

Abstract
Integrin cell adhesion receptors and fibronectin, one of their extracellular matrix ligands, have been demonstrated to be important for angiogenesis using functional perturbation studies and complete knockout mouse models. Here, we report on the roles of the α5 and αv integrins, which are the major endothelial fibronectin receptors, in developmental angiogenesis. We generated an integrin α5-floxed mouse line and ablated α5 integrin in endothelial cells. Unexpectedly, endothelial-specific knockout of integrin α5 has no obvious effect on developmental angiogenesis. We provide evidence for genetic interaction between mutations in integrin α5 and αv and for overlapping functions and compensation between these integrins and perhaps others. Nonetheless, in embryos lacking both α5 and αv integrins in their endothelial cells, initial vasculogenesis and angiogenesis proceed normally, at least up to E11.5, including the formation of apparently normal embryonic vasculature and development of the branchial arches. However, in the absence of endothelial α5 and αv integrins, but not of either alone, there are extensive defects in remodeling of the great vessels and heart resulting in death at ~E14.5. We also found that fibronectin assembly is somewhat affected in integrin α5 knockout endothelial cells and markedly reduced in integrin α5/αv double-knockout endothelial cell lines. Therefore, neither α5 nor αv integrins are required in endothelial cells for initial vasculogenesis and angiogenesis, although they are required for remodeling of the heart and great vessels. These integrins on other cells, and/or other integrins on endothelial cells, might contribute to fibronectin assembly and vascular development.
Keywords: Aortic arch remodeling defect, Compensation, Fibronectin, Integrin, Mouse, Tie2-Cre
INTRODUCTION
Vascular and congenital heart diseases are major causes of lethality (Bruneau, 2008) and much effort has been directed towards developing anti-angiogenic drugs (Ferrara and Kerbel, 2005). Therefore, there is considerable interest in understanding the roles of molecules that drive and control vascular development.
One important participant in developmental and pathological angiogenesis is fibronectin (Astrof and Hynes, 2009), a secreted glycoprotein that assembles into a fibrillar extracellular matrix. Fibronectin-null mice show lethality at embryonic day (E) 9.5 and have severe defects in the development of vasculature and somites (Francis et al., 2002; George et al., 1997; George et al., 1993). Integrins are heterodimeric cell adhesion receptors that mediate cell-matrix and cell-cell interactions (Hynes, 2002b; van der Flier and Sonnenberg, 2001). α5β1 integrin binds to the Arg-Gly-Asp (RGD) tri-peptide motif in fibronectin. This binding site is also recognized by αv integrins (i.e. αvβ1, αvβ3, αvβ5, αvβ6 and αvβ8) (Pankov and Yamada, 2002; Leiss et al., 2008). α5 integrin-null embryos show a similar phenotype to the fibronectin nulls, but die 1 day later, at ~E10.5 (Francis et al., 2002; Goh et al., 1997; Yang et al., 1993). Similarly, targeted inactivation of the RGD site in fibronectin (FnRGE) results in placental and cardiovascular defects and lethality at E10.5 (Takahashi et al., 2007; Takahashi et al., 2009). All these data point to a key role for the α5β1 integrin-fibronectin interaction in angiogenesis.
Complete knockout of all β1 integrins is lethal at the preimplantation stage (Fassler and Meyer, 1995; Stephens et al., 1995). Recently, several studies have shown that endothelial-specific knockout of β1 integrins results in severe vascular defects and lethality at E10 (Carlson et al., 2008; Lei et al., 2008; Tanjore et al., 2008; Zovein et al., 2010). This raises the question of which α subunit is involved in developmental angiogenesis. Deletion of α4 integrins is embryonic lethal (E10/11.5), with placental and cardiac defects (Yang et al., 1995). However, α4 endothelial-specific knockout mice are viable, with hematopoietic defects but no vascular phenotype (Priestley et al., 2007). α9 integrin-null mice have lymphatic defects and die of chylothorax soon after birth (Bazigou et al., 2009; Huang et al., 2000b), but lack other major vascular defects. No major vascular developmental defects have been reported for knockouts of the other ten α integrin subunits, including those of the laminin- and collagen-binding families.
The complete ablation of all five αv integrins does not block angiogenesis, but 80% of the embryos die at E11.5 due to placental defects and the remaining embryos succumb at birth with brain hemorrhage and cleft palate (Bader et al., 1998). The brain hemorrhage has been linked to defective activation of TGFβ by αvβ8 on glial cells supporting the brain vasculature (Cambier et al., 2005; McCarty et al., 2005; McCarty et al., 2002; Proctor et al., 2005). Although in vitro blocking experiments have suggested that αvβ3 and αvβ5 integrins promote angiogenesis, β3/β5-null mice are viable and show increased pathological angiogenesis (Hodivala-Dilke et al., 1999; Huang et al., 2000a; Reynolds et al., 2002). Furthermore, Tie2-Cre-mediated deletion of αv integrins in endothelial cells does not result in developmental vascular defects (McCarty et al., 2005), but instead leads to colitis through loss of αv integrins in hematopoietic cells causing defects in the activation of TGFβ (Lacy-Hulbert et al., 2007; Travis et al., 2007). These results suggest that αv integrins, although not essential, have some involvement in vascular development, but their exact roles remain uncertain.
To test the roles of endothelial α5β1 integrin in developmental angiogenesis we generated an integrin α5-floxed mouse line and ablated α5 in endothelial cells using Tie2-driven expression of Cre recombinase. Surprisingly, α5 conditional knockout mice were viable and lacked any immediately obvious phenotype. Characterization of α5 knockout endothelial cells suggested compensation by αv integrins, which we tested by generating endothelium-specific α5; αv double-knockout mice. Interestingly, these mice form normal vasculature initially but most die at mid-gestation due to vascular remodeling defects. These results shed further light on the complex interplay among integrins in controlling vascular development.
MATERIALS AND METHODS
Generation of α5 integrin-floxed mice
A conditional α5 integrin targeting vector (see Fig. S1B in the supplementary material) contained a thymidine kinase (TK) negative-selection cassette, an Frt-flanked PGK-neo cassette and the 255 bp exon 1 of α5 integrin flanked by loxP sites. R1 embryonic stem (ES) cells were electroporated, selected and screened for correct recombination and single integration. The PGK-neo cassette was removed by transient expression of Flip recombinase. Two karyotyped, correctly targeted ES cell clones (2H2 and 3G3) gave germline transmission and identical results. Cre-mediated excision of exon 1 was confirmed by PCR genotyping and Southern blotting (see Fig. S1B,C in the supplementary material).
Mouse strains
All mouse lines used were on a 129S4:C57BL/6 mixed background. Tie2-CreTg (Tek-Cre) (Kisanuki et al., 2001) (Jackson Laboratories) mice were crossed with α5+/− mice (Yang et al., 1993). α5-cKO crosses were set up as follows: α5flox/flox × α5+/−; Tie2-Cre or α5flox/flox × α5flox/+; Tie2-Cre. α5/αv double-floxed mice, i.e. α5flox/flox; αvflox/flox; R26RlacZ/lacZ and α5flox/flox; αvflox/flox; ImmortoTg mice, were generated by intercrossing α5flox/flox mice with αvflox mice (Lacy-Hulbert et al., 2007) and R26-lox-STOP-lox-lacZ reporter (R26R-lacZ) mice (Soriano, 1999) (Jackson Laboratories) or Immorto mice (Jat et al., 1991) (Charles River Laboratories). α5; αv conditional double-knockout crosses (α5/αv-cdKO) were generally made by crossing α5flox/flox; αvflox/flox; R26R-lacZ mice to α5+/−; αv+/−; Tie-CreTg/Tg mice. αv+/− mice have been reported previously (Bader et al., 1998). Genotyping was performed in-house (see Fig. S1D in the supplementary material) or by Transnetyx.
Histology, whole-mount staining and immunohistochemistry
E14.5 embryos were fixed in Bouin's (Sigma), paraffin embedded, serially sectioned and analyzed after Hematoxylin and Eosin (H&E) staining. Whole-mount ears or trachea were fixed overnight in 4% glutaraldehyde/PBS; embryos were fixed overnight in methanol:DMSO (4:1). Tissue was blocked with wash buffer (PBS/0.5% Tween 20) containing 5% normal goat serum. Antibody incubations were in 1:1 PBS-diluted blocking buffer overnight at 4°C, followed by six 1-hour washes. Tissues were embedded in Fluoromount-G (Southern-Biotech).
Tissues for sectioning were either cryofixed in Tissue-Tek OCT (Sakura Finetek) or tissues/embryos were fixed in IHC Zinc Fixative (BD-Pharmingen) and paraffin embedded. Cryosections were postfixed for 10 minutes in acetone at −20°C. Sections were blocked with PBS containing 5% normal goat serum (depleted of fibronectin by gelatin-Sepharose for fibronectin staining). Primary antibody incubations were overnight at 4°C. Secondary antibody incubations were 1 hour at room temperature. Antibodies were: from Millipore, integrin α4 (9C10), α5 (5H10-27), αv (AB1930), β1 (MB2.1) and PECAM1 (CD31) (390IHC); from Sigma, smooth-muscle actin-Cy3 (1A4), vinculin (hVin-1); from Abcam, LYVE1 (Ab14917) and pericentrin (Ab4448). Secondary goat anti-mouse, anti-rat and anti-rabbit antibodies conjugated with Alexa 488, Alexa 594 or Alexa 647 were from Invitrogen and goat anti-rat-HRP from Jackson ImmunoResearch. Staining of whole mouse embryos for β-galactosidase (lacZ) activity followed a standard protocol (Nagy, 2003).
