

Abstract
Background
In a mouse model of focal cerebral ischemia, infarct volume is highly variable and strain dependent, but the natural genetic determinants responsible for this difference remain unknown. To identify genetic determinants regulating ischemic neuronal damage and to dissect apart the role of individual genes and physiological mechanisms in infarction in mice, we performed quantitative trait locus analysis of surgically induced cerebral infarct volume.
Methods and Results
After permanent occlusion of the distal middle cerebral artery, infarct volume was determined for 16 inbred strains of mice, chromosome substitution strains, and for 2 intercross cohorts, F2 (B6×BALB/c) and F2 (B6×SWR/J). Genome-wide linkage analysis was performed for infarct volume as a quantitative trait. Infarct volume varied up to 30-fold between strains, with heritability estimated at 0.88. Overall, 3 quantitative trait locus were identified that modulate infarct volume, with a major locus (Civq1) on chromosome 7 accounting for >50% of the variation, with a combined LOD score of 21.7. Interval-specific single nucleotide polymorphism haplotype analysis for Civq1 results in 12 candidate genes.
Conclusions
The extent of ischemic tissue damage after distal middle cerebral artery occlusion in inbred strains of mice is modulated by genetic variation mapping to at least 3 different loci. A single locus on chromosome 7 determines the majority of the observed variation in the trait. This locus seems to be identical to LSq1, a locus conferring limb salvage and reperfusion in a mouse model of hindlimb ischemia. The identification of the genes underlying these loci may uncover novel genetic and physiological pathways that modulate cerebral infarction and provide new targets for therapeutic intervention in ischemic stroke, and possibly other ischemic diseases.
Keywords: cerebral infarction, genetics, quantitative trait loci, stroke
Stroke results in cerebral ischemia with subsequent neural tissue injury (infarct) in the perfusion territory of the occluded cerebral artery. In general, the severity of tissue damage depends on several factors related to the cerebrovascular circulation including the duration of occlusion, the site of occlusion along the affected vessel, the size of the territory perfused by the vessel, and the extent of the collateral circulation.1 Experimental studies of stroke in rodents have provided an extensive body of new data leading to the current understanding of the pathophysiological mechanisms underlying cerebral infarction. Several genes have been identified that alter the volume of the infarct when focal cerebral ischemia is surgically induced in genetically manipulated mouse models.2 Neuronal survival can be strongly influenced by genes involved in excitotoxicity, inflammation, and endogenous neuroprotection pathways.2-5 These combined data have provided a large and diverse set of genes and pathways that have the potential to modulate ischemic cell death.6 Unfortunately, evidence solely based from gene knockout or transgenic mice does not always provide insight into the mechanisms involved in the natural disease state. Experimental gene deletion or transgenic overexpression creates an artificial genetic and physiological state that can be far removed from that found in naturally occurring diseases. Thus, risk factors and the pathophysiological differences that determine the outcome of ischemic stroke in the natural state remain uncertain.
In the well-established mouse model of focal cerebral ischemia, innate sensitivity to focal cerebral ischemia has been shown to be highly variable and strain dependent.7-10 In this study, we have extended these observations to 16 inbred mouse strains, where we found a wide range of difference in infarct volume after permanent distal middle cerebral artery (MCA) occlusion, providing further evidence that the innate response to focal cerebral ischemia is under strong genetic control. Although the studies in genetically manipulated rodent models suggest various mechanisms that can influence the extent of ischemic tissue damage, the genetic determinants and the underlying pathophysiological mechanisms responsible for the differential outcome of ischemic stroke in the inbred strains of mice remain unknown. The observed phenotypic differences among strains may be attributable to multiple mechanisms, each driven by sequence variation at distinct genes. To identify genetic determinants critical to ischemic brain damage and to dissect apart the role of individual genes and physiological mechanisms in infarction in mice, we performed quantitative trait locus (QTL) analysis of surgically induced cerebral infarction.
