

Abstract
Cardiac tissues express constitutively an NADPH oxidase, which generates reactive oxygen species (ROS) and is involved in redox signaling. Myocardial metabolism generates abundant adenosine, which binds to its receptors and plays important roles in cardiac function. The adenosine A2A receptor (A2AR) has been found to be expressed in cardiac myocytes and coronary endothelial cells. However, the role of the A2AR in the regulation of cardiac ROS production remains unknown. We found that knockout of A2AR significantly decreased (39±8%) NADPH-dependent O2•− production in mouse hearts compared to age (10 weeks)-matched wild-type controls. This was accompanied by a significant decrease in Nox2 (a catalytic subunit of NADPH oxidase) protein expression, and down-regulation of ERK1/2, p38MAPK, and JNK phosphorylation (all P<0.05). In wild-type mice, intraperitoneal injection of the selective A2AR antagonist SCH58261 (3–10 mg/kg body weight for 90 min) inhibited phosphorylation of p47phox (a regulatory subunit of Nox2), which was accompanied by a down-regulated cardiac ROS production (48±8%), and decreased JNK and ERK1/2 activation by 54±28% (all P<0.05). In conclusion, A2AR through MAPK signaling regulates p47phox phosphorylation and cardiac ROS production by NADPH oxidase. Modulation of A2AR activity may have potential therapeutic applications in controlling ROS production by NADPH oxidase in the heart.
Keywords: Adenosine, Receptor, Free radicals, Myocardium, NADPH oxidase
Introduction
Adenosine, a metabolite of ATP abundantly produced by myocardial metabolism, is well known for its important regulatory effects on cardiac function such as heart rate, vasodilatation, and cardioprotection [1,36-38]. Adenosine exerts cardiovascular effects by means of cell surface adenosine receptors, which belong to the G-protein-coupled receptor superfamily. To date, four adenosine receptor (AR) subtypes, A1R, A2AR, A2BR, and A3R, have been identified and all of them have been cloned [14,45]. Among these adenosine receptors, the A2AR has been found to be expressed in vascular endothelial cells and is well known for its roles in mediating coronary vasodilation [13,35]. The A2AR has also been found to be expressed in the cardiac myocytes of several species (human, rat, and mouse), and to be involved in modulating spontaneous calcium release from the sarcoplasmic reticulum [16,19,34,41]. Activation of A2AR enhances cardiac contraction, and knockout or blocking the A2AR using selective antagonists increases the antiadrenergic effects of A1R activation in the heart [21,42,44]. A2AR also plays a key role in adenosine-mediated cardioprotection during ischemia–reperfusion or after myocardial infarction, although this is achieved partially through its anti-inflammatory effect on neutrophils [20].
Cardiac generation of ROS also plays important roles in the regulation of cardiac function including cellular development and remodelling. Several key proteins involved in myocyte excitation–contraction coupling, such as sarcolemmal ion channels and exchangers and sarcoplasmic reticulum calcium release channels, can undergo redox-sensitive alterations in activity which contribute to myocardial contractile dysfunction [23,48]. On the other hand, intracellular generation of ROS, in particular O2•−, may trigger delayed preconditioning and protect the heart from postischemic injury [3]. Although several enzymatic sources of ROS generation are expressed in the heart, recently substantial evidence has suggested that an O2•− generating NADPH oxidase is involved in the redox signaling regulation of cardiac function and development [48]. The NADPH oxidase comprises a cytochrome b, which can be further divided into one catalytic subunit (a member of the Nox family) and one p22phox, and at least 4 regulatory subunits (p47phox, p67phox, p40phox, and rac1). To date, 5 members of the Nox family have been identified (Nox1–5), each encoded by separate genes, Nox2 of which is gp91phox (the neutrophil isoform of Nox) [6].
A direct link between the A2AR and the NADPH oxidase can be found in neutrophils. For example, adenosine acting through the A2AR inhibits O2− production by NADPH oxidase in neutrophils and thereby protects tissues from inflammation [12,37]. As cardiac NADPH oxidase is an analogue of neutrophil NADPH oxidase, it is possible that the A2AR may also modulate ROS production from cardiac NADPH oxidase and therefore be involved in the regulation of cardiac function. In this study, we investigated the role of A2AR in the regulation of cardiac ROS production from NADPH oxidase and the possible downstream signaling pathways in wild-type and A2AR knockout mice. We also examined the mechanisms and the effects of in vivo blockade of the A2AR using a selective antagonist SCH58261 (7-(2-phenylethyl)-5-amino-2-(2-furyl)-pyrazole-[4,3-e]-1,2,4-mazolo[1,5-c] pyrimidine) on the cardiac ROS production in wild-type mice.
Methods
Reagents
The polyclonal antibodies against p22phox, Nox2, Nox4, p40phox, p47phox, p67phox, rac1, and cardiac-troponin I were from Santa Cruz Biotechnology; antibodies to phospho-ERK1/2, phospho-p38 MAPK, and phospho-JNK were from Cell Signalling Technology. SCH58261 and other chemicals were from Sigma unless stated otherwise.
A2AR KO mice and age-matched littermate wild-type controls
The A2AR knockout mice, generated on a CD1 background [22], and the wild-type mice were bred in our institution from heterozygote mice and genotyped [17]. All studies were performed in accordance with protocols approved by the Home Office under the Animals (Scientific Procedures) Act 1986 UK. Cardiac tissues were harvested from male mice at 10–12 weeks of age and 12–24 mice from each group were used for the study. Left ventricular (LV) tissues from these mice were dissected out and used for measuring O2− generation, immunoblotting, and immunocytochemistry.