FACS analysis
E10.5 yolk sacs or embryos were minced and incubated for 10 (yolk sac) or 40 (embryos) minutes at 37°C in Dulbecco's Modified Eagle's Medium containing 0.1% collagenase type I (Worthington) and 12 U/ml DNase. Subsequently, cells were incubated for 10 minutes in 1× Versene (Invitrogen) and strained through a 70-μm nylon mesh (Falcon). Endothelial cell lines (see below) were detached using sequential collagenase and Versene treatments. Antibody incubations were for 30 minutes at 4°C in PBS containing 2 mM EDTA and 0.5% BSA, using antibodies conjugated to either FITC, PE, APC or biotin, followed by conjugated streptavidin (BD-Pharmingen). Antibodies were obtained from BD-Pharmingen: integrin α4 (R1-2), α5 (5H10-27), αv (RMV-7), CD34 (RAM34); ICAM2 (CD102) (2C4), PECAM1 (CD31) (390), VE-cadherin (CD144) (11D4.1). Analysis was on a FACSCalibur high-throughput sampler (BD Biosciences) and data were analyzed using FlowJo software (Treestar).
Isolation of endothelial cell lines and cell culture
Endothelial cells were isolated from mice carrying the Immorto gene (Jat et al., 1991) with a 1-hour collagenase treatment. α5-KO and control lung (mLEC) or brain endothelial (mBEC) cells were isolated from adult α5flox/−; Tie2-Cre or α5flox/+; Tie2-Cre Immorto mice. Cells were grown to subconfluency on coated plates (see below) and immune cells were negatively selected with anti-CD18 (BD-Pharmingen, C71/16) followed by positive selection for endothelial cells with conjugated anti-ICAM2 antibodies using MACS beads (Miltenyi Biotec). After expansion, several mLEC preparations were selected as PECAM1+. Eventually, all endothelial cell lines were subcloned by FACS sorting for ICAM2+ cells followed by limited dilution cloning. α5/αv double-floxed mLEC clones (α5flox/flox; αvflox/flox), derived from adult lungs, were incubated with AdCre (Gene Transfer Vector Core, University of Iowa, USA) to excise the α5 and αv genes, and the α5/αv-dKO cells were isolated by FACS sorting for ICAM2+ and α5− αv− cells followed by limited dilution cloning. Embryonic endothelial cells (eECs) were isolated from the heads and tails of E13.5 embryos. α5-KO and control cell lines (mLEC and mBEC) were grown on 0.1% gelatin-coated plates. The eECs, α5flox/flox; αvflox/flox control and their AdCre-derived α5/αv-dKO mLECs were grown on plates coated with 20 μg/ml Matrigel basement membrane matrix (BD Biosciences).
Cells were maintained at 33°C in low-glucose DME/Ham's-F12 (1:1), 20% normal bovine serum, 50 μg/ml endothelial mitogen (Biomedical Technologies, MA, USA) and 20 U/ml mouse interferon-γ (Millipore). For experiments, cells were transferred to a 37°C incubator and depleted of interferon-γ. Endothelial cells were reconstituted by retroviral expression of human α5 integrin subcloned into LZRS-ms-IRES-zeo (Taverna et al., 1998; van der Flier et al., 2002).
Immunofluorescent staining of cells
Cells were grown overnight on coated glass coverslips: mLECs and mBECs were plated on 10 μg/ml fibronectin (BD Biosciences), whereas eECs were plated on a mix of 20 μg/ml Matrigel and 10 μg/ml human fibronectin. Cells were fixed for 10 minutes in 4% paraformaldehyde/PBS (or for 10 minutes in methanol at −20°C for αv integrin), washed and permeabilized for 10 minutes at room temperature with PBS containing 0.2% Triton X-100. Cells were blocked and incubated overnight at 4°C with primary antibody in PBS/2% BSA. Sections were incubated for 1 hour at room temperature with secondary antibodies and embedded in Vectashield mounting medium with DAPI (Vector Laboratories).
Fibronectin binding and assembly assays
Ninety-six-well tissue culture plates were coated with the indicated concentrations of fibronectin, washed and blocked with 5% BSA, and 20,000 endothelial cells/well were allowed to adhere for 2 hours in DMEM/0.2% BSA at 37°C. Plates were washed three times; adherent cells were fixed with 4% formaldehyde and stained with 0.1% Crystal Violet. After washes and permeabilization in 50 μl PBS/0.2% Triton X-100, the OD540 was measured in a plate reader. Two to three independent experiments were performed in triplicate.
Endothelial cells were seeded on Matrigel-coated or gelatin-coated 6-well plates (400,000 cells/well) in fibronectin-depleted medium. For incorporation of exogenous fibronectin, after culture overnight the medium was changed to fibronectin-depleted medium containing 10 μg/ml exogenous biotinylated human fibronectin. At the times indicated, medium was collected, cells were washed with PBS containing 1 mM Ca2+ and Mg2+ and solubilized in 0.5 ml DOC buffer [2 mM EDTA, 1% sodium deoxycholate, 20 mM Tris pH 8.5, Complete Mini-protease Inhibitors (Roche)]. After passing eight times through a 22G needle, DOC-insoluble material was spun down for 20 minutes at 20,000 g at 4°C and solubilized in 120 μl 2× reducing SDS-PAGE loading buffer. Reduced (100 mM DDT) samples were loaded on 4-12% gels. For fibronectin immunostaining, 15,000 cells/well were plated on coated 8-well Lab Tek Permanox coverslips (Nunc). Treatments and immunostainings were as described above.
Immunoblotting
Novex Tris-glycine precast gels (Invitrogen) were used and wet-transferred to nitrocellulose. Where indicated, samples were treated with PNGaseF (500 U/sample, New England Biolabs). Blots were blocked and incubated with antibodies in 5% non-fat dried milk, 0.2% NP40, Tris-buffered saline (pH 8). Primary antibodies were integrin α5 (AB1928), αv (AB1930) and GAPDH (MAB374) (all from Millipore), vimentin (Sigma), and rabbit anti-fibronectin (297.1; generated in our laboratory). HRP-conjugated secondary antibodies were from Jackson ImmunoResearch: goat anti-rabbit and sheep anti-mouse IgM and HRP-streptavidin. Blots were developed using Western-Lightning ECL (PerkinElmer)
RESULTS
Mice lacking endothelial α5 integrin exhibit no obvious vascular phenotypes
Since complete integrin α5 knockout (α5-KO) mice die at ~E11, preventing functional studies at later stages, we developed mouse lines with a floxed α5 integrin gene (see Fig. S1B-D in the supplementary material). To study the function of α5 integrin in angiogenesis we crossed α5flox/flox mice to those bearing a Tie2-Cre transgene, which is expressed in both endothelial and hematopoietic cells (Koni et al., 2001; Srinivasan et al., 2007). PCR genotyping and Southern blotting confirmed Cre-mediated excision of the floxed α5 allele (see Fig. S1C,D in the supplementary material). We obtained several mice with a maternal germline-excised α5-null allele, as Tie2-Cre is frequently expressed in the female germline and occasionally the male germline (de Lange et al., 2008; Koni et al., 2001). Intercrosses of those α5+/− mice confirmed the previously reported α5-KO phenotype (Yang et al., 1993). α5-null embryos at E10.5 (n=8) were severely growth retarded, posterior somites were incompletely formed and embryos showed reduced vascularization (data not shown). To exclude potential complications arising from germline-mediated excision of the α5flox allele in subsequent experiments, we intercrossed mice carrying the Tie2-Cre transgene and an α5-null allele with α5flox/flox mice.
Unexpectedly, we obtained normal Mendelian ratios of offspring from such α5flox/flox × α5+/−; Tie2-CreTg/+ crosses (χ2 test, P>0.3; see Table S1 in the supplementary material). FACS analysis for α5 integrin expression in cell populations of freshly isolated samples from E10.5 embryos (Fig. 1A,B) showed complete loss of α5 integrin protein in both hematopoietic and endothelial cells derived from the yolk sacs or from the integrin α5 conditional knockout (α5-cKO) embryos themselves. This confirmed that α5 integrin was indeed efficiently depleted early during embryonic development.
Fig. 1.

Tie2-Cre-mediated depletion of α5 integrin in endothelial cells results in no obvious vascular phenotypes. (A) FACS analysis of cells dissociated from E10.5 mouse yolk sacs, gated on immune cells (SSClow, PECAM1+) and endothelial cells (SSChigh, PECAM1+, VE-Cad+). Black line, isotype control antibody; green, α5 integrin on cells from α5flox/+; Tie2-Cre embryos; red, α5 integrin on cells from α5flox/−; Tie2-Cre embryos. Note the complete loss of α5 integrin from endothelial and hematopoietic cells but not from control ‘other’ cells (PECAM1−). For details of FACS analysis, see Figs S2 and S4 in the supplementary material. (B) Bar chart indicating loss of α5 integrin expression on greater than 95% of endothelial cells (SSChigh, PECAM1+ VE-Cad+) isolated from E10.5 α5flox/−; Tie2-Cre embryos. (C) Apparently normal vasculature in integrin α5 conditional knockout (α5-cKO) mice (α5f/− Tie versus α5f/+ Tie control) as seen by whole-mount staining for endothelial (PECAM1) and smooth muscle (α-SMA) cells in (a-d) ear skin and (e,f) trachea. Arteries (a), veins (v) and lymphatics (asterisk) are indicated. Scale bars: 180 μm in a,b,e,f; 45 μm in c,d.
