

Abstract
Objective
HMG-CoA reductase inhibitors have been shown to upregulate GTP cyclohydrolase I (GTPCH-I), the key enzyme for tetrahydrobiopterin de novo synthesis and to normalize tetrahydrobiopterin levels in hyperglycemic endothelial cells. We sought to determine whether in vivo treatment with the HMG-CoA reductase inhibitor atorvastatin is able to upregulate the GTPCH-I, to recouple eNOS and to normalize endothelial dysfunction in an experimental model of diabetes mellitus.
Methods and results
In male Wistar rats, diabetes was induced by streptozotocin (STZ, 60 mg/kg). In STZ rats, atorvastatin feeding (20 mg/kg/d, 7 weeks), normalized vascular dysfunction as analyzed by isometric tension studies, levels of circulating endothelial progenitor cells (FACS-analysis), superoxide formation (assessed by lucigenin-enhanced chemiluminescence and dihydroethidium staining), vascular levels of the phosphorylated vasodilator-stimulated phosphoprotein (P-VASP), tyrosine nitration of the prostacyclin synthase, expression of GTPCH-I, dihydrofolate reductase and eNOS, translocation of regulatory NADPH oxidase subunits rac1, p47phox and p67phox (assessed by Western blot) and vascular tetrahydrobiopterin levels as measured by HPLC. Dihydroethidine staining revealed that the reduction of vascular superoxide was at least in part due to eNOS recoupling.
Conclusion
HMG-CoA reductase inhibition normalizes endothelial function and reduces oxidative stress in diabetes by inhibiting activation of the vascular NADPH oxidase and by preventing eNOS uncoupling due to an upregulation of the key enzyme of tetrahydrobiopterin synthesis, GTPCH-I.
Keywords: Diabetes mellitus, Vascular biology, Nitric oxide synthase, Reactive oxygen species, HMG-CoA reductase inhibitors
1. Introduction
Diabetes mellitus is a major risk factor for the development of cardiovascular disease. Endothelial dysfunction is encountered early during the development of vascular damage [1]. Animal and human studies have demonstrated that increased oxidative stress largely accounts for this phenomenon since vitamin C was able to correct endothelial dysfunction in patients with diabetes mellitus type 1 and 2 [2,3]. As predominant sources of superoxide, the vascular NADPH oxidase[4–6], an uncoupled endothelial nitric oxide synthase (eNOS)[5,7,8], xanthine oxidase [9] and mitochondria [10] have been identified. The uncoupled eNOS has gained growing attention, since a dysfunctional eNOS not only leads to a decreased NO bioactivity in the vasculature, thereby shifting the superoxide (O2•–)/nitric oxide (NO) equilibrium towards O2•–, but can also be a source of O2•– itself by transferring electrons to molecular oxygen in the uncoupled state [11]. The uncoupling reaction of eNOS is triggered largely by a peroxynitrite (ONOO–)-mediated oxidation of the eNOS co-factor tetrahydrobiopterin (BH4) leading to the formation of the BH3-radical and subsequently to dihydrobiopterin (BH2) [8]. Intracellular depletion of BH4 is counteracted mainly by the activity of the BH4-synthesizing enzyme GTP cyclohydrolase I (GTPCH-I) and the BH2-reducing enzyme dihydrofolate reductase (DHFR).
3-Hydroxy-3-methylglutaryl(HMG)-coenzyme(CoA) reductase inhibitors (statins) were initially designed to lower LDL-cholesterol levels. Today, they are established in the treatment of coronary artery disease even in patients even with normal LDL levels due to their beneficial pleiotropic effects. Many of these effects can be explained by the inhibition of isoprenylation of proteins, which consequently lack their lipid anchor. For example, statin-induced inhibition of protein-isoprenylation (e.g., of RhoGTPases) can prevent the assembly of the membrane bound NADPH oxidase [12], decrease the amount of membrane bound endothelin-1 receptor [13] and increase eNOS activity [14] and eNOS-mRNA stability [15]. Interestingly, statin therapy as well as diabetes mellitus may lead to increase in eNOS protein expression [16–18], which may be beneficial only when the enzyme is in its coupled state. Recently, two groups have demonstrated that HMG-CoA reductase inhibition increases the expression of the GTPCH-I and subsequently cellular BH4 levels in cultured endothelial cells and that inhibitory effects of high glucose on the GTPCH-I expression as well as the BH4 lowering effects were reversed by the HMG-CoA reductase inhibitor atorvastatin[19–21].
It remains to be established, however, whether GTPCH-I is downregulated in an in vivo model of diabetes mellitus, whether in vivo treatment with statins is able to recouple eNOS, whether this is due to upregulation of GTPCH-I and whether statin treatment is thereby able to prevent harmful events downstream of eNOS uncoupling mediated by decreased NO and increased O2•– and ONOO– formation, like reduction of circulating endothelial progenitor cells, inactivation of the prostacyclin synthase (PGI2S) by tyrosine nitration (PGI2S-3NT), the phenomenon of nitrate resistance and endothelial dysfunction.
2. Methods
2.1. Chemicals and reagents
Streptozotocin was from Fluka (Seelze, Germany), atorvastatin from Pfizer (New York, USA), nitroglycerin (glycerol trinitrate, GTN) was from Pohl-Boskamp (Hohenlockstedt, Germany). All other chemicals where of highest analytical grade and of highest purity available (Sigma–Aldrich, Seelze, Germany).
2.2. Animal model
Eighty-four male Wistar rats (6 weeks old, 250 g; Charles River Laboratories, Sulzfeld, Germany) were divided into four treatment groups: untreated controls (Ctr) versus atorvastatin (Ator) treatment (20 mg/day/kg bodyweight,) versus streptozotocin-induced diabetes mellitus type 1 (STZ) versus STZ/Ator. Animals were housed in a 12-h light–dark cycle and allowed free access to standard chow and water. Atorvastatin was mixed into the chow pellets by the company providing the animal diet (ssniff, Soest, Germany).
For induction of diabetes mellitus type 1, rats were anesthetized with ketamine/xylocain and injected with a single dose of STZ into the vena dorsalis penis (60 mg/kg bodyweight in 5 mM pH 4.5 citrate buffer). Animals from the other study arms were injected with the solvent. Animals were allowed to recover for 4 days before initiation of the feeding regimen; diabetes mellitus type 1 was verified by measuring levels of blood glucose using an Accu-check Sensor analyzer (Roche, Mannheim, Germany). Of the STZ-treated rats, only animals exceeding 300 mg/dl of blood glucose were considered hyperglycemic and included in the study.
After 7 weeks of treatment, rats were anesthetized by isoflurane inhalation (5% inhalant in room air) and killed by exsanguination. Blood was collected by right ventricular puncture. Aorta and heart were rapidly excised, transferred to 4 °C Krebs–HEPES solution (pH 7.35, containing 99.01 mM NaCl, 4.69 mM KCl, 2.50 mMCaCl2, 1.20 mM MgSO4, 25.0 mM NaHCO3, 1.03 mM K2HPO4, 20.0 mM Na–HEPES, 11.1 mM D-glucose) and cleaned of adhesive tissue. Aortas were carefully rinsed prior to further handling.