Drug treatment of wild-type mice and cultured cardiac myocytes with SCH58261
Drug treatment using SCH58261 on wild-type CD1 mice was performed exactly as described previously [17]. Briefly, SCH58261 was dissolved in DMSO and further diluted using phosphate-buffered saline (PBS) to achieve a final concentration of 10% DMSO in the drug injection solution. SCH58261 was injected intraperitoneally at a dose of 3 and 10 mg/kg. Control mice were injected with vehicle (10% DMSO/PBS). Nine mice were used for each group and all received either drug or vehicle control in an injection volume of 10 ml/kg. Cardiac tissues were collected 90 min after drug treatment. For the experiments with H9C2 cardiac myocytes (ATCC), cells were cultured in 5% FCS/DMEM medium with or without SCH58261 (100 nM) for 30 and 60 min. Cells were then washed with PBS, frozen immediately, and detached by scraping. The cell homogenates were used for ROS detection.
Protein extraction, immunoprecitation, and immunoblotting
Protein samples were prepared from LV tissues (200 mg/ml) as described previously [25]. Soluble protein concentrations were determined by using a Bio-Rad kit (Bio-Rad Laboratories, UK). Immunoblotting (40 μg protein per sample) was performed as described previously [25]. The protein extract from human phagocytic U937 cells after phorbol-12-myristate-13-acetate (PMA 100 ng/ml) stimulation was used as the positive control for the detection of NADPH oxidase subunits. Immunoprecipitation was performed as described previously [28]. Briefly, protein samples (250 μg in a final volume of 750 μl) were diluted in immunoprecipitation buffer containing Tris-HCl 0.05 M (pH 7.4), NaCl 0.25 M, Nonidet P-40 0.1% (v/v), and a cocktail of proteinase inhibitors and phosphatase inhibitors (Sigma). Proteins were immunoprecipitated down with antibodies to p47phox coupled to protein G agarose beads (Sigma, UK) overnight at 4°C. Normal rabbit IgG-coupled protein G agarose beads were used as negative controls. Immunocomplex-bound beads were washed 4 times with immunoprecipitation buffer and resuspended in 25 μl of 2X Laemmli buffer. Samples were boiled for 3 min, and proteins were separated by 10% SDS-PAGE for immunoblotting.
Immunofluorescence confocal microscopy
Sample preparation and confocal microscopy were performed as described previously [25]. Briefly, frozen sections were first treated with a Biotin Blocking kit (DAKO) according to the manufacturer’s instructions. Primary antibodies were used at 1:250 dilution in PBS with 0.1% BSA for 30 min at room temperature. Biotin-conjugated anti-rabbit or anti-goat (1:1000 dilution) were used as secondary antibodies. Specific binding was detected by extravidin-FITC or streptavidin-Cy3. Normal rabbit or goat IgG (5 μg/ml) was used instead of primary antibody as a negative control. Images were acquired on a Zeiss LS510 confocal microscopy system. Optical sections were taken at 1-μm intervals, and images were captured and stored digitally for analysis. Fluorescence intensity was quantified from at least 3 random fields (1024×1024 pixels; 269.7×269.2 μm) per slide, from 3 slides per experimental condition and repeated 3 times using separate hearts.
Measurement of cardiac ROS production
O2− production by LV tissue homogenate (n=9 hearts per group) was measured using lucigenin (5 μM)-enhanced chemiluminescence (BMG Lumistar, Germany) as described previously [25]. O2− production was expressed as arbitrary mean light units (MLU/min) over 20 min. The specificity of O2− thus measured was confirmed either by adding superoxide dismutase (SOD, 100 units/ml) or tiron (10 mM), a nonenzymatic scavenger of O2−, to quench the O2− dependent chemiluminescence. Other enzymatic sources of O2− production were also identified by preincubation of cardiac homogenates with inhibitors such as Nω-nitro-L-arginine methyl ester (L-NAME, 100 μM), rotenone (50 μM), oxypurinol (100 μM), and diphenyleneiodonium (DPI, 20 μM). All studies were performed in triplicate. ROS generation within the cardiac tissues in situ was also measured using dihydroethidium (DHE) fluorescence as described previously [30]. Fluorescence intensity was quantified under confocal microscopy from at least 5 random fields (1024×1022 pixels; 269.7×269.2 μm) per slide, 3 slides per animal, and 6 animals per group.
Statistics
Data were presented as mean±SD of 6–24 mice per group. Comparisons were made by unpaired Student’s t test, with Bonferonni correction for multiple testing. P<0.05 was considered statistically significant.
Results
Differences in levels of ROS production and the expression of NADPH oxidase subunits between wild-type and A2AR knockout hearts
To begin to investigate whether A2AR deficiency per se may cause changes in cardiac ROS production, we isolated hearts from WT and A2AR knockout mice and examined the O2•− production in cardiac homogenate by lucigenin (5 μM)-chemiluminescence (Fig. 1A). We found no significant difference in the basal (without adding NADPH) levels of ROS production between wild-type and A2AR knockout mice. However, the tiron (an O2•− scavenger)-inhibitable, NADPH-dependent O2•− production was significantly decreased in the A2AR knockout hearts compared to WT controls (*P<0.03, n=12) (Figs. 1A and B). The O2− production thus measured could be significantly inhibited by apocynin (NADPH oxidase inhibitor), DPI (flavoprotein inhibitor) and superoxide dismutase, but not by rotenone (mitochondrial complex I enzyme inhibitor) or L-NAME (nitric oxidase synthase inhibitor) (Fig. 1C). As an alternative approach, we also examined the in situ ROS production by DHE fluorescence on cardiac sections to confirm the results obtained with lucigenin-chemiluminescence. A basal level of DHE fluorescence was detected throughout the sections of wild-type hearts (Fig. 2). Compared to wild-type controls, DHE fluorescence was significantly reduced in the cardiac sections of A2AR knockout mice as seen by fluorescence quantification (Fig. 2, left panel). The DHE fluorescence was significantly inhibited by tiron in both groups, which confirmed that the ROS detected were mainly O2•−. These data suggested that deficiency of A2AR might down-regulate the activity of NADPH oxidase and reduce the levels of ROS production in the knockout hearts.
Fig. 1.

