- Sandgren E. P., Luetteke N. C., Palmiter R. D., Brinster R. L., Lee D. C. (1990). Overexpression of TGF alpha in transgenic mice: induction of epithelial hyperplasia, pancreatic metaplasia, and carcinoma of the breast. Cell 61, 1121-1135 [DOI] [PubMed] [Google Scholar]
- Sharma A., Zangen D. H., Reitz P., Taneja M., Lissauer M. E., Miller C. P., Weir G. C., Habener J. F., Bonner-Weir S. (1999). The homeodomain protein IDX-1 increases after an early burst of proliferation during pancreatic regeneration. Diabetes 48, 507-513 [DOI] [PubMed] [Google Scholar]
- Solar M., Cardalda C., Houbracken I., Martin M., Maestro M. A., De Medts N., Xu X., Grau V., Heimberg H., Bouwens L., et al. (2009). Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev. Cell 17, 849-860 [DOI] [PubMed] [Google Scholar]
- Song S. Y., Gannon M., Washington M. K., Scoggins C. R., Meszoely I. M., Goldenring J. R., Marino C. R., Sandgren E. P., Coffey R. J., Jr, Wright C. V., et al. (1999). Expansion of Pdx1-expressing pancreatic epithelium and islet neogenesis in transgenic mice overexpressing transforming growth factor alpha. Gastroenterology 117, 1416-1426 [DOI] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70-71 [DOI] [PubMed] [Google Scholar]
- Srinivas S., Watanabe T., Lin C. S., William C. M., Tanabe Y., Jessell T. M., Costantini F. (2001). Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev. Biol. 1, 4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strobel O., Dor Y., Alsina J., Stirman A., Lauwers G., Trainor A., Castillo C. F., Warshaw A. L., Thayer S. P. (2007). In vivo lineage tracing defines the role of acinar-to-ductal transdifferentiation in inflammatory ductal metaplasia. Gastroenterology 133, 1999-2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez-Pinzon W. L., Lakey J. R., Brand S. J., Rabinovitch A. (2005a). Combination therapy with epidermal growth factor and gastrin induces neogenesis of human islet {beta}-cells from pancreatic duct cells and an increase in functional {beta}-cell mass. J. Clin. Endocrinol. Metab. 90, 3401-3409 [DOI] [PubMed] [Google Scholar]
- Suarez-Pinzon W. L., Yan Y., Power R., Brand S. J., Rabinovitch A. (2005b). Combination therapy with epidermal growth factor and gastrin increases beta-cell mass and reverses hyperglycemia in diabetic NOD mice. Diabetes 54, 2596-2601 [DOI] [PubMed] [Google Scholar]
- Teta M., Rankin M. M., Long S. Y., Stein G. M., Kushner J. A. (2007). Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev. Cell 12, 817-826 [DOI] [PubMed] [Google Scholar]
- Tokui Y., Kozawa J., Yamagata K., Zhang J., Ohmoto H., Tochino Y., Okita K., Iwahashi H., Namba M., Shimomura I., et al. (2006). Neogenesis and proliferation of beta-cells induced by human betacellulin gene transduction via retrograde pancreatic duct injection of an adenovirus vector. Biochem. Biophys. Res. Commun. 350, 987-993 [DOI] [PubMed] [Google Scholar]
- Wang R. N., Kloppel G., Bouwens L. (1995). Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia 38, 1405-1411 [DOI] [PubMed] [Google Scholar]
- Wang S., Jensen J. N., Seymour P. A., Hsu W., Dor Y., Sander M., Magnuson M. A., Serup P., Gu G. (2009). Sustained Neurog3 expression in hormone-expressing islet cells is required for endocrine maturation and function. Proc. Natl. Acad. Sci. USA 106, 9715-9720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., D'Hoker J., Stange G., Bonne S., De Leu N., Xiao X., Van de Casteele M., Mellitzer G., Ling Z., Pipeleers D., et al. (2008). Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 132, 197-207 [DOI] [PubMed] [Google Scholar]
- Zhang H., Fujitani Y., Wright C. V., Gannon M. (2005). Efficient recombination in pancreatic islets by a tamoxifen-inducible Cre-recombinase. Genesis 42, 210-217 [DOI] [PubMed] [Google Scholar]
- Zhou Q., Brown J., Kanarek A., Rajagopal J., Melton D. A. (2008). In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature 455, 627-632 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}