2.3. Serum parameters
Seven millilitres of venous blood were transferred into serum syringes, left on ice for 30 min and centrifuged for 10 min at 2000 × g. The supernatant (serum) was stored at −80 °C. Insulin levels were measured using a rat insulin ELISA (DRG Instruments GmbH, Marburg, Germany) following the manufacturer’s instructions. Cholesterol and triglyceride levels were analyzed in the Department for Clinical Chemistry, University Hospital Mainz, Germany, employing the facilities in use for daily routine in patient care.
2.4. Determination of endothelial progenitor cell (EPC) numbers and cellular characterization
Peripheral blood mononuclear cells (PBMCs) were isolated by Ficoll density centrifugation [22]. PBMCs were cultured on fibronectin-precoated 6-wells in EBM-2 culture medium, characterized and counted by appropriate flow cytometric analyses as previously described [23]. Briefly, PBMCs (2 × 105) were cultured in EBM-2 culture medium supplemented with EBM SingleQuots (Clonetics, Germany) and 20% FCS for 3 days. To exclude contamination with mature circulating endothelial cells, we carefully removed non-adherent cells 8 h after initial seeding and placed them on new fibronectin-precoated chamber slides. After dilution of 1,1′-dioctadecyl-3,3,3′,3-tetramethyl-indocarbocyanine perchlorate-labeled acetylated LDL (dil-acLDL; Molecular Probes, Eugene, USA) and fluorescein isothiocyanate (FITC)-conjugated lectin from Ulex europeus (UEA-1; Sigma, Germany) in serum-free EBM2 media, cells were washed twice and incubated for 4 h at 37 °C in EBM2 medium containing 10 μg/ml dil-acLDL and 20 μg/ml UEA-1. After washing, cells were determined by appropriate flow cytometric analyses. It is clear to the authors that various types of EPC exist. Therefore, EPC investigated in the present study belong to monocytic-derived EPC[24,25].
2.5. Isometric tension studies
Isolated aortas were cut into 4-mm segments and mounted on force transducers (Kent, USA; Powerlab, USA) in organ chambers containing Krebs–Henseleit solution (37 °C pH 7.35, containing 118.3 mM NaCl, 4.69 mM KCl, 1.87 mM CaCl2, 1.2 mM MgSO4, 1.03 mM K2HPO4, 25 mM NaHCO3, 11.1 mM D-glucose) bubbled with carbogen gas (95% O2, 5% CO2). To test for vasorelaxation in response to acetylcholine (ACh) and GTN, aortic segments were stretched gradually over 1 h to reach a resting tension of 3.0 g. Following preconstriction with phenylephrine (1 μM) to reach 50–80% of maximal tone induced by KCl, concentration–response curves to increasing concentrations of ACh and NTG were recorded [5].
2.6. Vascular ROS formation in isolated aortic rings
Vascular O2•– formation was measured in 4-mm segments of isolated aorta using lucigenin (5 μmol/l)-enhanced chemiluminescence (ECL) as previously described [26].
2.7. NADPH oxidase activity in heart membrane fractions
Membrane fractions were prepared and measured as previously described [27,28]. NADPH oxidase activity (200 μM NADPH) of the membrane suspensions (0.2 mg/ml protein in PBS) was measured by lucigenin (5 μM) ECL. Chemiluminescence was detected in a Lumat LB 9507 (Berthold Techn., Bad Wildbad, Germany). Results were normalized for protein content and expressed as counts/mg/min after 5 min.
2.8. Oxidative fluorescent microtopography
Isolated aorta was cut into 3 mm rings and incubated in Krebs–HEPES solution for 15 min at 37 °C in the presence or absence of NG-nitro-L-arginine (L-NNA, 10 μM), embedded in aluminium cups of about 1 ml of a polymeric resin (Tissue Tek, USA) and frozen in liquid nitrogen. Cryosections (6 μm) were stained with the superoxide-sensitive dye dihydroethidium (DHE, 1 μM in PBS) and incubated for 30 min at 37 °C. Green and red fluorescence was detected using a Zeiss Axiovert 40 CFL Camera (Zeiss, Oberkochen, Germany). Sections of all four study arms were analyzed in parallel with identical imaging parameters.
2.9. Protein expression
Rat aortic tissue was homogenized in liquid nitrogen following an established protocol. Sample preparation and Western blotting was performed as described previously [29]. To determine translocation of rac1, p47phox and p67phox, protein homogenates of aortic tissue were divided by ultracentrifugation (1 h, 100,000 × g, 4 °C) to obtain cytosolic (supernatant) and membrane fractions (pellet, resuspended in buffer [29] containing 1% Triton X; re-centrifuged for 0.5 h, 100,000 × g, 4 °C to remove debris). For detection of membrane-bound NADPH oxidase subunits in hearts, snap-frozen samples of the membrane fractions obtained for detection of NADPH oxidase activity (see above) were used. Probes were subjected to SDS–PAGE and electroblotting on nitrocellulose membranes (BioRad, Hercules, USA). Immunoblotting was performed with antibodies against alpha actinine (rabbit polyclonal, dilution 1:5000; Sigma–Aldrich, Seelze, Germany), p47phox (rabbit polyclonal, dilution 1:500), p67phox (mouse monoclonal, dilution 1:500), eNOS (dilution 1:1000, all three: Transduction Laboratories, Lexington, USA), nox1 (goat polyclonal, dilution 1:100; Santa Cruz Biotechnologies, Santa Cruz, USA), nox2 (i.e., gp91phox, mouse monoclonal, dilution 1:1000), rac1 (mouse monoclonal, dilution 1:1000, both BD Bioscience, San Jose, USA), VASP-Ser239-P (mouse monoclonal, dilution 1,5 μg/ml, Calbiochem, San Diego, USA), dihydrofolate reductase (DHFR, mouse monoclonal, dilution 1:250, RDI division of Fitzgerald, Concord, USA), GTPCH (mouse monoclonal, 1:1000, Abnova, Taipei, Taiwan). Alpha actinine served as loading control. We used secondary antibodies directed towards mouse, goat and rabbit IgG (anti-mouse, anti-goat and anti-rabbit-IgG peroxidase-labeled; Vector, Burlingame, USA). Immunodetections were accomplished with either SuperSignal Substrate (Pierce, Rockford, USA) or ECL Reagent (Amersham, Piscataway, USA). The bands were evaluated by densitometry.
To determine translocation of rac1, p47phox and p67phox, protein homogenates of aortic tissue were divided by ultracentrifugation (1 h, 100,000 × g, 4 °C) to obtain cytosolic (supernatant) and membrane fractions (pellet, resuspended in buffer [29] containing 1% Triton X; re-centrifuged for 0.5 h, 100,000 × g, 4 °C to remove debris).
2.10. Assessment of the activity of the NO/sGC/cGMP pathway
Phosphorylation of the vasodilator-stimulated phosphoprotein (P-VASP) is a surrogate parameter for the activity of the cGMP-dependent kinase I and is suitable to measure NO bioactivity and the integrity of the NO/cGMP signalling pathway.
Prior to snap freezing the aortic segments selected for P-VASP analysis, rings were incubated for 15 min in Krebs–HEPES buffer either with or without acetylcholine (10−6.5 M). Sample preparation and evaluation was conducted according to a published protocol [29].