No obvious vascular defects were visualized by whole-mount staining for platelet endothelial adhesion molecule [PECAM1 (CD31)] and smooth muscle α-actin (α-SMA; ACTA2) of vasculature of the inner-ear skin or trachea of adult α5-cKO mice (Fig. 1C). Similarly, immunohistochemical staining of sections from various other vascular beds showed no clear differences from controls (data not shown).
The α5flox/−; Tie2-Cre (α5-cKO) mice did not develop any obvious spontaneous vascular or hematopoietic phenotypes for a period of 18 months. In addition, FACS analysis of the immune system showed no disparities in subsets of hematopoietic cells in adult α5-cKO mice (data not shown). These unexpected results suggested that deletion of α5 integrin from endothelial cells does not replicate the global α5-null phenotype. Together, these data show that early developmental deletion of α5 integrin in endothelial cells has no obvious effects on developmental vasculogenesis or angiogenesis.
Integrin α5-deficient endothelial cells adhere to fibronectin in vitro but redistribute integrin αv to focal adhesions
We isolated immortalized endothelial cell lines from adult lungs (mLEC) and brains (mBEC) using α5-cKO crosses into which a conditional SV40 large T antigen was introduced (Jat et al., 1991). FACS analysis and immunofluorescence staining showed that the clones were positive for ICAM2 and often PECAM1 and VE-cadherin (cadherin 5) (data not shown). FACS analysis and immunoblots confirmed complete loss of α5 integrin in these endothelial cell lines.
α5-KO endothelial cells showed reduced adhesion to fibronectin, which was rescued by ectopic (over)expression of human α5 integrin (see Fig. S2 in the supplementary material). Extended adhesion times or increased fibronectin coating abolished this difference in adhesion between α5-KO and control cells (see Fig. S2C in the supplementary material). Immunofluorescence staining for vinculin of α5-KO endothelial cell cultures (mBECs, mLECs) plated on fibronectin consistently showed fewer, but larger and more peripherally localized, focal adhesions (see Fig. S3 in the supplementary material). We also observed relocalization of αv integrins from a diffuse surface expression in control cells to a focal adhesion localization in α5-KO cells (see Fig. S3 in the supplementary material). FACS analysis revealed no, or minor, upregulation of αv (see Fig. S2B in the supplementary material) or β3 (not shown) integrin in α5-KO endothelial cells. The relocalization and modest surface upregulation of αv integrins in α5-KO endothelial cells suggested that αv integrin heterodimers (i.e. αvβ1, β3, β5, β6 or β8) might be functionally compensating for loss of α5 integrins in endothelial cells. To explore this hypothesis, we knocked out both α5 and αv integrins in endothelial cells using Tie2-Cre (α5/αv-dcKO).
Mice lacking both α5 and αv integrins in endothelium die at ~E14.5 and have organizational defects in their great vessels
We crossed doubly homozygous integrin α5;αv floxed mice carrying the R26R-lacZ reporter gene (α5flox/flox; αvflox/flox; R26RlacZ/lacZ) to doubly heterozygous α5 and αv integrin-null mice carrying the Tie2-Cre allele (α5+/−; αv+/−; Tie2-CreTg/Tg). Whenever possible, we used mice homozygous for the Tie2-Cre transgene to increase the numbers of offspring carrying a Tie2-Cre allele. These crosses generate four potentially informative genotypes: α5/αv conditional doubly hemizygous control mice (α5flox/+; αvflox/+; Tie2-Cre, hereafter designated α5/αv-cdHemi), α5-cKO and αv conditional hemizygous (α5flox/−; αvflox/+; Tie2-Cre, hereafter α5-cKO/αv-cHemi), α5 conditional hemizygous and αv-cKO (α5flox/+; αvflox/−; Tie2-Cre, hereafter α5-cHemi/αv-cKO) and the α5/αv conditional double knockout (α5flox/−; αvflox/−; Tie2-Cre, hereafter α5/αv-cdKO mice).
Table 1 shows that embryos lacking both α5 and αv integrins in endothelial cells exhibit embryonic lethality and also that fewer than half of the expected α5-cKO/αv-cHemi mice survive (Table 1). Strikingly, there were no obvious vascular defects in the α5-cHemi/αv-cKO mice, suggesting that hemizygosity for α5 integrin is sufficient for survival in the absence of αv integrins. As expected, α5-cHemi/αv-cKO mice phenocopied the Tie2-Cre αv-cKO mice and developed colitis at several months of age (Lacy-Hulbert et al., 2007). These α5/αv Tie2-Cre crosses thus showed a genetic interaction between α5 and αv mutations, which could indicate overlap, compensation or convergence of the functions of these integrins. FACS analyses of E10.5 embryos confirmed efficient loss of both integrins (see Fig. S4 in the supplementary material).
Table 1.
Genotypes of progeny from α5flox/flox; αvflox/flox × α5+/−; αv+/−; Tie2-Cre crosses

Analysis of timed matings confirmed that endothelial deletion of both the α5 and αv integrin genes causes embryonic lethality. Most embryos died at ~E14.5 and showed severe dorsal edema and sometimes hemorrhage (Fig. 2A). Similarly, a subgroup of α5-cKO/αv-cHemi embryos was embryonic lethal (Table 1). Strikingly, all the embryos, as well as their vasculature, seemed normal until at least E11.5, as determined by whole-mount staining for PECAM1 (Fig. 2Ba-d; see Fig. S5 in the supplementary material) and for β-galactosidase expressed from the R26R-lacZ reporter in α5/αv-cdKO embryos (Fig. 2Be-f′).
Fig. 2.

Integrin α5/αv endothelial conditional double-knockout (α5/αv-cdKO) embryos develop severe edema at ~E14.5 but have normal vasculature at earlier stages. (Aa-c) Most mice doubly deficient for α5 and αv integrins in endothelial and immune cells show edema (arrows) and hemorrhage at ~E14.5 (α5f/− αvf/− Tie versus α5f/+ αvf/+ Tie control). (B) Whole-mount vascular staining for (a,b) PECAM1 (HRP) or for (c-f′) β-galactosidase (X-gal) using the R26R-lacZ reporter strain crossed into the experimental crosses in control conditional doubly hemizygous and conditional double-knockout embryos (as labeled) at E10.5 and E11.5. Neither method reveals any obvious vascular defects in early embryos deficient for integrins. The boxed regions in e and f are shown at higher magnification in e′ and f′. Scale bars: 1 mm.
A small number of α5/αv-cdKO mice survived (<4% of expected) and were found to lack both α5 and αv integrins on their hematopoietic cells as determined by FACS analysis (data not shown). These adult α5/αv-cdKO mice appeared to have normal vasculature and lymphatics, compared with control littermates, when examined by whole-mount vascular staining of ear skin and trachea (see Fig. S6 in the supplementary material) or by sectioning (data not shown). The results on early embryos and on the rare surviving α5/αv-cKO mice show that it is possible to construct apparently normal vasculature in the absence of any α5 or αv integrins in the endothelium.
Despite normal vasculogenesis and angiogenesis, the majority of α5/αv-cdKO embryos die by E14.5. This prompted us to analyze the formation of the heart and great vessels. Serial sectioning of three E14.5 litters revealed severe cardiovascular defects in α5/αv-cdKO and α5-cKO/αv-cHemi embryos (Fig. 3B and see Table S2 in the supplementary material). In normal embryos at this stage (~E14.5), extensive branchial arch artery remodeling has occurred and the embryonic circulation is established, with the symmetrical branchial arch arteries reorganized into the aortic arch and the major arteries supplying the head and neck (carotid) and upper limbs (subclavian) (Fig. 3A). The left sixth branchial artery forms the temporary ductus arteriosus (DA) that shunts 80% of the blood from the pulmonary arteries directly into the descending aorta, bypassing the non-functional lungs in the embryo. All the α5/αv-cdKO and α5-cKO/αv-cHemi embryos displayed ventricular septation defects (VSDs), whereas the ventricles were separated in the control (α5/αv-cdHemi) and α5-cHemi/αv-cKO embryos at this stage. α5/αv-cdKO embryos also frequently had aortic arch abnormalities, including a retro-oesophageal subclavian artery, vascular ring and absent ascending aorta (Fig. 3B and see Table S2 in the supplementary material). These results indicate that the endothelial α5 and αv integrins together play roles in the development or remodeling of the branchial arch arteries.
Fig. 3.