O2•− production in wild-type and A2AR knockout heart homogenates detected by lucigenin chemiluminescence. (A) Kinetic measurement of O2•− at every minute for a total of 30 min. NADPH was added after 15 min of measurement. (B) Difference in NADPH-dependent O2•− production between wild-type and A2AR knockout heart homogenates. MLU: mean light unit of 15 measurements. (C) Effects of enzyme inhibitors on the level of O2•− production by wild-type heart homogenates. The results were presented as percentage of the control (without inhibitor). *P<0.05 for indicated values versus the control value in the same figure. n=12.
Fig. 2.
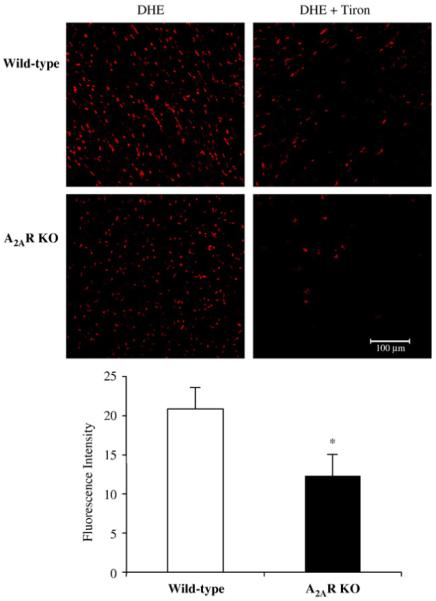
Dihydroethidium (DHE) fluorescence detection of the ROS production in cardiac sections. Cardiac sections from wild-type and A2AR were incubated with DHE in the presence or absence of tiron (an O2•− scavenger). The DHE fluorescence was visualised under confocal microscopy and quantified. *P<0.05 for A2AR knockout versus wild-type controls. n=18 sections from 6 hearts/per group.
We then examined the protein expression of NADPH oxidase subunits in the cardiac tissues by Western blotting (Fig. 3). There was no significant difference in the protein levels of Nox4, p67phox, p47phox, p40phox, p22phox, and rac1 expression between wild-type and A2AR knockout hearts. However, the level of Nox2 (a major isoform of the Nox family) was significantly decreased in A2AR knockout hearts compared to the wild-type controls (Figs. 3A and B). It is therefore possible that decreased Nox2 expression found in A2AR knockout hearts may contribute to the lower level of cardiac ROS production in knockout mice.
Fig. 3.
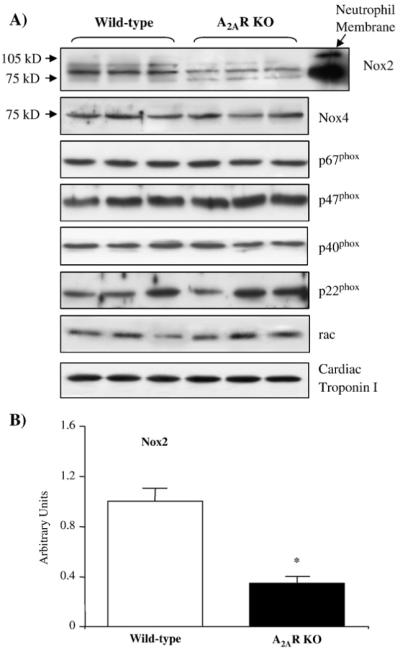
Immunoblotting for the protein expression of NADPH oxidase subunits in wild-type and A2AR knockout hearts. (A) A neutrophil membrane preparation was used as a positive control for the detection of Nox2. Cardiac troponin I was used as a loading control. (B) Protein bands were quantified densitometrically and normalised to the expression of cardiac troponin I in the same sample. The results were expressed as arbitrary units. *P<0.05 for A2AR knockout versus wild-type controls. n=12.
Changes in MAPK activation and heart/body weight ratio in A2AR knockout mice
Mitogen-activated protein kinases (MAPKs) have been reported to be redox-sensitive and involved in NADPH oxidase signaling [24,29]. MAPKs are also important downstream signaling molecules of G-coupled proteins including A2AR [14,39]. Therefore, we investigated possible changes in the levels of ERK1/2, p38MAPK, and JNK phosphorylation using phospho-specific monoclonal antibodies. The results were normalised to the levels of total protein expression of these molecules in the same samples (Fig. 4A). We found that there was no significant difference in the protein levels of total ERK1/2, p38MAPK, and JNK between the two groups. However, the levels of phosphorylated ERK1/2, p38MAPK, and JNK were significantly reduced in A2AR knockout hearts compared to wild-type controls (Figs. 4A and B, P<0.05). Since MAPKs are important regulatory signaling molecules involved in the development and differentiation of cardiac cells, we examined the body weight, heart weight, and the heart/body weight ratio between wild-type and A2AR knockout mice (Fig. 4C). There was no significant difference in the body and heart weights at 10 weeks of age between the two groups. However, the heart/body weight ratio was slightly but significantly smaller (3.7±0.3) for A2AR knockout mice compared to wild-type controls (4.5±0.4, P<0.05) (Fig. 4C).
Fig. 4.

Differences in cardiac MAPK activation and heart/body weight ratio between wild-type and A2AR knockout mice. (A) Immunoblotting for the expression of total and phosphorylated ERK1/2, p38MAPK, and JNK detected by phospho-specific monoclonal antibodies. (B) Protein bands were quantified densitometrically and the levels of MAPK phosphorylation were normalised to the total protein levels of these molecules in the same samples and expressed as arbitrary units. n=12 hearts. (C) Body weights, heart weights, and heart/body weight ratio of wild-type and A2AR knockout mice. n=24 mice. *P<0.05 for A2AR knockout versus wild-type controls.