2.11. Protein tyrosine nitration of prostacycline synthase
Detection of PGI2S nitration was performed as previously described [30]. Briefly, solubilized proteins (3 mg) from the homogenized rat aortic tissue were precleared with protein A sepharose CL-4B. The resulting supernatant was incubated with monoclonal anti-PGI2S antibody (Oxford Biomedical Research, Oxford, MI, USA). Immune complexes were precipitated with 30 μl protein A sepharose CL-4B and washed with 0.5 ml SNNTE (0.5% sucrose, 1% NP-40, NaCl (0.5 mM), Tris (50 mM), EDTA, pH 7.4 (5 mM)). The pellet complex was subjected to Western blot probed with anti-PGI2S antibody and anti-nitrotyrosine antibody (Upstate Biotechnology/Milipore, Billerica, MA, USA).
2.12. Measurement of aortic levels of (6R)-5,6,7,8-tetrahydro-L-biopterin (BH4) and 7,8-dihydrobiopterin (BH2)
Four centimetre of isolated and rinsed aorta was homogenized in ice-cold lysis buffer (0.1 mol/l Tris–HCl, ph 7.8, containing 5 mmol/l ethylenediamine tetra acetic acid, 0.3 mol/l KCl, 5 mmol/l 1,4-dithioerythritol, 0.5 mM Pefabloc, and 0.01% saponin). Samples were oxidised under either acidic conditions (with 0.2 mol/l HCl containing 50 mmol/l I2) or alkaline conditions (with 0.2 mol/l NaOH containing 50 mmol/l I2). Biopterin content was assessed using high-performance liquid chromatography with fluorescence detection (350 nm excitation, 450 nm emission). BH4 concentration was calculated as fmol/μg protein by subtracting the biopterin peak obtained after alkaline oxidation (accounting for BH2) from the biopterin peak obtained after acidic oxidation (accounting for both BH2 and BH4).
2.13. Statistical analysis
Results are expressed as mean±S.E.M. One-way ANOVA (with Bonferroni′s or Dunn’s correction for comparison of multiple means) was used for comparisons of serum parameters, weight gain, vascular responses (EC50 and maximum relaxation), protein expression levels, chemiluminescence counts, biopterin levels and number of EPCs. In tension recordings, the EC50 value for each experiment was obtained by log-transformation. One-way RM ANOVA using all pairwise multiple comparison procedures (HolmSidak method) was used for concentration–relaxation curves in Fig. 1. Each experiment was carried out with 6 to 12 animals per study group. P < 0.05 was considered significant.
Fig. 1.

Vascular function and NO/sGC/cGMP-signalling is improved by HMG-CoA reductase inhibition. (A) Isolated aortic rings (4 mm) were mounted in organ chambers to carry out isometric tension studies. Concentration–relaxation curves in response to acetylcholine (ACh) and nitroglyerin (NTG) were obtained (logarithmic scale of increasing concentration on the x-axis). Percentage of maximal relaxation is denoted on the y-axis. Symbols: broken lines, Ctr (open circles) and Ator (filled circles); continuous lines: STZ (open triangles) and STZ/Ator (filled triangles). (*) p <0.05 STZ vs. Ctr; (†) p <0.05 STZ/Ator vs. STZ (one-way RM ANOVA). (B) Prior to snap freezing, isolated aortic rings were incubated for 15 min either in the presence (black † bars) or absence (grey bars) of acetylcholine (ACh, 0.5 μM). Phosphorylation of vasodilator-stimulated phosphoprotein (P-VASP) was measured using an antibody specific for phosphorylation at serin239. Y-axis: expression as % of Ctr. (*) p <0.05 vs. Ctr; (†) p <0.05 vs. STZ-ACh. Top panel depicts representative original Western blot of P-VASP (ACh stimulated vs. Ctr buffer) levels. Data are mean±S.E.M. of 12–24 (tension studies) and 5–17 (P-VASP) independent experiments.
3. Results
3.1. Serum parameters and body weight
After 7 weeks of diabetes mellitus type 1, STZ-injected animals (STZ) had a significant decrease of plasma insulin levels and a strong increase of blood glucose levels compared to control. STZ-treated animals gained significantly less weight as compared to controls and had a significant increase in triglyceride levels. Atorvastatin had no significant effect on insulin and blood glucose levels, although the latter was slightly reduced in STZ/Ator as compared to STZ. Importantly, levels of low-density and high-density lipoprotein were not different at all in all treatment groups (for exact values and statistics, see Table 1).
Table 1.
Effects of atorvastatin on STZ-induced diabetes mellitus
| Ctr | Ator | STZ | STZ/Ator | |
|---|---|---|---|---|
| Blood glucose (mg/dl) | 190.0±14.01 | 189.0±14.39 | 772.8±64.03* | 653.17±35.81* |
| Insulin (μg/l) | 1.82±0.36 | 2.03±0.49 | 0.05±0.03* | 0.28±0.17* |
| Weight gain (g) | 248.75±12.06 | 227.9±9.77 | 107±18.31* | 122.91±26.2* |
| LDL (mg/dl) | 8.75±0.75 | 11.5±1.55 | 10.6±1.91 | 15.8±0.49 |
| HDL (mg/dl) | 52.5±8.26 | 48.25±1.93 | 63.6±3.29 | 76.4±4.12 |
| Triglycerides (mg/dl) | 89.25±9.47 | 80.25±11.42 | 249.2±71.92* | 354.6±167.1* |
| Circulating EPCs (% of total PBMCs) | 0.63±0.16 | 1.20±0.32 | 0.28±0.03* | 0.60±0.1** |
| Vascular superoxide formationa (counts/min/mg) | 305.74±15.31 | 333.75±20.37 | 496.0±25.78* | 392.74±20.37** |
| NADPH oxidase activityb (counts/min/mg/1000) | 36.20±1.66 | 41.30±3.56* | 73.38±4.81* | 59.1±3.78** |
| eNOS dimer:monomer ratio | 0.32±0.07 | 0.39±0.11 | 0.11±0.03* | 0.32±0.07** |
Lucigenin (5 μM) enhanced chemiluminescence.
Lucigenin (5 μM) enhanced chemiluminescence in the presence of NADPH (200 μM).
p <0.05 vs. Ctr.
p <0.05 vs. STZ.
3.2. Vascular function: isometric tension studies, phosphorylation of VASP and circulating endothelial progenitor cells
In STZ, a marked degree of desensitization to acetylcholine (ACh, endothelial dysfunction) and to nitroglycerin (NTG, nitrate resistance) was established (Fig. 1A, Table 2). Atorvastatin treatment corrected these abnormalities completely, while having no effect at all on the concentration relaxation curve to the endothelium-dependent vasodilator ACh and NTG in control animals. However, since ACh and NTG response are impaired in diabetic animals it is impossible to separate endothelial from smooth muscle dysfunction and accordingly, we will use the term vascular dysfunction in the discussion. It should be noted that impaired NTG potency in diabetic animals could be due to inhibition of mitochondrial aldehyde dehydrogenase (ALDH-2), the redox-sensitive nitrate reductase [31]. However, it was also shown that the potency of the direct NO donor diethylamine NONOate was impaired in diabetic animals compatible with smooth muscular dysfunction [32].