Cardiovascular defects in α5/αv-cdKO embryos. (A) Schematic of the embryonic remodeling process of the symmetric branchial arch arteries into the aortic arch and great vessels (E14.5). Some vessels regress (gray dashed lines), while others are stabilized. (Ba-n) Sections and schematics illustrating the heart and great-vessel defects found in integrin conditional double-knockout (cdKO) and hemizygous (cdHemi) mice. Genotypes (in endothelial cells) are indicated and color-coded for this and subsequent figures as follows: green, α5flox/+; αvflox/+; Tie2-Cre (α5/αv-cdHemi); blue, α5flox/−; αvflox/+; Tie2-Cre (α5-cKO; αv-cHemi); red, α5flox/−; αvflox/−; Tie2-Cre (α5/αv-cdKO). Schematics illustrate the observed phenotypes and the blue dashed lines refer to the locations of the corresponding sections. Control hearts and great arteries (a,c,e,h,k,m); atrophic (or interrupted, not shown) aortic arch and ventricular septation defect (VSD) (b,d); interrupted aortic arch and retro-oesophageal right subclavian artery (RERSA) (f,i); vascular ring (g,j); interrupted aortic arch, loss of ascending aorta and RERSA (l,n). (a) Control aortic arch; (b) atrophic aortic arch; (c) separated left and right ventricles; (d) VSD; (e) normal position of great arteries ventral to the trachea; (f) retro-oesophageal right subclavian artery (arrowhead); (g) vascular ring around trachea (arrowhead), which results from maintenance of left sixth branchial arch artery and left dorsal aorta in A; (h) aorta in plane of ductus arteriosus (DA); (i) tiny remnant of ascending aorta (arrowhead) in plane of DA; (j) maintenance of both left and right sixth branchial arch arteries resulting in vascular ring; (k) control ascending aorta in plane of DA; (l) absence of ascending aorta in plane of DA; (m) control ascending aorta in plane of pulmonary artery; (n) remnant of ascending aorta in plane of pulmonary artery (arrowhead). AA, aortic arch; AAo, ascending aorta; B, bronchi; Dao, descending aorta; E, oesophagus; lca, left carotid artery; lsa, left subclavian artery; LV, left ventricle; rca, right carotid artery; rsa, right subclavian artery; RV, right ventricle; T, trachea; Th, thymus. Scale bars: 200 μm.
In two α5/αv-cdKO mice that survived, we checked castings of the major arteries for aortic arch abnormalities. We found a patent ductus arteriosus (PDA) in one of the two adult α5/αv-cdKO mice (see Fig. S7E in the supplementary material) and PDA in several of the α5-cKO/αv-cHemi mice (data not shown), all of mixed background. This led us to check 10- to 20-week-old α5-cKO mice (α5flox/flox; Tie2-Cre) of which 9/10 had PDA (see Fig. S7A-D in the supplementary material). These mice were from α5flox/flox × α5flox/+; Tie2-Cre/+ crosses on a C57BL/6 N7 background and about half of the α5flox/flox; Tie2-Cre (α5-cKO) from this cross seemed to be lost before weaning (see Table S3 in the supplementary material). However, in other crosses using a mixed 129S4:C57BL/6 background and α5flox/−; Tie2-Cre mice the DA was closed from postnatal day (P) 1 onwards, indicating a role of potential genetic background modifiers. These results indicate that endothelial α5 or αv integrins play roles in the development or remodeling of the vasculature.
Integrin α5/αv-cdKO mice have normal branchial arch and cardiac cushion development
We next sought to define when and why defects arise in the great arteries of α5/αv-cdKO embryos. We first investigated whether the development of branchial arch arteries was normal (Fig. 4). The branchial arch arteries developed symmetrically and normally between E9.5 and E11.5 in α5/αv-cdKO embryos, as shown by whole-mount staining for PECAM1 and expression of the R26R-lacZ reporter (Fig. 4). Intracardial India ink injections also indicated normal symmetrical branchial arch formation (data not shown), as did histological analysis of branchial arch artery development in cdKO mice up to E11.5 (Fig. 4B and see Fig. S8G,H in the supplementary material). Furthermore, we detected comparable levels of staining for fibronectin around the vasculature of control and cdKO embryos (Fig. 4B).
Fig. 4.
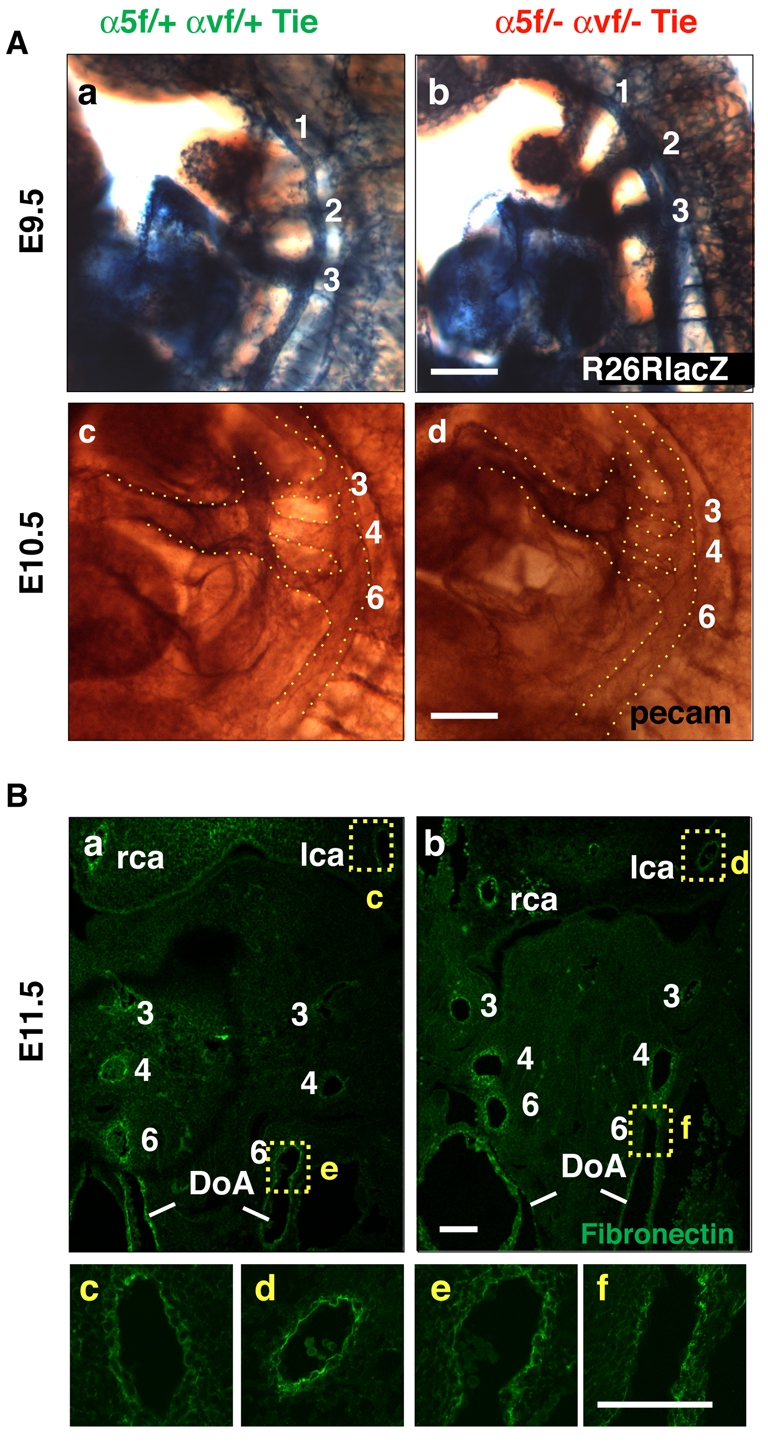
Normal development of branchial arch arteries and fibronectin deposition until E11.5. (Aa-d) Whole-mount β-galactosidase reporter expression at E9.5 (a,b) and whole-mount PECAM1 staining at E10.5 (c,d) showing normal symmetric formation of branchial arch arteries in α5/αv-cdKO (b,d) as compared with control cHemi (a,c) mouse embryos. (Ba-f) Frontal sections of E11.5 embryos showing symmetric branchial arch formation and similar fibronectin staining in (a) cHemi and (b) α5/αv-cdKO embryos. Higher magnification images are shown of (c,d) carotid fibronectin staining and (e,f) the left sixth arch artery. Note the similar fibronectin staining pattern of several cell layers around the arteries. lca, left carotid artery; rca, right carotid artery; DoA, doral aorta. Scale bars: 100 μm.
We also checked whether the cardiac cushions formed normally in α5/αv-cdKO mice. Endothelial cells migrate out of the endothelium into the cardiac jelly to form cardiac cushions, which contribute to the formation of the cardiac valves and closure of the septum (Kisanuki et al., 2001; Savolainen et al., 2009; Webb et al., 1998) and affect the hemodynamics in the embryo, which could play a crucial role in the vascular remodeling process (Yashiro et al., 2007). However, whole-mount R26R-lacZ reporter staining of E10.5 and E11.5 α5/αv-cdKO embryos did not show any differences in cardiac cushion formation (see Fig. S8 in the supplementary material). In the outflow tract cushions, most cells are derived from the neural crest and thus are not labeled by Tie2-Cre; R26R-lacZ (Kisanuki et al., 2001). However, the outflow tract cushions also appeared to have normal numbers of cells and exhibited comparable immunohistochemical staining for fibronectin (data not shown).
Great-artery remodeling phenotypes are found in many mutants in which neural crest cell migration or differentiation of neural crest cells into smooth muscle cells covering the branchial arch arteries is affected. However, we did not detect consistent differences in smooth muscle cell coverage of the branchial arch arteries between E10.5 and E11.5, indicating that the neural crest cells reach the branchial arches and, at least at these stages, differentiate into smooth muscle cells in a fashion comparable to that of their control littermates (data not shown). Therefore, no obvious defects were found in the formation of the branchial arches or of the cardiac cushions that could readily explain the subsequent arterial remodeling defects.