The effect of in vivo treatment of wild-type mice with a selective A2AR antagonist (SCH58261) on cardiac ROS production and redox signaling
Our results obtained so far strongly suggested that the A2AR is involved in the regulation of ROS production by NADPH oxidase, and the redox-sensitive MAPK signaling pathways in heart. In order to investigate further the inhibitory role of the A2AR on the activity and expression of NADPH oxidase in the heart, we injected intraperitoneally SCH58261 into wild-type mice at doses of 3 and 10 mg/kg body weight. Ninety minutes after the injection, hearts were harvested and the effect of pharmacological A2AR blockage on NADPH activity was assessed by lucigenin-chemiluminescence (Fig. 5A). Compared to mice injected with vehicle only, treatment with SCH58261 at 3 mg/kg body weight significantly inhibited O2•− production by NADPH oxidase (Fig. 5A). When SCH58261 was used at 10 mg/kg body weight, it reduced levels of NADPH-dependent ROS production still further to 48±8% of the control value (Fig. 5B, P<0.05). The specificity of the assay was confirmed by adding apocynin (a NADPH oxidase inhibitor) and tiron (an O2•− scavenger). In order to confirm that the effect of SCH58261 on ROS production that we observed in the whole hearts was indeed an effect on cardiac myocyte, we cultured H9C2 cardiac myocytes and incubated these cells with SCH58261 (100 nM) for 30 and 60 min. We then examined the ROS production by cell homogenates (Fig. 5C). Compared to control cells (without SCH58261), 30 min of incubation of H9C2 myocytes with SCH58261 significantly inhibited (20±3%) the ROS production, and the inhibitory effect was further increased (39±7%) after 60 min of incubation.
Fig. 5.

The effect of SCH58261 on cardiac ROS production. (A) Kinetic measurement of NADPH-dependent O2•− production detected by lucigenin chemiluminescence in cardiac homogenates from wild-type mice treated in vivo with vehicle or SCH58261 (3 mg/kg body weight). Tiron (an O2•− scavenger) and apocynin (an NADPH oxidase inhibitor) were used to confirm the specificity of the assay. (B) Difference in NADPH-dependent O2•− production between heart homogenates from vehicle-treated (0 mg of SCH58261) and SCH58261 (3 and 10 mg/kg body weight)-treated mice. n=12 animals/per group. (C) The effect of SCH58261 on ROS production by cultured H9C2 cardiac myocytes. The cell homogenate from 2×106 cells was used for each measurement. n=3 independent cell cultures. MLU: mean light unit of 15 measurements. *P<0.05 for indicated values versus control value (0 mg SCH58261).
The mechanisms of SCH58261 inhibition of NADPH oxidase activity in the hearts were also investigated. There was no significant change in cardiac expression of NADPH oxidase subunits after 90 min of SCH58261 treatment (data not shown). However, we found that SCH58261 treatment significantly decreased the levels of p47phox serine phosphorylation as revealed by immunoprecipitation of p47phox followed by immunoblotting with a phospho-serine-specific monoclonal antibody (Fig. 6A). The p47phox is a major regulatory subunit of NADPH oxidase and p47phox serine phosphorylation is a pre-requisite for Nox2 oxidase activation [26]. It is possible that A2AR activity and signaling are necessary for optimal p47phox phosphorylation.
Fig. 6.
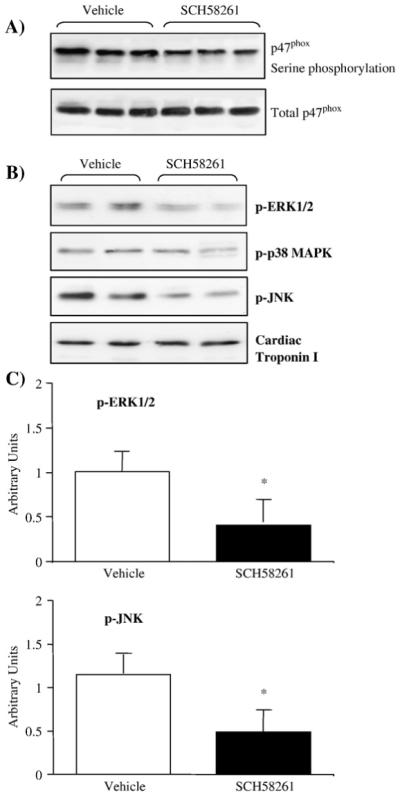
The effect of SCH58261 treatment on cardiac p47phox phosphorylation and MAPK activation. (A) Top panel: p47phox was immunoprecipitated down and detected by a phospho-serine-specific monoclonal antibody. Total p47phox was detected in parallel as loading controls. (B) ERK1/2, p38MAPK, and JNK phosphorylation detected by phospho-specific monoclonal antibodies. (C) Protein bands were quantified densitometrically and the results were normalised to the total protein levels of these molecules in the same samples and expressed as arbitrary units. n=12 hearts. *P<0.05 for indicated value versus vehicle control.
The effects of SCH58261 treatment on cardiac MAPK phosphorylation were also examined in these mice (Figs. 6B and C). As expected, SCH58261 treatment significantly reduced the levels of ERK1/2 and JNK phosphorylation to 54±28% of the levels found in vehicle-treated controls (P<0.05). However, the levels of p38MAPK phosphorylation showed no significant change after 90 min of SCH58261 treatment. It is possible that changes in p38MAPK phosphorylation found in A2AR knockout hearts were due to long-term lack of A2AR activation.
Cellular expression of NADPH oxidase in the heart
Although we showed changes in activity and expression of NADPH oxidase in mouse heart deficiency of A2AR, we had no information on the expression of NADPH oxidase in cardiac myocytes and coronary endothelial cells, where the A2AR was found to be expressed in the heart [13,16,19,34,35,41]. In order to investigate this, we examined the cellular expression of NADPH oxidase using antibodies to p47phox (a major regulatory subunit of NADPH oxidase) and Nox2 (catalytic subunit of NADPH oxidase) in cardiac sections of wild-type mice by confocal microscopy (Fig. 7). We found that NADPH oxidase (labeled by p47phox) was constitutively expressed in coronary endothelial cells, which were labeled by CD31, an endothelial cell surface maker (red fluorescence). NADPH oxidase (labeled by Nox2) was also detected in cardiomyocytes, which were outlined by laminin, a myocyte sarcolemmal membrane component (green fluorescence). The yellow fluorescence in the superimposed images (bottom panels) clearly indicated the expression of NADPH oxidase in both coronary endothelial cells (left panel), and cardiomyocytes (right panel).