Table 2.
Potency and efficacy of ACh and GTN in isolated aortic rings
| ACh | Ctr | Ator | STZ | STZ/Ator |
|---|---|---|---|---|
| EC50 (10−x M) | 7.42±0.09 | 7.39±0.12 | 7.07±0.12* | 7.36±0.06** |
| Maximum relaxation (%) | 92.9±1.6 | 93.7±1.5 | 82.9±3.6* | 92.0±1.8** |
| GTN | Ctr | Ator | STZ | STZ/Ator |
|---|---|---|---|---|
| EC50 (10−x M) | 7.50±0.04 | 7.32±0.08 | 6.89±0.23* | 7.20±0.17** |
| Max. relaxation (%) | 99.5±0.27 | 97.73±0.37 | 96.81±1.05 | 99.7±0.26 |
p <0.05 vs. Ctr.
p <0.05 vs. STZ/Ator.
We also determined levels of VASP phosphorylated at Ser239 (P-VASP) (Fig. 1B) at baseline and following stimulation with ACh. Basal levels of P-VASP (%±S.E.M.) were not different between the four study groups. Following incubation with 0.5 μM ACh, P-VASP levels increased significantly in Ctr, Ator and STZ/Ator. No increase of P-VASP levels was observed in STZ animals.
We calculated the percentage of circulating EPCs (% of total PBMCs) as an additional marker for endothelial function and the regenerative capacity of the vasculature (see Table 1). In Ctr, EPCs were 0.63±0.16% and dropped in STZ-treated animals to 0.28±0.03% of total PBMCs. Atorvastatin treatment elevated significantly the amount of circulating EPCs in hyperglycemic animals (0.60±0.1%) and to an even greater extent in control animals (1.20±0.32%).
3.3. Oxidative stress: vascular superoxide production, NADPH oxidase activity and expression/assembly, oxidative fluorescent microtopography and tyrosine nitration of prostacyclin synthase
To asses vascular oxidative stress, we measured O2•– production of isolated aortic rings in a single photon counter using lucigenin-enhanced chemiluminescence (see Table 1). Superoxide production in vessels from STZ-treated animals was higher compared to control. Atorvastatin treatment markedly decreased superoxide production in these, but not in control vessels.
In membrane fractions from STZ, NADPH oxidase activity was markedly higher compared to controls (see Table 1). Atorvastatin treatment reduced the chemiluminescence signal in STZ/Ator while having no effect on the NADPH oxidase activity of control vessels. The chemiluminescence signal in response to NADPH was completely blocked by adding diphenyliodonium (DPI, 150 μM, data not shown) and decreased to the level of control by adding low dose DPI (15 μM, data not shown). Importantly, NADH was not able to stimulate the superoxide signal in STZ (5.4±0.25) since vascular Nox-isoforms prefer NADPH as the cofactor. We use NADH to exclude lucigenin-dependent redox cycling, which is most pronounced in the presence of NADH.
To further characterize the topographic localisation of O2•–, we prepared cryosections of isolated aortic segments and stained them with the superoxide sensitive flourescent dye, dihydroethidium (DHE) to obtain fluorescent photomicrographs (see Fig. 2A). STZ treatment increased O2•– formation throughout the vessel wall, in the endothelial cell layer, media and adventitia. Atorvastatin treatment strikingly reduced vascular superoxide. In controls, the NOS inhibitor NG-nitro-L-arginine (L-NNA) increased superoxide staining in the endothelial cell layer due to the quenching of baseline NO. In contrast, the increased staining for O2•– in the endothelial cell layer was blocked by L-NNA in STZ, compatible with an uncoupled eNOS as a significant source of superoxide in diabetes mellitus.
Fig. 2.

Dihydroethidium staining reveals involvement of eNOS uncoupling and prostacyclin synthase nitration the formation of peroxynitrite. (A) Fluorescent photomicrographs of aortic segments of vessels from control animals, streptozotocin-treated animals with and without atorvastatin treatment with (right panel) or without (left panel) preincubation with the eNOS inhibitor NG-nitro-L-arginine (L-NNA, 10 μM). L-NNA increases the DHE staining within endothelial cells in vessels from control rats while decreasing it in vessels from STZ animals, compatible with eNOS uncoupling. Incubation of vessels from STZ animals treated with atorvastatin with L-NNA increased rather than decreased the superoxide signal, indicating that statin treatment prevented eNOS uncoupling. Autofluorescence of the laminae yields a green signal; red fluorescence reflects superoxide. Arrows indicate the endoluminal side (endothelium, E) of the cryosection. Representative photomicrographs of 6 to 8 independent experiments are shown. (B) The burden of peroxynitrite was assessed by measuring protein tyrosine nitration. Therefore, aortic homogenates were immunoprecipitated against prostacyclin synthase (PGI2S) and immunoblotted with an antibody specific for nitrotyrosine adducts (3-NT). Original blots shown are representative for 6 to 8 independent experiments.
The ratio of membranous versus cytosolic localisation of rac1, p47phox and p67phox in homogenates of aortic tissue was significantly increased in STZ as compared to controls and was normalized by atorvastatin treatment (see Fig. 3). The expression of the NADPH oxidase subunits nox1, nox2 and p67phox in heart membrane fractions of STZ was increased in STZ as compared to controls and normalized by atorvastatin treatment (see supplement Fig. i).
Fig. 3.
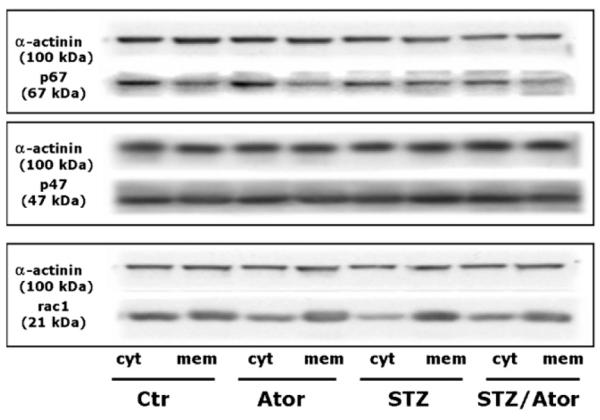
Translocation of NADPH oxidase subunits is prevented by HMGCoA reductase inhibition. Cytosolic and membranous fractions of aortic homogenate were prepared by ultracentrifugation. Expression of rac1, p47phox and p67phox was assessed by Western blot in both fractions. The ratio of membranous cytosolic expression is depicted after densitometric analysis of the respective blots. Top left panel depicts representative original Western blots of the cytosolic and membranous fractions. Data are mean±S.E.M. of 5-12 independent experiments. (*) p < 0.05 vs. Ctr; (†) p <0.05 vs. STZ.
In an additional experiment, we assessed nitrosative stress by analyzing the levels of PGI2S-3NT (see Fig. 2B). We detected a marked increase of tyrosine nitration in STZ as compared to control, all of which was normalized by atorvastatin treatment.