Integrin α5/αv-dKO endothelial cells do not assemble normal fibronectin fibrils
To analyze further the endothelial phenotype of α5/αv-cdKO mice, we isolated dKO cell lines using two approaches. Adult lung-derived dKO endothelial (dKO-mLEC) cells were generated in vitro from α5flox/flox; αvflox/flox Immorto endothelial cells by AdCre-mediated excision of both α5 and αv integrins. We also isolated ECs from E13.5 experimental cdKO crosses carrying the Immorto gene. All α5/αv-dKO cell lines failed to adhere effectively to fibronectin (see Fig. S9B in the supplementary material; data not shown) and were therefore maintained on Matrigel-coated tissue culture plates.
As mentioned above, integrin α5 plays a major role in fibronectin matrix assembly (Wierzbicka-Patynowski and Schwarzbauer, 2003), so we analyzed fibronectin fibrillogenesis by determining the amount of fibronectin incorporated into 1% DOC-insoluble matrix fractions. α5-KO cells assembled less fibronectin than did control cells, α5/αv-dKO ECs assembled even less DOC-insoluble fibronectin, and dKO-mLEC cells assembled almost none (Fig. 5A; see Fig. S10 in the supplementary material; data not shown). These differences were also seen in immunofluorescence analyses of endogenous fibronectin assembly. α5-KO endothelial cells synthesized an extensive fibrillar network from 24 hours onwards (Fig. 5Bb,e), but the fibrils appeared slightly thicker and shorter than those of the control cells (Fig. 5Ba,d). However, both dKO-eEC (Fig. 5Bc,f) and dKO-mLEC (not shown) cells deposited only small fibronectin aggregates at 24 hours and only some subsets of dKO-eEC assembled long wavy fibronectin fibrils after prolonged cell culture (Fig. 5Bc,f). Both the DOC-insolubility and immunofluorescence results were obtained for several independent cell lines, as well as for the incorporation of exogenously added soluble fibronectin (10 μg/ml) (data not shown). Interestingly, the different appearance of fibronectin fibers in long-term eEC cultures correlated with α4 integrin expression (see Fig. S9A in the supplementary material; data not shown). dKO cell lines completely negative for α4 integrin expression did not assemble fibrillar fibronectin (data not shown). Indeed, it has been shown that α4 integrin can assemble fibronectin in an RGD-independent manner (Sechler et al., 2000). Furthermore, we found that α4 integrin relocalized just as αv did (Fig. 6; see also Fig. S3 in the supplementary material) into focal adhesions in α5-KO-eEC and dKO-eEC. These findings suggest that, when both α5 and αv are depleted, α4 integrin, if expressed, can assemble fibronectin into a fibrillar network, although the fibronectin fibers seem qualitatively different from those formed by α5 and/or αv integrins.
Fig. 5.

Integrin α5-KO and α5/αv-dKO cells assemble less fibrillar fibronectin. (A) Immunoblot of 1% DOC-insoluble fibronectin matrix assembled by embryonic endothelial cell (eEC) lines plated for 24 hours on Matrigel. Note the slightly reduced insoluble fibronectin in α5-KO endothelial cells and even further reduced insoluble fibronectin in α5/αv-dKO cells. The right-hand lane shows that fibronectin-null cells do not produce endogenous (insoluble) fibronectin (negative control). (Ba-f) Fibronectin fibrillogenesis as detected by immunofluorescence analyses of control α5-KO and α5/αv-dKO endothelial cells after 24 and 96 hours plating on Matrigel. Similar results were obtained after plating on Matrigel containing 10 μg/ml fibronectin. Note the small aggregates of fibronectin instead of fibrils in the dKO cells at 24 hours (c) and the delayed formation of long, wavy fibronectin fibrils in the dKO cell lines at 96 hours (f). Scale bars: 30 μm.
Fig. 6.

Embryonic endothelial α5-KO and α5/αv-dKO cells show relocalization of αv and α4 integrins when plated on fibronectin. Cells were cultured overnight on a mixture of fibronectin (10 μg/ml) and Matrigel (20 μg/ml) and stained for integrins, vinculin or fibronectin. (A-C) α5 integrin staining showing loss of α5 in both α5-KO and α5/αv-dKO eEC lines. (D-F) Relocalization of αv integrin to focal contacts in α5-KO cells. (G-I′) Relocalization of α4 integrin to focal adhesions in cells plated on fibronectin in both α5-KO (H) and α5/αv-dKO cells (l) but not in control cells (G). (G′-I′) Merged images of α4 and fibronectin (same frames as G-I), showing colocalization of α4 in focal adhesions with fibrillar fibronectin. Arrows point to focal adhesions containing co-stained integrin and fibronectin. Scale bars: 30 μm.
DISCUSSION
The results presented here document several novel findings. First, there is the surprising result that mice lacking endothelial α5 integrin are viable and fertile and have no obvious defects in developmental angiogenesis. Second, we provide evidence for genetic interaction between mutations in the α5 and αv integrin genes and for overlapping functions and/or compensation between these integrins and perhaps others. Third, even in the absence of both α5 and αv integrins, initial vasculogenesis and angiogenesis proceed normally, at least up to E11.5, including the formation of intact and ostensibly normal yolk sac and embryonic vasculature and development of the branchial arches. Finally, in the absence of endothelial α5 and αv integrins, but not of either alone, there are extensive defects in remodeling of the great vessels and heart. These results have to be considered in light of cell biological data on the expression and distribution of these and other integrins and the assembly of their common ligand, fibronectin.
The dispensability of endothelial α5 integrin
Complete deletion of α5 in knockout mice (Francis et al., 2002; Yang et al., 1993), α5-KO teratomas (Taverna and Hynes, 2001) and ES cells (Francis et al., 2002) and several α5β1 integrin blocking studies (Bhaskar et al., 2007; Kim et al., 2000) have implicated α5β1 integrin in both developmental and pathological angiogenesis. To understand more about the role of endothelial α5β1 in angiogenesis, we depleted α5 integrin specifically in endothelial cells. In contrast to α5-KO mice, which die at E11 because of vascular defects, we found that mice lacking endothelial α5 integrin are viable and that α5, like αv (this study) (McCarty et al., 2005), is not required for developmental angiogenesis. This suggests that in α5-KO embryos, the reported somite and neural crest defects might also contribute to the lethal phenotype (Goh et al., 1997; Yang et al., 1993).
The lack of a vascular phenotype raises several questions and possibilities as to how endothelial α5 integrin could be dispensable for angiogenesis, whereas its major ligand, fibronectin, is essential. The data suggest that the timing, efficiency or delay in α5 protein depletion in endothelial cells are not responsible for the lack of developmental vascular defects. Therefore, endothelial α5β1 integrin is dispensable on endothelial cells, certainly after E10.5. Its role as a receptor and assembler of fibronectin must be substituted by another (integrin) receptor or by α5β1 expression on another cell type.
Genetic interactions and overlapping functions of endothelial α5 and αv integrins
We have reported previously that endothelial αv integrins are completely dispensable for angiogenesis (McCarty et al., 2005), even though they may play some role in its regulation. Here we show that mice lacking endothelial α5 and a single αv allele show increased lethality, whereas the homozygous deletion of both genes leads to almost complete embryonic lethality. This suggests that α5 and αv integrins either overlap or converge in function, or that they can compensate for one another in some way. These developmental genetic data are complemented by cell biological data suggesting redeployment of αv integrins in the absence of endothelial α5 integrin. When α5-null endothelial cells spread on fibronectin, αv integrins relocate to focal contacts, a position usually occupied by α5 integrins in wild-type cells. This happens in the absence of major upregulation of αv protein levels. The fact that αv integrins can also mediate adhesion to fibronectin and assemble fibronectin-rich matrices, albeit less well than does α5β1, is consistent with earlier work (Takahashi et al., 2007; Wennerberg et al., 1996; Yang and Hynes, 1996). Therefore, α5-null endothelial cells can interact with fibronectin through αv integrins and this presumably underlies the lack of obvious embryonic phenotypes in the absence of α5β1 integrin on endothelial cells. However, this is unlikely to be the full story, as discussed below.
Angiogenesis in the absence of endothelial α5 and αv integrins
The first 10 days of development proceed apparently normally in the absence of all the major fibronectin receptors on endothelial cells. Embryonic lethality does not commence until E14.5, after vasculogenesis and angiogenesis and the formation of the branchial arch network. This is in contrast to Tie2-Cre-mediated endothelial-specific ablation of all β1 integrins (Carlson et al., 2008; Lei et al., 2008; Tanjore et al., 2008) and to endothelial cKOs of several downstream signaling molecules, such as ILK (Friedrich et al., 2004) and FAK (PTK2) (Braren et al., 2006), all of which result in early embryonic death at ~E11 due to a variety of placental, yolk sac or vascular defects. These findings suggest that some endothelial β1 integrin(s) and integrin-mediated signaling are essential for early developmental angiogenesis. A few animals lacking α5 and αv integrins on endothelial cells complete development and survive. These mice appear to be healthy, although we have not obtained sufficient numbers to investigate their fertility and fecundity or their capabilities in pathological angiogenesis. Nonetheless, it is surprising, given the existing data implicating these integrins in angiogenesis, that they appear to be dispensable on endothelial cells for normal development, routinely to mid-gestation and occasionally into postnatal life.