Methods
Mice
All inbred strain of mice were obtained from the Jackson Laboratory (Bar Harbor, Me) either directly or bred locally breeding pairs of each strain. Mice were age-matched (12±1 week) and sex-matched for all experiments. Studies were performed under protocols approved by the Animal Care and Use Committee of Duke University.
Surgical Procedure
Focal cerebral ischemia was induced by direct occlusion of the distal MCA as detailed in previous publications,8,9 with the following modifications. Briefly, mice were anesthetized with ketamine (100 mg/kg) and xylazine (5 mg/kg), and the right MCA was exposed by a 0.5-cm vertical skin incision midway between the right eye and ear. After the temporalis muscle was split, a 2-mm burr hole was drilled at the junction of the zygomatic arch and the squamous bone. While visualizing with a stereomicroscope, the right MCA distal to the lenticulostriate branches was electrocauterized using a microcauterizer (Fine Science Tools). The coagulated MCA segment was then transected with Vannas scissors to verify that the occlusion was permanent. The surgical site was closed with 6-0 sterile nylon sutures, and 4% lidocaine cream was applied. Animals were maintained at 37°C during and after surgery until fully recovered from anesthesia, when they were returned to their cages and allowed free access to food and water. All mice were housed in an air-ventilated room with ambient temperature maintained at 24±0.5°C.
Measurement of Infarct Volume
Twenty-four hours after surgery, the animal was euthanized and the brain was removed and sliced into 1-mm coronal sections using a brain matrix. Slices were stained with TTC as previously described.11,12 Infarct volumes were calculated by measuring infarct areas on the separate slices, multiplying areas by slice thickness, and summing all slices; this “indirect” morphometric method corrects for edematous swelling.11
Statistical Analysis
Infarct volumes were expressed as mean±SD and compared among mouse strains using 1-way ANOVA or nonparametric Kruskal–Wallis tests. Heritability H2[(genetic (interstrain) variance)/(genetic variance+environmental (intrastrain) variance)] was estimated using 1-way ANOVA.
Genotyping
Single nucleotide polymorphism (SNP) genotyping was performed using the GoldenGate genotyping assay using the 377 genome-wide mouse SNP panel (Illumina). Reported genetic map positions were retrieved from the SNP database (build 37.1) of NCBI.
Linkage Analysis
Genome-wide scans were plotted using the J/QTL mapping program, version 1.2.1 (http://research.jax.org/faculty/churchill/). Suggestive (P=0.63) and significant (P=0.05) thresholds were established empirically for each phenotypic trait by 1000 permutation tests using all informative markers.13 QTLs over suggestive threshold value were named in accordance with the International Committee on Standard Genetic Nomenclature for Mice (http://www.informatics.jax.org/mgihome/nomen). The percentage of total trait variance attributable to each locus was determined using the Fit QTL function provided within the J/QTL software.
Interval-Specific SNP Haplotype Analysis
For the 31.2-Mb interval on chromosome 7, SNP data were obtained from the Mouse Phenome Database (http://phenome.jax.org/), Perlegen mouse SNP haplotype analysis browser (http://mouse.perlegen.com/mouse/mousehap.html), and the Center for Genome Dynamics (http://cgd.jax.org/). Physical map position was based on the genomic sequence from the NCBI build 37. Haplotype blocks were defined by 3 or more adjacent14 informative SNPs shared between the large infarct strains (BALB/c, A/J, and SWR/J), which differed from the haplotype for B6.