3.4. Uncoupling of endothelial nitric oxide synthase: vascular BH4 levels, vascular eNOS, GTPCH-I and DHFR expression
In aortas of STZ rats, BH4 levels (fmol/μg) were markedly reduced as compared to controls. Treatment with atorvastatin normalized BH4 levels. BH2 levels showed a reverse pattern: they were significantly elevated in aortas from STZ rats as compared to controls and normalized by atorvastatin treatment (see Fig. 4D).
Fig. 4.

eNOS coupling is preserved by HMG-CoA reductase inhibition. Protein expression of aortic endothelial nitric oxide synthase (eNOS) GTP cyclohydrolase I (GTPCH-I) and dihydrofolate reductase (DHFR) was measured using Western blot. Bands were normalized on the loading probe α-actinin and evaluated using densitometry. Original blots shown are representative for 5 to 11 independent experiments. Data are mean±S.E.M. of 5 to 11 independent experiments. (*) p <0.05 vs. Ctr; (†) p < 0.05 vs. STZ. Levels of (6R)-5,6,7,8-tetrahydro-L-biopterin (BH4) and 7,8-dihydrobiopterin (BH2) were measured by HPLC and expressed as fmol/μg protein. Data are mean±S.E.M. of 10 to 16 independent experiments. (*) p <0.05 vs. Ctr; (†) p < 0.05 vs. STZ.
We then determined aortic protein expression (%Ctr±S.E.M.) of eNOS, GTPCH-I and DHFR (Fig. 4A–C). Expression of eNOS was significantly increased in vessels from control animals treated with atorvastatin and STZ as compared to controls. Increased eNOS expression in vessels from STZ animals was reduced to normal by atorvastatin treatment. The eNOS dimer:monomer ratio as assessed by non-reducing SDS–PAGE and Western blot was diminished in STZ and recovered atorvastatin treatment (supplement, Fig. ii and Table 1). In vessels from STZ-treated animals, GTPCH-I expression levels decreased compared to controls. Atorvastatin treatment of STZ animals normalized GTPCH-I expression. Atorvastatin treatment of control animals had no effect on aortic GTPCH-I expression. STZ treatment increased vascular DHFR expression as compared to controls. In these vessels, statin treatment decreased DHFR expression, while having no significant effect on the expression of DHFR in control vessels.
4. Discussion
The results of the present studies demonstrate that eNOS uncoupling in the setting of diabetes mellitus is mediated at least in part by a downregulation of the key enzyme for the synthesis of BH4, the GTPCH-I. Importantly, chronic treatment with the HMG-CoA reductase inhibitor atorvastatin normalizes vascular GTPCH-I expression and simultaneously causes an inhibition of vascular superoxide production via prevention of eNOS uncoupling, thereby leading to a normalization of vascular (endothelial and smooth muscle) function, although plasma LDL levels were not modified by atorvastatin therapy at all. These findings add further mechanistic insight into the importance of pleiotropic mechanisms of HMG-CoA reductase inhibitors leading to improved prognosis in patients with diabetes mellitus.
4.1. Vascular dysfunction, oxidative stress and diabetes mellitus
Increased oxidative stress is a hallmark of cardiovascular disease and is evident in the vasculature of hyperglycemic patients and animal models of diabetes mellitus already at early stages of the disease. Besides xanthine oxidase [9] and mitochondria [10], the vascular NADPH oxidase [4–6] and an uncoupled eNOS [5,7,8] have been shown to represent the predominant sources of vascular and myocardial super-oxide production in diabetes mellitus. In the animal model of STZ diabetes, we were recently able to demonstrate that vascular dysfunction is associated with increased expression of the NADPH oxidase subunits nox1 and nox2 (gp91phox)in vascular and myocardial tissue along with increased activity of this enzyme [5,6]. Our group was also the first to provide evidence for an uncoupled eNOS in hyperglycemic vessels [5]. One of the most attractive concepts so far explaining eNOS uncoupling is an intracellular depletion of vascular BH4 levels. This concept is supported by experiments showing that the administration of the BH4 precursor sepiapterin or a BH4-repleting substance such as 5-methyltetrahydrofolate is able to reduce vascular superoxide production in cultured endothelial cells, in animal experiments but also in human tissue [33] associated with a restoration of NO production and endothelial function, respectively.
The results of the present studies further support this uncoupling concept as one of the reasons for vascular dysfunction and increased oxidative stress in the vessels of hyperglycemic animals. Vascular dysfunction in hyperglycemic rats was associated with increased superoxide production, and we could also show that the eNOS inhibitor L-NNA eliminated the DHE staining primarily in the endothelial cell layer (Fig. 2). These observations were in accordance with previous reports on increased endothelial superoxide formation in diabetic vessels from humans [4] as well as murine and rat aorta [5,34], which were based on quantitative methods. Importantly, we also established a marked decrease in vascular BH4 levels (Fig. 4), increased eNOS expression (Fig. 4) and found evidence for a decreased dimer:monomer ratio (supplement, Fig. iv), a condition which has also been demonstrated to be associated with or even causing the uncoupling phenomenon [35,36].
There is a continued discussion whether a biochemical destruction of BH4, e.g., due to an interaction with the NO/O2•– intermediate peroxynitrite and/or a diminished BH4 synthesis, e.g., due to a downregulation of the BH4-synthesizing enzyme GTPCH-I or both contributes primarily to the phenomenon of eNOS uncoupling. With the present study we focussed on the vascular GTPCH-I expression and BH4 levels since previous in vitro studies have demonstrated that hyperglycemic conditions lead to a downregulation of the GTPCH-I associated with increased endothelial superoxide production and that this condition could be corrected by endothelium targeted overexpression of GTPCH-I [34]. Indeed, in our hands 7 weeks of diabetes mellitus type 1 lead to a substantial downregulation of the expression of this BH4-synthesizing enzyme and to a substantial decrease in vascular BH4 levels (Fig. 4). Interestingly, the recovering enzyme dihydrofolate reductase (DHFR) was up- rather than downregulated in STZ diabetes (Fig. 4), but was apparently unable to prevent a depletion of BH4. This result is in line with previous observations showing that DHFR, representing part of the so-called “salvage pathway” of BH4, is not sufficient to counteract depletion of BH4 [37,38]. Bearing in mind that the BH2 levels were increased along with decreased BH4, the compensatory activation of the salvage pathway points towards an oxidation of tetrahydrobiopterin in STZ, most likely due to increased vascular NADPH oxidase activity as evidenced by our results (Figs. 2 and 3) and by recent studies in the literature [35].
Increased oxidative stress mediated by increased NADPH oxidase activity and eNOS uncoupling lead to the phenomenon of “nitrate resistance” in vessels from STZ rats (Fig. 1), to a decreased ACh-stimulated activation of the cGMP-dependent kinase (P-VASP) and to increased tyrosine nitration of the prostacyclin synthase-mediated by peroxynitrite [39,40]. We also established a substantial decrease in the levels of circulating EPCs, all of which may have also contributed to vascular dysfunction in the setting of diabetes mellitus [41]. Interestingly, in a recent paper Thum and coworkers showed eNOS uncoupling in EPCs as a feature of EPC malfunction in diabetes [42].