Vascular remodeling in the absence of endothelial α5 and αv integrins
The embryonic lethality in the great majority of mice lacking α5 and αv integrins in their endothelial cells appears to arise from defects in remodeling of the great vessels and heart from E11.5 onwards. The embryos exhibit a variety of defects including VSDs, misrouting and interruptions of the aorta and other vessels. We found a second defect related to branchial arch remodeling in α5-cKO mice, i.e. PDA in adult mice. PDA accounts for 14% of congenital heart defects in humans and the phenotype correlates with flow volume and symptoms may range from none to morbidity at birth (Schneider and Moore, 2006). Interestingly, one report has suggested that reduced fibronectin assembly in the vessel wall leads to PDA (Mason et al., 1999). This might correlate with the modest reduction in fibronectin assembly by α5-KO endothelial cells in vitro. We detected the PDA phenotype in mice backcrossed to C57BL/6J (N7), and about half of the expected α5flox/flox; Tie2-Cre mice are lost before weaning. Thus, the α5-cKO PDA phenotype seems to be background dependent, in line with other studies showing that 129S4 and C57BL/6 genetic modifiers play a significant role in α5β1 and fibronectin function (Astrof et al., 2007; George et al., 1997; Yang et al., 1999).
Two distinct classes of model could explain these vascular remodeling defects: local and systemic. Since it has been established previously that fibronectin is strongly expressed in the cardiac cushions that form the valves (ffrench-Constant and Hynes, 1988; Mjaatvedt et al., 1987; Peters and Hynes, 1996; Roman and McDonald, 1992) and apparently plays a role in the migration of cushion cells into the cardiac jelly (Icardo et al., 1992; Loeber and Runyan, 1990), we investigated the possibility that defects in cushion formation might lead to defects in vascular flow and thus to the remodeling defects seen in the α5/αv-cdKO embryos. However, we did not detect defects in cushion formation at E10.5/11.5; cells migrate into the cushions in apparently normal numbers and deposit fibronectin. These results do not rule out subsequent failures in valve development and/or systemic effects that could disrupt normal blood flow with consequential effects on vessel remodeling. A second group of hypotheses – that local defects in integrin-mediated adhesion and signaling, assembly of extracellular matrix or migration of vascular cells could underlie the remodeling defects – are also likely possibilities and receive some support from the cell biological analyses presented.
Fibronectin assembly in the absence of endothelial α5 and αv integrins
Our in vitro data show decreased fibronectin fibril formation by α5/αv-dKO endothelial cell lines, suggesting that a defect in matrix assembly might contribute to the in vivo phenotypes. However, we have been unable to detect any consistent reduction in assembled fibronectin in the basement membrane underlying the endothelial cell layer in any of our cKO mice. This could reflect technical limitations in immunohistochemical detection of assembled fibronectin. Furthermore, the in vitro experiments show that fibronectin aggregates still form in α5/αv-dKO ECs, indicating that one might not expect absolute loss of fibronectin but rather a quantitative or qualitative difference in fibril formation in the proximity of the α5-deficient endothelial cells. These might well be difficult or impossible to detect by immunohistochemical staining for fibronectin. Higher resolution methods, such as electron microscopy (EM) or immuno-EM, might be required.
Potential involvement of other receptors
The continued deposition of fibronectin fibrils by at least some α5/αv-dKO endothelial cell lines suggests that some other integrin or even a different class of receptor might be able to replace this function of α5 and αv integrins in vitro, and conceivably also in vivo. As mentioned above, deletion of β1 integrin in endothelial cells has significantly more severe effects on vascular development than the deletion of α5 and αv integrins that we report here. There are twelve possible β1 integrins, of which only two would be deleted in our experiments (α5β1 and αvβ1, along with αvβ3, αvβ5, αvβ6 and αvβ8, if present). It is not certain which of these integrins are present on endothelial cells at the relevant periods of development. Although integrin receptors for laminins and collagens could well play a role, we are particularly interested in receptors for fibronectin, given that it is known to be essential for angiogenesis and heart and vascular development (Astrof and Hynes, 2009). Among the β1 integrins, several have been reported as fibronectin receptors, most notably α4β1, α8β1 and α9β1. We have observed a correlation between the expression of α4β1 integrin by α5/αv-dKO endothelial cell lines and their ability to assemble abnormal fibronectin fibrils. The complete knockout of α4 integrin does not block angiogenesis but does lead to defects in coronary vessels, probably as a secondary consequence of defects in the formation of the epicardium (Sengbusch et al., 2002; Yang et al., 1995) and defective mural cell coverage of dilated cranial vessels at E10 (Grazioli et al., 2006). Embryos deficient in both α4 and α5 integrins show the α5 phenotype, without enhancement (Yang et al., 1999). Mice lacking α4 integrin in endothelial cells are viable and lack a vascular phenotype (Priestley et al., 2007). These data argue against a major role for endothelial α4 integrin in normal vascular development but do not rule out its potential to compensate for the absence of α5 and αv integrins in our experiments. More elaborate compound mouse models that lack several integrins will be needed to test these hypotheses further.
Potential roles of α5 and αv integrins in other vessel-associated cell types
Quite apart from any function of α5β1 in vascular endothelial cells, it is likely that α5 integrin also plays roles in pericytes and/or smooth muscle cells that support the vasculature. Indeed, staining of α5-cKO tissue cryosections of intestine and skeletal muscle for α5 integrin showed the persistence of vessel-associated α5 immunoreactivity (data not shown), making it difficult to confirm endothelial knockout of α5 integrin by histology, even though it can be demonstrated by isolation of endothelial cells from embryos or adult tissues. This probably represents α5 integrin expression in pericytes and smooth muscle cells. Therefore, we hypothesize that the α5β1 integrin of such vessel-associated cell types might contribute to fibronectin assembly. Recently, mural cell-specific β1 cKO using platelet-derived growth factor receptor β (PDGFRβ)-Cre resulted in a severe vascular phenotype: the mice showed hemorrhage and dilated vessels and were perinatal lethal (E18-P10) (Abraham et al., 2008). Another example of integrin function in perivascular cells comes from the depletion of αv integrin in glial cells, which normally stabilize the vasculature of the brain, resulting in cerebral hemorrhage (McCarty et al., 2005), whereas there is no cerebral vascular phenotype in endothelial αv-cKO mice. We did not observe a similar trans function for α5β1 integrin in the brain as Nestin-Cre-mediated knockout of α5 integrin in the nervous system showed no embryonic cerebral hemorrhage, nor any other obvious vascular phenotype (our unpublished results).
Implications for the inhibition of angiogenesis
It is intriguing that α5β1 antagonists, currently in clinical trials, have been shown to inhibit angiogenesis in vitro, whereas we report that developmental angiogenesis seems unaffected by loss of endothelial α5. Strikingly, a similar contradiction has been found for αvβ3 and αvβ5 antagonists that appear to be anti-angiogenic in vitro, whereas the respective integrin KO mice show an increased pathological angiogenesis response (Reynolds et al., 2002). One hypothesis in the latter case is that αvβ3 and αvβ5 act as negative regulators of angiogenesis (Hynes, 2002a; Hynes, 2007). The experimental timing of integrin depletion might also play a role. Acute inhibition of α5 integrin in adult mice might offer fewer opportunities for compensation or plasticity in the adult organism than does depletion during embryogenesis. Our observation that several embryo-derived EC lines expressed some α4 integrin and formed delayed fibronectin fibrils, whereas adult-derived and in vitro-depleted α5/αv-dKO ECs did not express α4 integrin nor assemble fibrils, might also hint at such a mechanism. These hypotheses deserve further testing; for instance, inducible endothelial α5-cKO could address the difference between depletion of endothelial α5 in the adult versus the embryo. Another possible explanation is that there might be a fundamentally different function for α5β1 in developmental as distinct from pathological angiogenesis. For example, α5β1 integrin has been implicated in tumor angiogenesis (Taverna and Hynes, 2001) and has been found to be upregulated in vivo in a cerebral hypoxia model (Milner et al., 2008). In addition, developmental lymphangiogenesis seems to be unaffected, in contrast to a recent study showing that α5β1 blockade affects pathological lymphangiogenesis (Okazaki et al., 2009). Therefore, it would be interesting to test the role of endothelial α5 integrin in pathological models of angiogenesis, in order to validate endothelial cells as targets for the blocking reagents in those models.
Conclusions
The results reported here reveal further complexity in the roles of integrins in vascular development. Although various lines of evidence implicate α5 and αv integrins in vascular development and angiogenesis, it is clear that these processes can proceed almost, or completely, normally in the absence of either one of these integrin subunits and, to a significant extent, in the absence of both. These results raise intriguing questions about overlapping functions or compensation among integrins. This possibility pertains most strongly to the fibronectin receptor integrins because that ligand is the most essential extracellular matrix protein for vascular development. Since none of the clinical trials using antagonists of these integrins has yet proven their efficacy as anti-angiogenic agents, it might be worth considering the possibility of targeting both subunits at the same time. Basic developmental questions about which integrins play important roles in vascular development, and in which cells and cellular functions, require further investigation. This will be challenging given the number of potential players, their embryonic lethality and possible overlap in function.