Fig. 7.
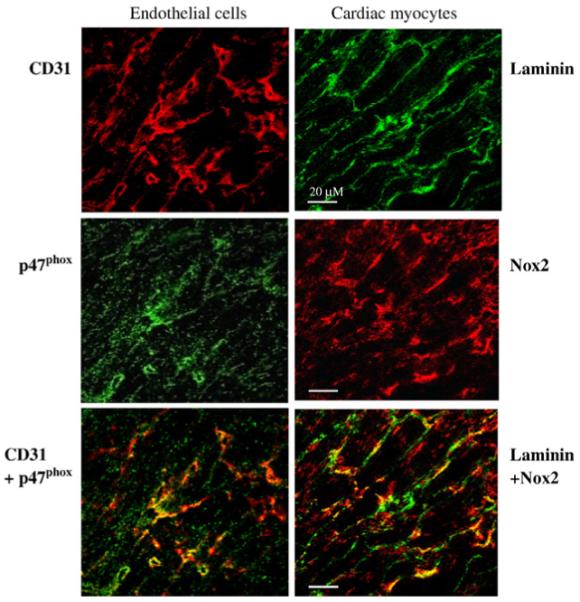
Confocal microscopy for the cellular expression of p47phox and Nox2 in cardiac sections. Endothelial cells (left panel) were labeled by CD31 (red). Cardiac myocytes (right panel) were outlined by laminin expressed in the sarcolemmal membrane (green). The yellow fluorescence in the lower panels shows the expression of p47phox (green, left middle panel) in endothelial cells, and the expression of Nox2 (red, right middle panel) in cardiac myocytes.
Discussion
Emerging evidence has revealed that adenosine through the A2AR plays important roles in modulating cardiovascular function. A2AR is the dominant adenosine receptor found in vascular endothelium and is involved in mediating coronary vessel dilatation [8,34,35,38]. Pharmacologically, the A2AR has been found in the myocardium and plays an important protective role against cardiac “stress”-related injuries such as ischemia/reperfusion, or pressure-overload-induced cardiac hypertrophy [3,15,20,42,47]. Activation of the A2AR attenuates cardiac oxidative damage by inhibiting ROS release from neutrophil NADPH oxidase [31,37,43]. As cardiac NADPH oxidase is an analogue of neutrophil NADPH oxidase, it is possible that the A2AR may also modulate ROS production from cardiac NADPH oxidase, and thereafter influence redox regulation of cardiac function. In this study, using a murine model of A2AR knockout and by treating wild-type mice with a selective A2AR antagonist (SCH58261), we provide direct evidence that A2AR is functionally involved in the regulation of ROS production from NADPH oxidase, which is constitutively expressed in the cardiac myocytes and coronary endothelial cells as shown in Fig. 6.
In contrast to what was reported for the role of A2AR in neutrophils [31,37,43], we found that knockout or pharmacological blockage of A2AR actually reduced ROS production in the heart. Although the mechanisms underlining these opposing roles of A2AR in the regulation of cardiac and neutrophil ROS production are largely unknown, a clear possibility is that the activation process (structural assembly) for the NADPH oxidase in the cardiovascular system is different from that found in neutrophils. In neutrophils, the NADPH oxidase is inactive in resting cells. In response to various stimuli, the cytosolic subunits (p47phox, p67phox, p40phox, and rac) translocate to and associate with membrane-bound cytochrome b558 (a heterodimer of p22phox and Nox2), a process that results in neutrophil oxidase activation and the production of large amounts of O2•− to kill invading bacteria [2]. However, in the cardiovascular system, the NADPH oxidase is preassembled even in the absence of agonist stimulation. It is located intracellularly and produces continuously a low basal level of O2•− that serves as a signaling molecule involved in the regulation of cardiovascular function [4,27].
Unlike the neutrophil NADPH oxidase, which contains only Nox2 (also called gp91phox), nonphagocytic NADPH oxidase (Nox) has at least 5 isoforms namely Nox1 to 5 [4,6]. Nox2 activity is dependent on the phosphorylation of p47phox and requires the presence of other cytosolic components [40], whereas Nox4 is constitutively active and its activity is not regulated by rac or any other known cytosolic regulatory components [32]. Both Nox2 and Nox4 have been found to be expressed in the heart [7]. With more than one Nox isoform expressed simultaneously in the heart, it is possible that ROS generated from Nox2 and Nox4 might be differently regulated and involved differently in cardiac function. We found that the levels of tiron (O2•− scavenger)-inhibitable NADPH-dependent (O2•− production from A2AR knockout hearts was significantly lower than from wild-type controls. Reduced levels of ROS production in A2AR knockout hearts were due to decreases in NADPH oxidase activity and this was confirmed by our experiments using different enzyme inhibitors. Moreover, we demonstrated by immunoblotting that A2AR deficiency actually reduced the expression of Nox2 (but not Nox4) in the hearts, indicating that A2AR is functionally involved in the regulation of Nox2 expression and activity.
The involvement of the A2AR in the regulation of cardiac ROS production was further supported by our experiments using wild-type mice treated with SCH58261, which is a highly selective A2AR antagonist [14,18]. Intraperitoneal injection of SCH58261 significantly reduced the levels of cardiac ROS production from NADPH oxidase. The specificity of the assay was confirmed by adding apocynin (a NADPH oxidase inhibitor) and tiron (an O2•− scavenger). More importantly, using a combination of immunoprecipitation, immunoblotting with phospho-serine-specific monoclonal antibody, we successfully showed that SCH58261 inhibited both the p47phox phosphorylation and the cardiac ROS production. The regulatory mechanisms of cardiac NADPH oxidase are complicated and far from clear. Nevertheless, a key feature of nonphagocytic Nox2 oxidase activation is similar to the neutrophil NADPH oxidase in that it requires p47phox phosphorylation [29]. It is important to know that blocking A2AR affects p47phox phosphorylation and thereby reduce ROS production from Nox2 oxidase in normal heart. In the brain, A2AR deficiency [9], the A2AR antagonist SCH58261 [33], or selective inactivation of A2AR on bone marrow cells [46] have all been shown to provide neuroprotection from ischemic injury. The mechanism involved was suggested at least in part to be due to suppression of ROS generation [9,33,46].