4.2. Effects of statin therapy on vascular function and eNOS uncoupling in hyperglycemic animals
It is known that statin therapy has a profound impact on vascular function, but also on prognosis in patients with diabetes mellitus. Mechanisms responsible for these beneficial effects include reductions in LDL levels but also pleiotropic effects which include stimulatory effects on eNOS expression and NO production [14,15,43,44], but also simultaneously inhibitory effects on vascular superoxide production, e.g., by an inhibition of the activity and expression of the vascular NADPH oxidase [12].
Indeed, when treating hyperglycemic animals with the HMG-CoA reductase inhibitor atorvastatin we found a quite remarkable effect of statin treatment on vascular dysfunction, nitrate resistance, P-VASP levels, PGI2S nitration and circulating EPCs levels. These findings were associated with a substantial statin-induced reduction in oxidative stress due to an inhibition of NADPH oxidase expression and activity, and due to a reversal of eNOS uncoupling. It should be noted that statins increase the adhesion of EPCs, which may contribute to the detection of an increased number of EPCs under statin therapy [23,45].
4.3. By which mechanism does atorvastatin prevent eNOS uncoupling?
Recently Ding et al. investigated the effects of high glucose on NO and superoxide formation on the expression of GTPCH-I and the endothelial NADPH oxidase [19]. High glucose concentrations increased superoxide, decreased BH4 levels and markedly decreased GTPCH-I expression and activity, all of which was corrected by pretreatment of these cells with atorvastatin. The results of the present studies go along with this observation. Atorvastatin treatment of hyperglycemic rats in a concentration of 20 mg/kg for a 7-week period resulted in a restoration of vascular BH4 levels and a normalization of the expression of GTPCH-I. Accordingly, eNOS uncoupling as assessed by the DHE method was prevented (Fig. 2). Further evidence for a prevention for eNOS uncoupling was provided by an increase in the eNOS dimer:monomer ratio (see supplement, Fig. iv).
The mechanisms underlying the prevention of a downregulation of GTPCH-I by statin therapy has been addressed recently by Hattori et al. [20]. With their studies with cultured vascular endothelial cells, the authors found that statins elevate GTPCH-I at the transcriptional level [20].
Another mechanism how statins may prevent eNOS uncoupling is the reduction in oxidative stress due to an inhibition of NADPH oxidase expression and activity. More recently, we have proposed that increased NADPH oxidasemediated superoxide production may lead to increased formation of the NO-superoxide reaction product peroxynitrite, which may cause BH4 oxidation and subsequently lead to increased formation of the so called BH3 radical. Thus NADPH oxidase-mediated superoxide may act as a kindling radical favoring eNOS uncoupling via stimulating increased formation of vascular ONOO– [5], a concept that has recently been strengthened by Xu and coworkers [35]. Indeed, we were able to demonstrate that atorvastatin is able to decrease vascular levels of tyrosine nitrated PGI2S, which can be used as a marker for decreased vascular ONOO– formation. In addition, reduction of PGI2S-3NT may also improve vascular function by increasing the formation of PGI22.
Statin therapy has been reported to cause an increase in the expression of eNOS in endothelial cells but also in the endothelial cell layer of in vivo-treated animals. The results of the present studies go along with this concept, because statin therapy increased eNOS expression in vessels from non-hyperglycemic animals by about 100% (Fig. 4). As described before, the condition of vascular (endothelial) dysfunction in diabetes is associated with a paradoxical increase rather than decrease of a functionally uncoupled eNOS. The increase in eNOS expression has been attributed to the increased formation of the superoxide dismutation product hydrogen peroxide (H2O2), which increases eNOS expression at the transcriptional and also translational level [16–18]. In our study, treatment of STZ rats with atorvastatin led to a reduction in eNOS expression, which might be explained by the reduction in oxidative stress and the subsequently decreased formation of the eNOS expression stimulus H2O2.
Another interesting observation was that treatment of hyperglycemic animals with atorvastatin was able to correct the decreased number of circulating EPCs. This is in line with previous observations [42,45–48] and may also reflect one mechanism by which statin therapy improves vascular dysfunction in the setting of diabetes, especially when bearing in mind, that eNOS uncoupling seems to be an important source of superoxide in EPCs, too [42].
It is important to note that these positive effects on vascular function by statins have been achieved, although the plasma LDL levels were not modified, suggesting that pleiotropic effects as mentioned above rather than effects secondary to lipid lowering cause these beneficial phenomena. Recent data suggest that diabetes triggers proteasome-dependent degradation of GTPCH-I [49] and statins may interfere with this break-down.
5. Conclusion and clinical implications
The results of the present studies demonstrate for the first time that in an in vivo model of diabetes, vascular dysfunction is associated with an uncoupled eNOS in the presence of a marked reduction of the BH4-synthesizing enzyme GTPCH-I. Statin treatment improved vascular function, reduced oxidative stress, prevented upregulation of the NADPH oxidase and eNOS uncoupling and normalized vascular BH4 levels and the expression of GTPCH-I (Fig. 5). Since at the same time plasma LDL levels were not modified at all, we conclude that predominantly pleiotropic effects of atorvastatin account for the improvement of vascular function and may indicate that in the setting of diabetes mellitus, statin therapy should be prescribed irrespective of the plasma LDL levels. Future clinical studies, however, have to be performed to test whether this concept may be translated into the clinical situation.
Fig. 5.

Scheme depicting the proposed mechanisms underlying improvement of vascular dysfunction by HMG-CoA reductase inhibition in the setting of diabetes mellitus. Hyperglycemia induces an increase in NADPH oxidase activity and expression, most probably in a protein kinase C-dependent fashion, leading to an increase in superoxide (O2•–) formation. After dismutation to hydrogen peroxide (H2O2), eNOS mRNA expression is enhanced. Intracellular tetrahydrobiopterin (BH4) content is depleted, e.g., via oxidation of BH4 to dihydrobiopterin (BH2) and/or by downregulation of the key enzyme in the de novo synthesis of BH4, the GTP cyclohydrolase I (GTPCH-I) subsequently leading to eNOS uncoupling. This further increases the O2•– and ONOO– formation in a positive feedback fashion, thereby causing a depletion of circulating endothelial progenitor cells (EPCs), a reduction of bioactive nitric oxide (NO), tyrosine nitration of the prostacyclin synthase (PGI2S-3NT) and vascular dysfunction. HMG-CoA reductase inhibition normalizes vascular dysfunction and reduces oxidative stress by upregulating GTPCH-I expression and by inhibiting activation of the vascular NADPH oxidase.
Supplementary Material
Acknowledgement
The expert technical assistance of Jöorg Schreiner, Nicole Schramm and Alice Habermaier is gratefully acknowledged. This study contains results that are part of the doctoral thesis work of Moritz Brandt. This study was supported by a research grant from Pfizer, Inc., USA, to Thomas Müunzel and by the European Vascular Genomic Network, a Network of Excellence supported by the European Community’s sixth Framework Program (Contract LSHMCT-2003-503254). The work was also supported in part by the grants from National Institutes of Health NIH grants (HL079584, HL080499, and HL074399), a research award from the American Diabetes Association and a research award from the Juvenile Diabetes Research Foundation (M.Z.).
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.atherosclerosis.2007.10.003.