Supplementary Material
Acknowledgements
We acknowledge the Koch Institute Fannie E. Rippel Transgenics Facility and the Flow Cytometry Core Facility for excellent technical assistance. We thank our colleagues from the R.O.H. laboratory for critically reading the manuscript. This work was supported by grants from the National Institutes of Health (PO1-HL66105 and the NIGMS Cell Migration Consortium, GC11451.126452, PI, A. F. Horwitz) and by the Howard Hughes Medical Institute of which R.O.H. is an Investigator. Deposited in PMC for release after 6 months.
Footnotes
Competing interests statement
The authors declare no competing financial interests.
Supplementary material
Supplementary material for this article is available at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.049551/-/DC1
References
- Abraham S., Kogata N., Fassler R., Adams R. H. (2008). Integrin beta1 subunit controls mural cell adhesion, spreading, and blood vessel wall stability. Circ. Res. 102, 562-570 [DOI] [PubMed] [Google Scholar]
- Astrof S., Hynes R. O. (2009). Fibronectins in vascular morphogenesis. Angiogenesis 12, 165-175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astrof S., Kirby A., Lindblad-Toh K., Daly M., Hynes R. O. (2007). Heart development in fibronectin-null mice is governed by a genetic modifier on chromosome four. Mech. Dev. 124, 551-558 [DOI] [PubMed] [Google Scholar]
- Bader B. L., Rayburn H., Crowley D., Hynes R. O. (1998). Extensive vasculogenesis, angiogenesis, and organogenesis precede lethality in mice lacking all alpha v integrins. Cell 95, 507-519 [DOI] [PubMed] [Google Scholar]
- Bazigou E., Xie S., Chen C., Weston A., Miura N., Sorokin L., Adams R., Muro A. F., Sheppard D., Makinen T. (2009). Integrin-alpha9 is required for fibronectin matrix assembly during lymphatic valve morphogenesis. Dev. Cell 17, 175-186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhaskar V., Zhang D., Fox M., Seto P., Wong M. H., Wales P. E., Powers D., Chao D. T., Dubridge R. B., Ramakrishnan V. (2007). A function blocking anti-mouse integrin alpha5beta1 antibody inhibits angiogenesis and impedes tumor growth in vivo. J. Transl. Med. 5, 61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braren R., Hu H., Kim Y. H., Beggs H. E., Reichardt L. F., Wang R. (2006). Endothelial FAK is essential for vascular network stability, cell survival, and lamellipodial formation. J. Cell Biol. 172, 151-162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruneau B. G. (2008). The developmental genetics of congenital heart disease. Nature 451, 943-948 [DOI] [PubMed] [Google Scholar]
- Cambier S., Gline S., Mu D., Collins R., Araya J., Dolganov G., Einheber S., Boudreau N., Nishimura S. L. (2005). Integrin alpha(v)beta8-mediated activation of transforming growth factor-beta by perivascular astrocytes: an angiogenic control switch. Am. J. Pathol. 166, 1883-1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson T. R., Hu H., Braren R., Kim Y. H., Wang R. A. (2008). Cell-autonomous requirement for beta1 integrin in endothelial cell adhesion, migration and survival during angiogenesis in mice. Development 135, 2193-2202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange W. J., Halabi C. M., Beyer A. M., Sigmund C. D. (2008). Germ line activation of the Tie2 and SMMHC promoters causes noncell-specific deletion of floxed alleles. Physiol. Genomics 35, 1-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassler R., Meyer M. (1995). Consequences of lack of beta 1 integrin gene expression in mice. Genes Dev. 9, 1896-1908 [DOI] [PubMed] [Google Scholar]
- Ferrara N., Kerbel R. S. (2005). Angiogenesis as a therapeutic target. Nature 438, 967-974 [DOI] [PubMed] [Google Scholar]
- ffrench-Constant C., Hynes R. O. (1988). Patterns of fibronectin gene expression and splicing during cell migration in chicken embryos. Development 104, 369-382 [DOI] [PubMed] [Google Scholar]
- Francis S. E., Goh K. L., Hodivala-Dilke K., Bader B. L., Stark M., Davidson D., Hynes R. O. (2002). Central roles of alpha5beta1 integrin and fibronectin in vascular development in mouse embryos and embryoid bodies. Arterioscler. Thromb. Vasc. Biol. 22, 927-933 [DOI] [PubMed] [Google Scholar]
- Friedrich E. B., Liu E., Sinha S., Cook S., Milstone D. S., MacRae C. A., Mariotti M., Kuhlencordt P. J., Force T., Rosenzweig A., et al. (2004). Integrin-linked kinase regulates endothelial cell survival and vascular development. Mol. Cell. Biol. 24, 8134-8144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George E. L., Georges-Labouesse E. N., Patel-King R. S., Rayburn H., Hynes R. O. (1993). Defects in mesoderm, neural tube and vascular development in mouse embryos lacking fibronectin. Development 119, 1079-1091 [DOI] [PubMed] [Google Scholar]
- George E. L., Baldwin H. S., Hynes R. O. (1997). Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of precursor cells. Blood 90, 3073-3081 [PubMed] [Google Scholar]
- Goh K. L., Yang J. T., Hynes R. O. (1997). Mesodermal defects and cranial neural crest apoptosis in alpha5 integrin-null embryos. Development 124, 4309-4319 [DOI] [PubMed] [Google Scholar]
- Grazioli A., Alves C. S., Konstantopoulos K., Yang J. T. (2006). Defective blood vessel development and pericyte/pvSMC distribution in alpha 4 integrin-deficient mouse embryos. Dev. Biol. 293, 165-177 [DOI] [PubMed] [Google Scholar]
- Hodivala-Dilke K. M., McHugh K. P., Tsakiris D. A., Rayburn H., Crowley D., Ullman-Cullere M., Ross F. P., Coller B. S., Teitelbaum S., Hynes R. O. (1999). Beta3-integrin-deficient mice are a model for Glanzmann thrombasthenia showing placental defects and reduced survival. J. Clin. Invest. 103, 229-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X., Griffiths M., Wu J., Farese R. V., Jr, Sheppard D. (2000a). Normal development, wound healing, and adenovirus susceptibility in beta5-deficient mice. Mol. Cell. Biol. 20, 755-759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X. Z., Wu J. F., Ferrando R., Lee J. H., Wang Y. L., Farese R. V., Jr, Sheppard D. (2000b). Fatal bilateral chylothorax in mice lacking the integrin alpha9beta1. Mol. Cell. Biol. 20, 5208-5215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes R. O. (2002a). A reevaluation of integrins as regulators of angiogenesis. Nat. Med. 8, 918-921 [DOI] [PubMed] [Google Scholar]
- Hynes R. O. (2002b). Integrins: bidirectional, allosteric signaling machines. Cell 110, 673-687 [DOI] [PubMed] [Google Scholar]
- Hynes R. O. (2007). Cell-matrix adhesion in vascular development. J. Thromb. Haemost. 5, 32-40 [DOI] [PubMed] [Google Scholar]
- Icardo J. M., Nakamura A., Fernandez-Teran M. A., Manasek F. J. (1992). Effects of injecting fibronectin and antifibronectin antibodies on cushion mesenchyme formation in the chick. An in vivo study. Anat. Embryol. (Berl.) 185, 239-247 [DOI] [PubMed] [Google Scholar]
- Jat P. S., Noble M. D., Ataliotis P., Tanaka Y., Yannoutsos N., Larsen L., Kioussis D. (1991). Direct derivation of conditionally immortal cell lines from an H-2Kb-tsA58 transgenic mouse. Proc. Natl. Acad. Sci. USA 88, 5096-5100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S., Bell K., Mousa S. A., Varner J. A. (2000). Regulation of angiogenesis in vivo by ligation of integrin alpha5beta1 with the central cell-binding domain of fibronectin. Am. J. Pathol. 156, 1345-1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki Y. Y., Hammer R. E., Miyazaki J., Williams S. C., Richardson J. A., Yanagisawa M. (2001). Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol. 230, 230-242 [DOI] [PubMed] [Google Scholar]
- Koni P. A., Joshi S. K., Temann U. A., Olson D., Burkly L., Flavell R. A. (2001). Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J. Exp. Med. 193, 741-754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy-Hulbert A., Smith A. M., Tissire H., Barry M., Crowley D., Bronson R. T., Roes J. T., Savill J. S., Hynes R. O. (2007). Ulcerative colitis and autoimmunity induced by loss of myeloid alphav integrins. Proc. Natl. Acad. Sci. USA 104, 15823-15828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei L., Liu D., Huang Y., Jovin I., Shai S. Y., Kyriakides T., Ross R. S., Giordano F. J. (2008). Endothelial expression of beta1 integrin is required for embryonic vascular patterning and postnatal vascular remodeling. Mol. Cell. Biol. 28, 794-802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiss M., Beckmann K., Giros A., Costell M., Fassler R. (2008). The role of integrin binding sites in fibronectin matrix assembly in vivo. Curr. Opin. Cell Biol. 20, 502-507 [DOI] [PubMed] [Google Scholar]
- Loeber C. P., Runyan R. B. (1990). A comparison of fibronectin, laminin, and galactosyltransferase adhesion mechanisms during embryonic cardiac mesenchymal cell migration in vitro. Dev. Biol. 140, 401-412 [DOI] [PubMed] [Google Scholar]