Results
Variability in the Outcomes of Focal Cerebral Ischemia in Different Genetic Backgrounds
To maximize survival and the reproducibility of the extent of ischemic tissue damage that is critical for any measurement of a quantitative trait, we chose direct occlusion of the right distal MCA, which exclusively and permanently eliminates blood flow to tissue distal to the occlusion.15,16 In contrast to proximal artery occlusion models, disruption of the distal MCA limits ischemic tissue damage to frontal and parietal cortex, reducing ischemia-associated death and providing a compact territory of tissue damage for quantitative measurements.16 Distal occlusion of the MCA beyond the Circle of Willis also eliminates any potential effects of anatomic variation in the anterior and posterior commissural arteries.3,8,17 Permanent occlusion of distal MCA does not directly affect flow in any other major artery, maximizes postsurgical survival, and avoids the variable size of the cortical infarct that can result from the suture ligation method.8,9,16 Infarct volume was measured at 24 hours, the time point showing the maximum size.16
As previously described for these 2 strains,7-10 we observed that infarct volumes 24 hours after MCA occlusion were significantly different between inbred strains C57BL/6J (hereafter B6) and BALB/cByJ (BALB/c; Figure 1A). Infarct volume of BALB/c mice was 6-fold larger (27.3 mm3) than B6 mice (4.4 mm3). We then determined infarct volumes for an additional 14 inbred strains encompassing the classical inbred strains recently used for mouse genome resequencing.18,19 Infarct volumes were highly reproducible among individual animals of the same inbred strain. Across strains, we observed a wide range of infarct volume (Figure 1B), with as much as 30-fold difference between the strain pair at the phenotypic extremes (C57BLKS/J versus SWR/J). Heritability (H2) of the trait of infarct volume after permanent middle cerebral artery occlusion was estimated to be 0.88. Postsurgical survival did not vary among strains, as only a single animal from a total of >100 did not survive the ischemic insult.
Figure 1.

Infarct volume after surgically induced focal cerebral ischemia differs widely among inbred mouse strains. A, Focal cerebral ischemia was induced and ischemic tissue damage was measured by TTC staining 24 hours after permanent distal MCA occlusion. The posterior faces of representative 1-mm coronal sections from the B6 and BALB/c mouse strains are shown. The size of the infarct is much larger in BALB/c than in B6 mice. B, Distribution of infarct volumes across 16 inbred strains of mice. Values represent mean±SD. The value in parentheses refers to number of animals used per strain.
A Chromosome 7 Locus Contributes the Predominant Effect on Infarct Volume
These marked differences in sensitivity to focal cerebral ischemia between inbred strains confirm a strong genetic influence on underlying mechanisms of cerebral infarction. To exploit these differences for identification of natural genetic determinants of infarct volume, we performed a reciprocal F1 intercross between strains B6 and BALB/c. Infarct volumes in F2 progeny exhibited a large variance, ranging from 0.2 to 43.6 mm3 (Supplemental Figure 1A). Infarct volume was not correlated with either body size or sex20 in the F2 cohort (Supplemental Figure 1B). Similarly, infarct volume was not correlated with the strain origin of the males or females in the (F1) cross, indicating that neither the mitochondrial genome nor the Y-chromosome contributes to the phenotypic differences.
A total of 105 F2 mice were genotyped for 239 SNP markers that were informative in this cross. The average spacing between fully informative SNP markers was 6.8±1.6 cM, affording complete coverage of the mouse genome. We identified a highly significant locus (LOD score=11.9) mapping to distal chromosome 7 influencing infarct volume. In addition, we identified 2 suggestive QTL mapping to chromosome 1 and 8 (Figure 2). We designated these loci Civq (Cerebral infarct volume QTL). Table 1 shows the characteristics of the 3 QTL, including peak SNP marker location, LOD score, effect size, and mode of inheritance. Civq1 is the strongest QTL that accounts for 56% of the observed variance in infarct volume. As predicted from the parental and F1 strain phenotypes, the B6 allele shows a codominant protective effect on infarct volume. To determine the allelic contribution of the effect of Civq1, infarct volumes were evaluated against genotype at the highest LOD score among all of the informative SNP markers (Figure 3). There was no statistical difference in infarct volume between F2 animals homozygous for the B6 and BALB/c allele at rs13479513 when compared with that of the parental strains. There was also no difference between F2 animals heterozygous at rs13479513 and F1 (B6/BALB) animals. Therefore, even though other loci are also present across the genome, Civq1 alone is able to explain nearly all of the phenotypic difference in infarct volume observed between the 2 inbred strains.