References
- [1].Jay D, Hitomi H, Griendling KK. Oxidative stress and diabetic cardiovascular complications. Free Radic Biol Med. 2006;40:183–92. doi: 10.1016/j.freeradbiomed.2005.06.018. [DOI] [PubMed] [Google Scholar]
- [2].Heitzer T, Finckh B, Albers S, et al. Beneficial effects of alpha-lipoic acid and ascorbic acid on endothelium-dependent, nitric oxidemediated vasodilation in diabetic patients: relation to parameters of oxidative stress. Free Radic Biol Med. 2001;31:53–61. doi: 10.1016/s0891-5849(01)00551-2. [DOI] [PubMed] [Google Scholar]
- [3].Ting HH, Timimi FK, Boles KS, et al. Vitamin C improves endothelium-dependent vasodilation in patients with non-insulindependent diabetes mellitus. J Clin Invest. 1996;97:22–8. doi: 10.1172/JCI118394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Guzik TJ, Mussa S, Gastaldi D, et al. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–62. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- [5].Hink U, Li H, Mollnau H, et al. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:E14–22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- [6].Wendt MC, Daiber A, Kleschyov AL, et al. Differential effects of diabetes on the expression of the gp91phox homologues nox1 and nox4. Free Radic Biol Med. 2005;39:381–91. doi: 10.1016/j.freeradbiomed.2005.03.020. [DOI] [PubMed] [Google Scholar]
- [7].Du XL, Edelstein D, Dimmeler S, et al. Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J Clin Invest. 2001;108:1341–8. doi: 10.1172/JCI11235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Kuzkaya N, Weissmann N, Harrison DG, Dikalov S. Interactions of peroxynitrite, tetrahydrobiopterin, ascorbic acid, and thiols: implications for uncoupling endothelial nitric-oxide synthase. J Biol Chem. 2003;278:22546–54. doi: 10.1074/jbc.M302227200. [DOI] [PubMed] [Google Scholar]
- [9].Desco MC, Asensi M, Marquez R, et al. Xanthine oxidase is involved in free radical production in type 1 diabetes: protection by allopurinol. Diabetes. 2002;51:1118–24. doi: 10.2337/diabetes.51.4.1118. [DOI] [PubMed] [Google Scholar]
- [10].Bindokas VP, Kuznetsov A, Sreenan S, et al. Visualizing superoxide production in normal and diabetic rat islets of Langerhans. J Biol Chem. 2003;278:9796–801. doi: 10.1074/jbc.M206913200. [DOI] [PubMed] [Google Scholar]
- [11].Forstermann U, Munzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation. 2006;113:1708–14. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- [12].Wassmann S, Laufs U, Baumer AT, et al. HMG–CoA reductase inhibitors improve endothelial dysfunction in normocholesterolemic hypertension via reduced production of reactive oxygen species. Hypertension. 2001;37:1450–7. doi: 10.1161/01.hyp.37.6.1450. [DOI] [PubMed] [Google Scholar]
- [13].Maeso R, Aragoncillo P, Navarro–Cid J, et al. Effect of atorvastatin on endothelium-dependent constrictor factors in dyslipidemic rabbits. Gen Pharmacol. 2000;34:263–72. doi: 10.1016/s0306-3623(00)00074-4. [DOI] [PubMed] [Google Scholar]
- [14].Zhou MS, Jaimes EA, Raij L. Atorvastatin prevents end-organ injury in salt-sensitive hypertension: role of eNOS and oxidant stress. Hypertension. 2004;44:186–90. doi: 10.1161/01.HYP.0000136395.06810.cf. [DOI] [PubMed] [Google Scholar]
- [15].Laufs U, La Fata V, Plutzky J, Liao JK. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation. 1998;97:1129–35. doi: 10.1161/01.cir.97.12.1129. [DOI] [PubMed] [Google Scholar]
- [16].Cai S, Khoo J, Mussa S, Alp NJ, Channon KM. Endothelial nitric oxide synthase dysfunction in diabetic mice: importance of tetrahydrobiopterin in eNOS dimerisation. Diabetologia. 2005;48:1933–40. doi: 10.1007/s00125-005-1857-5. [DOI] [PubMed] [Google Scholar]
- [17].Drummond GR, Cai H, Davis ME, Ramasamy S, Harrison DG. Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression by hydrogen peroxide. Circ Res. 2000;86:347–54. doi: 10.1161/01.res.86.3.347. [DOI] [PubMed] [Google Scholar]
- [18].Zanetti M, Barazzoni R, Stebel M, et al. Dysregulation of the endothelial nitric oxide synthase-soluble guanylate cyclase pathway is normalized by insulin in the aorta of diabetic rat. Atherosclerosis. 2005;181:69–73. doi: 10.1016/j.atherosclerosis.2005.01.011. [DOI] [PubMed] [Google Scholar]
- [19].Ding QF, Hayashi T, Packiasamy AR, et al. The effect of high glucose on NO and O2through endothelial GTPCH1 and NADPH oxidase. Life Sci. 2004;75:3185–94. doi: 10.1016/j.lfs.2004.06.005. [DOI] [PubMed] [Google Scholar]
- [20].Hattori Y, Nakanishi N, Akimoto K, Yoshida M, Kasai K. HMG-CoA reductase inhibitor increases GTP cyclohydrolase I mRNA and tetrahydrobiopterin in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2003;23:176–82. doi: 10.1161/01.atv.0000054659.72231.a1. [DOI] [PubMed] [Google Scholar]
- [21].Hattori Y, Nakanishi N, Kasai K. Statin enhances cytokine-mediated induction of nitric oxide synthesis in vascular smooth muscle cells. Cardiovasc Res. 2002;54:649–58. doi: 10.1016/s0008-6363(02)00266-3. [DOI] [PubMed] [Google Scholar]
- [22].Hill JM, Zalos G, Halcox JP, et al. Circulating endothelial progenitor cells, vascular function, and cardiovascular risk. N Engl J Med. 2003;348:593–600. doi: 10.1056/NEJMoa022287. [DOI] [PubMed] [Google Scholar]
- [23].Thum T, Fraccarollo D, Galuppo P, et al. Bone marrow molecular alterations after myocardial infarction: Impact on endothelial progenitor cells. Cardiovasc Res. 2006;70:50–60. doi: 10.1016/j.cardiores.2006.01.002. [DOI] [PubMed] [Google Scholar]
- [24].Romagnani P, Annunziato F, Liotta F, et al. CD14 + CD34low cells with stem cell phenotypic and functional features are the major source of circulating endothelial progenitors. Circ Res. 2005;97:314–22. doi: 10.1161/01.RES.0000177670.72216.9b. [DOI] [PubMed] [Google Scholar]
- [25].Thum T, Hoeber S, Froese S, et al. Age-dependent impairment of endothelial progenitor cells is corrected by growth-hormone-mediated increase of insulin-like growth-factor-1. Circ Res. 2007;100:434–43. doi: 10.1161/01.RES.0000257912.78915.af. [DOI] [PubMed] [Google Scholar]
- [26].Skatchkov MP, Sperling D, Hink U, et al. Validation of lucigenin as a chemiluminescent probe to monitor vascular superoxide as well as basal vascular nitric oxide production. Biochem Biophys Res Commun. 1999;254:319–24. doi: 10.1006/bbrc.1998.9942. [DOI] [PubMed] [Google Scholar]