- Mason C. A., Bigras J. L., O'Blenes S. B., Zhou B., McIntyre B., Nakamura N., Kaneda Y., Rabinovitch M. (1999). Gene transfer in utero biologically engineers a patent ductus arteriosus in lambs by arresting fibronectin-dependent neointimal formation. Nat. Med. 5, 176-182 [DOI] [PubMed] [Google Scholar]
- McCarty J. H., Monahan-Earley R. A., Brown L. F., Keller M., Gerhardt H., Rubin K., Shani M., Dvorak H. F., Wolburg H., Bader B. L., et al. (2002). Defective associations between blood vessels and brain parenchyma lead to cerebral hemorrhage in mice lacking alphav integrins. Mol. Cell. Biol. 22, 7667-7677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarty J. H., Lacy-Hulbert A., Charest A., Bronson R. T., Crowley D., Housman D., Savill J., Roes J., Hynes R. O. (2005). Selective ablation of alphav integrins in the central nervous system leads to cerebral hemorrhage, seizures, axonal degeneration and premature death. Development 132, 165-176 [DOI] [PubMed] [Google Scholar]
- Milner R., Hung S., Erokwu B., Dore-Duffy P., LaManna J. C., del Zoppo G. J. (2008). Increased expression of fibronectin and the alpha 5 beta 1 integrin in angiogenic cerebral blood vessels of mice subject to hypobaric hypoxia. Mol. Cell. Neurosci. 38, 43-52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mjaatvedt C. H., Lepera R. C., Markwald R. R. (1987). Myocardial specificity for initiating endothelial-mesenchymal cell transition in embryonic chick heart correlates with a particulate distribution of fibronectin. Dev. Biol. 119, 59-67 [DOI] [PubMed] [Google Scholar]
- Nagy A. (2003). Manipulating the Mouse Embryo: a Laboratory Manual Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; [Google Scholar]
- Okazaki T., Ni A., Ayeni O. A., Baluk P., Yao L. C., Vossmeyer D., Zischinsky G., Zahn G., Knolle J., Christner C., et al. (2009). alpha5beta1 Integrin blockade inhibits lymphangiogenesis in airway inflammation. Am. J. Pathol. 174, 2378-2387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pankov R., Yamada K. M. (2002). Fibronectin at a glance. J. Cell Sci. 115, 3861-3863 [DOI] [PubMed] [Google Scholar]
- Peters J. H., Hynes R. O. (1996). Fibronectin isoform distribution in the mouse. I. The alternatively spliced EIIIB, EIIIA, and V segments show widespread codistribution in the developing mouse embryo. Cell Adhes. Commun. 4, 103-125 [DOI] [PubMed] [Google Scholar]
- Priestley G. V., Ulyanova T., Papayannopoulou T. (2007). Sustained alterations in biodistribution of stem/progenitor cells in Tie2Cre+ alpha4(f/f) mice are hematopoietic cell autonomous. Blood 109, 109-111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proctor J. M., Zang K., Wang D., Wang R., Reichardt L. F. (2005). Vascular development of the brain requires beta8 integrin expression in the neuroepithelium. J. Neurosci. 25, 9940-9948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds L. E., Wyder L., Lively J. C., Taverna D., Robinson S. D., Huang X., Sheppard D., Hynes R. O., Hodivala-Dilke K. M. (2002). Enhanced pathological angiogenesis in mice lacking beta3 integrin or beta3 and beta5 integrins. Nat. Med. 8, 27-34 [DOI] [PubMed] [Google Scholar]
- Roman J., McDonald J. A. (1992). Expression of fibronectin, the integrin alpha 5, and alpha-smooth muscle actin in heart and lung development. Am. J. Respir. Cell Mol. Biol. 6, 472-480 [DOI] [PubMed] [Google Scholar]
- Savolainen S. M., Foley J. F., Elmore S. A. (2009). Histology atlas of the developing mouse heart with emphasis on E11.5 to E18.5. Toxicol. Pathol. 37, 395-414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider D. J., Moore J. W. (2006). Patent ductus arteriosus. Circulation 114, 1873-1882 [DOI] [PubMed] [Google Scholar]
- Sechler J. L., Cumiskey A. M., Gazzola D. M., Schwarzbauer J. E. (2000). A novel RGD-independent fibronectin assembly pathway initiated by alpha4beta1 integrin binding to the alternatively spliced V region. J. Cell Sci. 113, 1491-1498 [DOI] [PubMed] [Google Scholar]
- Sengbusch J. K., He W., Pinco K. A., Yang J. T. (2002). Dual functions of [alpha]4[beta]1 integrin in epicardial development: initial migration and long-term attachment. J. Cell Biol. 157, 873-882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet. 21, 70-71 [DOI] [PubMed] [Google Scholar]
- Srinivasan R. S., Dillard M. E., Lagutin O. V., Lin F. J., Tsai S., Tsai M. J., Samokhvalov I. M., Oliver G. (2007). Lineage tracing demonstrates the venous origin of the mammalian lymphatic vasculature. Genes Dev. 21, 2422-2432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens L. E., Sutherland A. E., Klimanskaya I. V., Andrieux A., Meneses J., Pedersen R. A., Damsky C. H. (1995). Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 9, 1883-1895 [DOI] [PubMed] [Google Scholar]
- Takahashi S., Leiss M., Moser M., Ohashi T., Kitao T., Heckmann D., Pfeifer A., Kessler H., Takagi J., Erickson H. P., et al. (2007). The RGD motif in fibronectin is essential for development but dispensable for fibril assembly. J. Cell Biol. 178, 167-178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S., Moser M., Montanez E., Nakano T., Seo M., Backert S., Inoue I., Awata T., Katayama S., Komoda T., et al. (2009). The fibronectin RGD motif is required for multiple angiogenic events during early embryonic development. Arterioscler. Thromb. Vasc. Biol. 30, e1 [DOI] [PubMed] [Google Scholar]
- Tanjore H., Zeisberg E. M., Gerami-Naini B., Kalluri R. (2008). Beta1 integrin expression on endothelial cells is required for angiogenesis but not for vasculogenesis. Dev. Dyn. 237, 75-82 [DOI] [PubMed] [Google Scholar]
- Taverna D., Hynes R. O. (2001). Reduced blood vessel formation and tumor growth in alpha5-integrin-negative teratocarcinomas and embryoid bodies. Cancer Res. 61, 5255-5261 [PubMed] [Google Scholar]
- Taverna D., Disatnik M. H., Rayburn H., Bronson R. T., Yang J., Rando T. A., Hynes R. O. (1998). Dystrophic muscle in mice chimeric for expression of alpha5 integrin. J. Cell Biol. 143, 849-859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travis M. A., Reizis B., Melton A. C., Masteller E., Tang Q., Proctor J. M., Wang Y., Bernstein X., Huang X., Reichardt L. F., et al. (2007). Loss of integrin alpha(v)beta8 on dendritic cells causes autoimmunity and colitis in mice. Nature 449, 361-365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier A., Sonnenberg A. (2001). Function and interactions of integrins. Cell Tissue Res. 305, 285-298 [DOI] [PubMed] [Google Scholar]
- van der Flier A., Kuikman I., Kramer D., Geerts D., Kreft M., Takafuta T., Shapiro S. S., Sonnenberg A. (2002). Different splice variants of filamin-B affect myogenesis, subcellular distribution, and determine binding to integrin [beta] subunits. J. Cell Biol. 156, 361-376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb S., Brown N. A., Anderson R. H. (1998). Formation of the atrioventricular septal structures in the normal mouse. Circ. Res. 82, 645-656 [DOI] [PubMed] [Google Scholar]
- Wennerberg K., Lohikangas L., Gullberg D., Pfaff M., Johansson S., Fassler R. (1996). Beta 1 integrin-dependent and -independent polymerization of fibronectin. J. Cell Biol. 132, 227-238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicka-Patynowski I., Schwarzbauer J. E. (2003). The ins and outs of fibronectin matrix assembly. J. Cell Sci. 116, 3269-3276 [DOI] [PubMed] [Google Scholar]
- Yang J. T., Hynes R. O. (1996). Fibronectin receptor functions in embryonic cells deficient in alpha 5 beta 1 integrin can be replaced by alpha V integrins. Mol. Biol. Cell 7, 1737-1748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. T., Rayburn H., Hynes R. O. (1993). Embryonic mesodermal defects in alpha 5 integrin-deficient mice. Development 119, 1093-1105 [DOI] [PubMed] [Google Scholar]
- Yang J. T., Rayburn H., Hynes R. O. (1995). Cell adhesion events mediated by alpha 4 integrins are essential in placental and cardiac development. Development 121, 549-560 [DOI] [PubMed] [Google Scholar]
- Yang J. T., Bader B. L., Kreidberg J. A., Ullman-Cullere M., Trevithick J. E., Hynes R. O. (1999). Overlapping and independent functions of fibronectin receptor integrins in early mesodermal development. Dev. Biol. 215, 264-277 [DOI] [PubMed] [Google Scholar]
- Yashiro K., Shiratori H., Hamada H. (2007). Haemodynamics determined by a genetic programme govern asymmetric development of the aortic arch. Nature 450, 285-288 [DOI] [PubMed] [Google Scholar]
- Zovein A. C., Luque A., Turlo K. A., Hofmann J. J., Yee K. M., Becker M. S., Fassler R., Mellman I., Lane T. F., Iruela-Arispe M. L. (2010) Beta1 integrin establishes endothelial cell polarity and arteriolar lumen formation via a Par3-dependent mechanism. Dev. Cell 18, 39-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.