The MAPK family consists of the extracellular regulated kinases (ERK) such as ERK1/2 and the stress-activated protein kinases, such as p38MAPK and jun-N-terminal kinase (JNK). These kinases have been shown to be activated by G-protein-coupled receptors, such as A2AR, via Gs/cAMP-dependent [14] or -independent pathways [39]. In accordance with these reports, we found reduced levels of MAPK phosphorylation in A2AR knockout hearts and in hearts from wild-type mice treated with a selective A2AR antagonist. Interestingly, both ERK and p38MAPK have been found to phosphorylate p47phox in vitro [10,11]. Therefore, reduced MAPK activity due to A2AR blockage might be responsible for decreased p47phox phosphorylation. On the other hand, as MAPKs are redox sensitive and have also been shown to be involved in Nox2 signaling [5,30], it is also possible that reduced MAPK activation may result from reduced levels of ROS production caused by A2AR blockage. Put together, our data strongly suggested that MAPKs might serve as a bridge linking A2AR and ROS signaling from NADPH oxidase in the heart.
Oxidative stress has multiple effects on cell function, depending on the amount and subcellular location of ROS generated. Increased cardiac generation of ROS is implicated in the development of cellular hypertrophy and remodeling, at least in part through activation of redox-sensitive protein kinases such as the MAPK superfamily [23,25,48]. A2AR, on the other hand, has an important inotropic effect on the myocardium directly and indirectly by facilitating a greater response to β-adrenergic stimulation in the murine heart [42]. Several factors linked to A2AR deficiency such as decreased response to adrenergic stimulation, lack of sufficient ROS production, and reduced MAPK signaling may all affect cardiac development after birth. This was supported by our data that at 10 weeks of age, the body weight was not significantly different, but the A2AR knockout mice had a slightly but significantly smaller heart/body weight ratio than age-matched littermates. Although there was no sign of cardiac dysfunction in A2AR knockout mice at that age, the long-term consequences and significance after middle age needs further investigation. In a previous study, the A2AR knockout mice were found to have higher body weights and heart weights than their age-matched littermates [34]. The discrepancy between our results and the previous study may due to the age differences. The mice used in our study were 10–12 weeks of age, and in the previous study they were 16–18 weeks of age. Nevertheless, absence of A2AR has been shown to promote body weight increase after 3 months of age in an initial study of these mice [22].
In conclusion, we report for the first time that A2AR is functionally involved in the regulation of cardiac ROS production under physiological conditions. Genetic knockout or pharmacological blockade of the A2AR in vivo was associated with decreased levels of p47phox phosphorylation, Nox2 expression, and MAPK activation in the heart. Fully understanding the role of the A2AR may provide insight into the control of ROS production by NADPH oxidase in the heart.
Acknowledgments
This work is supported by a grant from the British Heart Foundation (PG/06/073/21118) and a grant from the Biotechnology and Biological Sciences Research Council, UK (BB/D009510/1), to J.-M. Li.
Abbreviations
- A2AR
adenosine 2A receptor
- DMSO
dimethyl sulfoxide
- DPI
diphenyleneiodonium
- L-NAME
Nω-nitro-L-arginine methyl ester
- LV
left ventricular
- MAPK
mitogen-activated protein kinase
- PBS
phosphate-buffered saline
- ROS
reactive oxygen species
References
- [1].Ashton KJ, Peart JN, Morrison RR, Matherne GP, Blackburn MR, Headrick JP. Genetic modulation of adenosine receptor function and adenosine handling in murine hearts: insights and issues. J. Mol. Cell. Cardiol. 2007;42:693–705. doi: 10.1016/j.yjmcc.2006.12.012. [DOI] [PubMed] [Google Scholar]
- [2].Babior BM, Lambeth JD, Nauseef W. The neutrophil NADPH oxidase. Arch. Biochem. Biophys. 2002;397:342–344. doi: 10.1006/abbi.2001.2642. [DOI] [PubMed] [Google Scholar]
- [3].Baxter GF. Role of adenosine in delayed preconditioning of myocardium. Cardiovasc. Res. 2002;55:483–494. doi: 10.1016/s0008-6363(02)00280-8. [DOI] [PubMed] [Google Scholar]
- [4].Bedard K, Krause KH. The Nox family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol. Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- [5].Bendall JK, Rinze R, Adlam D, Tatham AL, Joe de Bono T, Channon KM. Endothelial Nox2 overexpresison potentiates vascular oxidative stress and hemodynamic response to angiotensin II: study in endothelial-targeted Nox2 transgenic mice. Circ. Res. 2007;100:1016–1025. doi: 10.1161/01.RES.0000263381.83835.7b. [DOI] [PubMed] [Google Scholar]
- [6].Bengtsson SHM, Gulluyan LM, Dusting G, Drummond GR. Novel isoforms of NADPH oxidase in vascular physiology and pathophysiology. Clin. Exp. Pharmacol. Physiol. 2003;30:849–854. doi: 10.1046/j.1440-1681.2003.03929.x. [DOI] [PubMed] [Google Scholar]