- [27].Daiber A, August M, Baldus S, et al. Measurement of NAD(P)H oxidase-derived superoxide with the luminol analogue L-012. Free Radic Biol Med. 2004;36:101–11. doi: 10.1016/j.freeradbiomed.2003.10.012. [DOI] [PubMed] [Google Scholar]
- [28].Rajagopalan S, Kurz S, Munzel T, et al. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–23. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Oelze M, Mollnau H, Hoffmann N, et al. Vasodilator-stimulated phosphoprotein serine 239 phosphorylation as a sensitive monitor of defective nitric oxide/cGMP signaling and endothelial dysfunction. Circ Res. 2000;87:999–1005. doi: 10.1161/01.res.87.11.999. [DOI] [PubMed] [Google Scholar]
- [30].Zou M, Jendral M, Ullrich V. Prostaglandin endoperoxide-dependent vasospasm in bovine coronary arteries after nitration of prostacyclin synthase. Br J Pharmacol. 1999;126:1283–92. doi: 10.1038/sj.bjp.0702434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wenzel P, Hink U, Oelze M, et al. Role of reduced lipoic acid in the redox regulation of mitochondrial aldehyde dehydrogenase (ALDH2) activity. Implications for mitochondrial oxidative stress and nitrate tolerance. J Biol Chem. 2007;282:792–9. doi: 10.1074/jbc.M606477200. [DOI] [PubMed] [Google Scholar]
- [32].Schafer A, Flierl U, Vogt C, et al. Telmisartan improves vascular function and reduces platelet activation in rats with streptozotocin-induced diabetes mellitus. Pharmacol Res. 2007;56:217–23. doi: 10.1016/j.phrs.2007.05.009. [DOI] [PubMed] [Google Scholar]
- [33].Antoniades C, Shirodaria C, Warrick N, et al. 5-methyltetrahydrofolate rapidly improves endothelial function and decreases superoxide production in human vessels: effects on vascular tetrahydrobiopterin availability and endothelial nitric oxide synthase coupling. Circulation. 2006;114:1193–201. doi: 10.1161/CIRCULATIONAHA.106.612325. [DOI] [PubMed] [Google Scholar]
- [34].Alp NJ, Mussa S, Khoo J, et al. Tetrahydrobiopterin-dependent preservation of nitric oxide-mediated endothelial function in diabetes by targeted transgenic GTP-cyclohydrolase I overexpression. J Clin Invest. 2003;112:725–35. doi: 10.1172/JCI17786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Xu J, Xie Z, Reece R, Pimental D, Zou MH. Uncoupling of endothelial nitric oxidase synthase by hypochlorous acid: role of NAD(P)H oxidase-derived superoxide and peroxynitrite. Arterioscler Thromb Vasc Biol. 2006;26:2688–95. doi: 10.1161/01.ATV.0000249394.94588.82. [DOI] [PubMed] [Google Scholar]
- [36].Zou MH, Shi C, Cohen RA. Oxidation of the zinc-thiolate complex and uncoupling of endothelial nitric oxide synthase by peroxynitrite. J Clin Invest. 2002;109:817–26. doi: 10.1172/JCI14442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hasegawa H, Sawabe K, Nakanishi N, Wakasugi OK. Delivery of exogenous tetrahydrobiopterin (BH4) to cells of target organs: role of salvage pathway and uptake of its precursor in effective elevation of tissue BH4. Mol Genet Metab. 2005;86(Suppl. 1):S2–10. doi: 10.1016/j.ymgme.2005.09.002. [DOI] [PubMed] [Google Scholar]
- [38].Moens AL, Kass DA. Tetrahydrobiopterin and cardiovascular disease. Arterioscler Thromb Vasc Biol. 2006;26:2439–44. doi: 10.1161/01.ATV.0000243924.00970.cb. [DOI] [PubMed] [Google Scholar]
- [39].Hink U, Oelze M, Kolb P, et al. Role for peroxynitrite in the inhibition of prostacyclin synthase in nitrate tolerance. J Am Coll Cardiol. 2003;42:1826–34. doi: 10.1016/j.jacc.2003.07.009. [DOI] [PubMed] [Google Scholar]
- [40].Zou MH, Shi C, Cohen RA. High glucose via peroxynitrite causes tyrosine nitration and inactivation of prostacyclin synthase that is associated with thromboxane/prostaglandin H(2) receptor-mediated apoptosis and adhesion molecule expression in cultured human aortic endothelial cells. Diabetes. 2002;51:198–203. doi: 10.2337/diabetes.51.1.198. [DOI] [PubMed] [Google Scholar]
- [41].Fadini GP, Miorin M, Facco M, et al. Circulating endothelial progenitor cells are reduced in peripheral vascular complications of type 2 diabetes mellitus. J Am Coll Cardiol. 2005;45:1449–57. doi: 10.1016/j.jacc.2004.11.067. [DOI] [PubMed] [Google Scholar]
- [42].Thum T, Fraccarollo D, Schultheiss M, et al. Endothelial nitric oxide synthase uncoupling impairs endothelial progenitor cell mobilization and function in diabetes. Diabetes. 2007;56:666–74. doi: 10.2337/db06-0699. [DOI] [PubMed] [Google Scholar]
- [43].Kureishi Y, Luo Z, Shiojima I, et al. The HMG-CoA reductase inhibitor simvastatin activates the protein kinase Akt and promotes angiogenesis in normocholesterolemic animals. Nat Med. 2000;6:1004–10. doi: 10.1038/79510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Laufs U, Liao JK. Post-transcriptional regulation of endothelial nitric oxide synthase mRNA stability by Rho GTPase. J Biol Chem. 1998;273:24266–71. doi: 10.1074/jbc.273.37.24266. [DOI] [PubMed] [Google Scholar]
- [45].Vasa M, Fichtlscherer S, Adler K, et al. Increase in circulating endothelial progenitor cells by statin therapy in patients with stable coronary artery disease. Circulation. 2001;103:2885–90. doi: 10.1161/hc2401.092816. [DOI] [PubMed] [Google Scholar]
- [46].Assmus B, Urbich C, Aicher A, et al. HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circ Res. 2003;92:1049–55. doi: 10.1161/01.RES.0000070067.64040.7C. [DOI] [PubMed] [Google Scholar]
- [47].Dimmeler S, Aicher A, Vasa M, et al. HMG-CoA reductase inhibitors (statins) increase endothelial progenitor cells via the PI 3-kinase/Akt pathway. J Clin Invest. 2001;108:391–7. doi: 10.1172/JCI13152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Llevadot J, Murasawa S, Kureishi Y, et al. HMG-CoA reductase inhibitor mobilizes bone marrow-derived endothelial progenitor cells. J Clin Invest. 2001:399–405. doi: 10.1172/JCI13131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Xu J, Wu Y, Song P, et al. Proteasome-dependent degradation of guanosine 5′-triphosphate cyclohydrolase I causes tetrahydrobiopterin deficiency in diabetes mellitus. Circulation. 2007;116:944–53. doi: 10.1161/CIRCULATIONAHA.106.684795. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.