- [7].Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lamberth JD, Cave AC, Shah AM. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ. Res. 2003;93:802–804. doi: 10.1161/01.RES.0000099504.30207.F5. [DOI] [PubMed] [Google Scholar]
- [8].Cerqueira MD. The future of pharmacologic stress: selective A2A adenosine receptor agonists. Am. J. Cardiol. 2005;94(suppl.):33D–42D. doi: 10.1016/j.amjcard.2004.04.017. [DOI] [PubMed] [Google Scholar]
- [9].Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D, Moskowitz MA, Fink JS, Schwarzschild MA. A2A adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J. Neurosci. 1999;19:9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Dang PMC, Stensballe A, Boussetta T, Raad H, Dewas C, Kroviarski Y, Hayem G, Jensen ON, Gougerot-Pocidalo MA, El-Benna J. A specific p47phox-serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J. Clin. Invest. 2006;116:2033–2043. doi: 10.1172/JCI27544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dewas C, Fay M, Gougerot-Pocidalo M-A, El-Benna J. The mitogen-activated protein kinase extracellular signal-regulated kinase 1/2 pathway is involved in formyl-methionyl-leucyl-phenylalanine-induced p47phox phosphorylation in human neutrophils. J. Immunol. 2000;165:5238–5244. doi: 10.4049/jimmunol.165.9.5238. [DOI] [PubMed] [Google Scholar]
- [12].Ernens I, Rouy D, Velot E, Devaux Y, Wagner DR. Adenosine inhibits matrix metalloproteinase-9 secretion by neutrophils: implication of A2a receptor and cAMP/PKA/Ca2+ pathway. Circ. Res. 2006;99:590–597. doi: 10.1161/01.RES.0000241428.82502.d4. [DOI] [PubMed] [Google Scholar]
- [13].Flood A, Headrick JP. Functional characterization of coronary vascular adenosine receptors in the mouse. Br. J. Pharmacol. 2005;133:1063–1072. doi: 10.1038/sj.bjp.0704170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fredholm BB, Ijzerman AP, Jacobson KA, Klotz KN, Linden J. Nomenclature and classification of adenosine receptors. Pharmacol. Rev. 2001;53:527–552. [PMC free article] [PubMed] [Google Scholar]
- [15].Gan XT, Rajapurohitam V, Haist JV, Chidiac P, Cook MA, Karmazyn M. Inhibition of phenylephrine-induced cardiomyocyte hypertrophy by activation of multiple adenosine receptor subtypes. J. Pharmacol. Exp. Ther. 2005;312:27–34. doi: 10.1124/jpet.104.073122. [DOI] [PubMed] [Google Scholar]
- [16].Hove-Madsen L, Prat-Vidal C, Liach A, Ciruela F, Casado V, Liuis C, Bayes-Genis A, Cinca J, Franco R. Adenosine A2A receptors are expressed in human atrial myocytes and modulate spontaneous sarcoplasmic reticulum calcium release. Cardiovasc. Res. 2006;72:292–302. doi: 10.1016/j.cardiores.2006.07.020. [DOI] [PubMed] [Google Scholar]
- [17].Hussey MJ, Clarke GD, Ledent C, Hourani SMO, Kitchen I. Reduced response to the formalin test and lowered spinal NMDA glutamate receptor binding in adenosine A2A receptor knockout mice. Pain. 2007;129:287–294. doi: 10.1016/j.pain.2006.10.014. [DOI] [PubMed] [Google Scholar]
- [18].Jacobson KA, Gao ZG. Adenosine receptors as therapeutic targets. Nat. Rev. Drug Discov. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jenner TL, Mellick AS, Harrison GJ, Griffith LR, Rose’Meyer RB. Age-related changes in cardiac adenosine receptor expression. Mech. Ageing Dev. 2004;125:211–217. doi: 10.1016/j.mad.2003.11.016. [DOI] [PubMed] [Google Scholar]
- [20].Kin H, Zatta AJ, Lofye MT, Amerson BS, Halkos ME, Kerendi F, Zhao ZQ, Guyton RA, Headrick JP, Vinten-Johansen J. Postconditioning reduces infarct size via adenosine receptor activation by endogenous adenosine. Cardiovasc. Res. 2007;67:124–133. doi: 10.1016/j.cardiores.2005.02.015. [DOI] [PubMed] [Google Scholar]
- [21].Lasley RD, Kristo G, Keith BJ, Mentzer RM., Jr. The A2a/A2b receptor antagonist ZM-241385 blocks the cardioprotective effect of adenosine agonist pretreatment in in vivo rat myocardium. Am. J. Physiol. Heart Circ. Physiol. 2007;292:H426–H431. doi: 10.1152/ajpheart.00675.2006. [DOI] [PubMed] [Google Scholar]
- [22].Ledent C, Vaugeois JM, Schiffmann SN, Pedrazzini T, El Yacoubi M, Vanderhaeghen JJ, Costentin J, Heath JK, Vassart G, Parmentier M. Aggressiveness, hypoalgesia and high blood pressure in mice lacking the adenosine A2A receptor. Nature. 1997;388:674–678. doi: 10.1038/41771. [DOI] [PubMed] [Google Scholar]
- [23].Lefer DJ, Granger DN. Oxidant stress and cardiac disease. Am. J. Med. 2000;109:315–323. doi: 10.1016/s0002-9343(00)00467-8. [DOI] [PubMed] [Google Scholar]
- [24].Li J-M, Fan LM, Christie MR, Shah AM. Acute tumor necrosis factor alpha signaling via NADPH oxidase in microvascular endothelial cells: Role of p47phox phosphorylation and binding to TRAF4. Mol. Cell. Biol. 2005;25:2320–2330. doi: 10.1128/MCB.25.6.2320-2330.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li J-M, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40:477–484. doi: 10.1161/01.hyp.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- [26].Li J-M, Mullen AM, Yun S, Wientjes F, Brouns GY, Thrasher AJ, Shah AM. Essential role of the NADPH oxidase subunit p47phox in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-α. Circ. Res. 2002;90:143–150. doi: 10.1161/hh0202.103615. [DOI] [PubMed] [Google Scholar]
- [27].Li J-M, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004;287:R1014–R1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- [28].Li J-M, Shah AM. Intracellular localization and pre-assembly of the NADPH oxidase complex in cultured endothelial cells. J. Biol. Chem. 2002;277:19952–19960. doi: 10.1074/jbc.M110073200. [DOI] [PubMed] [Google Scholar]
- [29].Li J-M, Shah AM. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II: role of the p47phox subunit. J. Biol. Chem. 2003;278:12094–12100. doi: 10.1074/jbc.M209793200. [DOI] [PubMed] [Google Scholar]
- [30].Li J-M, Wheatcroft S, Fan LM, Kearney MT, Shah AM. Opposing role of p47phox in basal versus angiotensin II-stimulated alteration in vascular O2•− production, vascular tone, and mitogen-activated protein kinase activation. Circulation. 2004;109:1307–1313. doi: 10.1161/01.CIR.0000118463.23388.B9. [DOI] [PubMed] [Google Scholar]
- [31].Linden J. Molecular approach to adenosine receptors: receptor-mediated mechanisms of tissue protection. Annu. Rev. Pharmacol. Toxicol. 2001;41:775–787. doi: 10.1146/annurev.pharmtox.41.1.775. [DOI] [PubMed] [Google Scholar]
- [32].Martyn KD, Frederick LM, von Loeheysen K, Dinauer MC, Knaus UK. Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidase. Cell. Signal. 2006;18:69–82. doi: 10.1016/j.cellsig.2005.03.023. [DOI] [PubMed] [Google Scholar]
- [33].Monopoli A, Lozza G, Forlani A, Mattavelli A, Ongini E. Blockade of adenosine A2A receptors by SCH 58261 results in neuroprotective effects in cerebral ischemia in rats. Neuroreport. 1998;9:3955–3959. doi: 10.1097/00001756-199812010-00034. [DOI] [PubMed] [Google Scholar]
- [34].Morrison RR, Talukder MAH, Ledent C, Mustafa SJ. Cardiac effects of adenosine in A2A receptor knockout hearts: uncovering A2B receptors. Am. J. Physiol. Heart Circ. Physiol. 2002;282:H437–H444. doi: 10.1152/ajpheart.00723.2001. [DOI] [PubMed] [Google Scholar]
- [35].Olanrewaju HA, Mustafa SJ. Adenosine A2A and A2B receptors mediated nitric oxide production in coronary artery endothelial cells. Gen. Pharmacol. 2000;35:171–177. doi: 10.1016/s0306-3623(01)00107-0. [DOI] [PubMed] [Google Scholar]
- [36].Peart JN, Headrick JP. Adenosinergic cardioprotection: multiple receptors, multiple pathways. Pharmacol. Ther. 2007;114:208–221. doi: 10.1016/j.pharmthera.2007.02.004. [DOI] [PubMed] [Google Scholar]
- [37].Ramkumar V, Hallam DM, Nie Z. Adenosine, oxidative stress and cytoprotection. Jpn. J. Pharmacol. 2001;86:265–274. doi: 10.1254/jjp.86.265. [DOI] [PubMed] [Google Scholar]
- [38].Sands WA, Palmer TM. Adenosine receptors and the control of endothelial cell function in inflammatory disease. Immunol. Lett. 2005;101:1–11. doi: 10.1016/j.imlet.2005.04.005. [DOI] [PubMed] [Google Scholar]
- [39].Schulte G, Fredholm BB. Signaling from adenosine receptor to mitogen-activated protein kinases. Cell. Signal. 2003;15:813–827. doi: 10.1016/s0898-6568(03)00058-5. [DOI] [PubMed] [Google Scholar]
- [40].Sumimoto H, Miyano K, Takeya R. Molecular composition and regulation of the Nox family NADPH oxidase. Biochem. Biophys. Res. Commun. 2005;338:677–686. doi: 10.1016/j.bbrc.2005.08.210. [DOI] [PubMed] [Google Scholar]
- [41].Talukder MAH, Morrison RR, Mustafa SJ. Comparison of the vascular effects of adenosine in isolated mouse heart and aorta. Am. J. Physiol. Heart Circ. Physiol. 2002;282:H49–H57. doi: 10.1152/ajpheart.2002.282.1.H49. [DOI] [PubMed] [Google Scholar]
- [42].Tikh EI, Fenton RA, Dobson JG., Jr. Contractile effects of adenosine A1 and A2A receptors in isolated murine hearts. Am. J. Physiol. Heart Circ. Physiol. 2007;290:H348–H356. doi: 10.1152/ajpheart.00740.2005. [DOI] [PubMed] [Google Scholar]
- [43].Varani K, Manfredini R, Iannotta V, Pancaldi C, Cattabriga E, Uluoglu C, Borea PA, Portaluooi F. Effects of doxazosin and propranolol on A2A adenosine receptors in essential hypertension. Hypertension. 2002;40:909–913. doi: 10.1161/01.hyp.0000039505.79741.15. [DOI] [PubMed] [Google Scholar]
- [44].Woodiwiss AJ, Honeyman TW, Fenton RA, Dobson JG., Jr. Adenosine A2a-receptor activation enhances cardiomyocyte shortening via Ca2+-independent and-dependent mechanisms. Am. J. Physiol. Heart Circ. Physiol. 1999;276:H1434–H1441. doi: 10.1152/ajpheart.1999.276.5.H1434. [DOI] [PubMed] [Google Scholar]
- [45].Yaar R, Jones MR, Chen J-F, Ravid K. Animal models for the study of adenosine receptor function. J. Cell. Physiol. 2005;202:9–20. doi: 10.1002/jcp.20138. [DOI] [PubMed] [Google Scholar]
- [46].Yu L, Huang Z, Mariani J, Wang Y, Moskowitz M, Chen J-F. Selective inactivation or reconstitution of adenosine A2A receptors in bone marrow cells reveals their significant contribution to the development of ischemic brain injury. Nat. Med. 2004;10:1081–1087. doi: 10.1038/nm1103. [DOI] [PubMed] [Google Scholar]
- [47].Zatta AJ, Matherne GP, Headrick JP. Adenosine receptor-mediated coronary vascular protection in post-ischemic mouse heart. Life Sci. 2006;78:2426–2437. doi: 10.1016/j.lfs.2005.09.035. [DOI] [PubMed] [Google Scholar]
- [48].Zimmet JM, Hare JM. Nitroso-redox interaction in the cardiovascular system. Circulation. 2006;114:1531–1544. doi: 10.1161/CIRCULATIONAHA.105.605519. [DOI] [PubMed] [Google Scholar]