Figure 2.

A major locus for infarct volume maps to distal chromosome 7. The graph presents the results of a genome-wide linkage scan for infarct volume 24 hours after permanent MCA occlusion in 105 (B6×BALB/c) F2 progeny. Chromosomes 1 through X are represented numerically on the x axis. The relative width of the space allotted for each chromosome reflects the relative length of each chromosome. The y axis represents the LOD score. The significant (P<0.05) and suggestive (P<0.63) levels of linkage were determined by 1000 permutation tests. One region of the genome, distal chromosome 7, displays highly significant linkage to the trait, with a LOD score of 11.9. Two loci mapping to chromosome 1 and 8 only reached the P=0.63 suggestive level.
Table 1. Chromosomal Location, LOD Score, Effect Size, and Nearest Markers for QTL.
| QTL | Location | LOD Score | Effect Size, %* | Protective Allele | Mode of Inheritance | Marker at Peak | Overlapping QTL† |
|---|---|---|---|---|---|---|---|
| Civq1 | Chr 7 | 11.9 | 56 | B6 | Additive | rs13479513 | LSq1 (Ref. 21) |
| Civq2‡ | Chr 1 | 2.7‡ | 7 | B6 | Dominant | rs13475783 | |
| Civq3‡ | Chr 8 | 3.2‡ | 12 | BALB/c | Additive | rs8253516 |
The percentage of the total trait variance attributable to this locus.
Overlapping QTL identified in previous studies.
Suggestive QTL.
Figure 3.
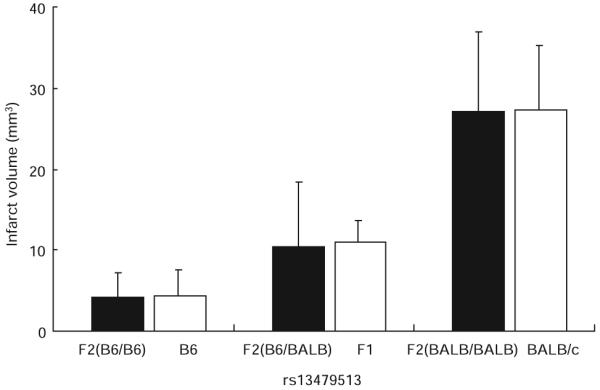
The chromosome 7 QTL contributes the predominant effect to the infarct volume trait. The histogram displays the phenotypic effect of the allele at SNP rs13479513 (in parenthesis) on infarct volume in comparison with the overall phenotype of the parental strains. F2 animals homozygous for the BALB/c allele (n=29) exhibit higher mean infarct volume than mice homozygous for the B6 allele (n=32). Infarct volumes for F2 animals heterozygous at the SNP (n=44) are intermediate to those of the homozygous animals. Significantly, there is no difference in infarct volume between F2 animals homozygous for the B6 or BALB alleles at rs13479513 when compared with that of the respective B6 or BALB/c parental strains. Similarly, there is no difference between F2 heterozygotes at rs13479513 and F1 (B6/BALB) animals. Thus, the parental origin of the allele at this locus effectively determines infarct volume, regardless of the genotype throughout the remainder of the genome. Error bars represent SD.
The 2 other QTL located on chromosome 1 (Civq2) and 8 (Civq3) explain 7% and 12%, respectively, of the variation in the trait. At Civq2, the B6 allele displayed a dominantly acting protective effect on infarct volume. By contrast, although the BALB/c strain shows larger infarct volume, the BALB/c allele at Civq3 conferred a protective additive effect to the trait (Table 1). These opposing phenotypic effects of the B6 (or BALB/c) alleles at the minor loci would counteract each other in the parental strains, and this may explain the robust correlation between overall phenotype in the F2 cohort and genotype at the major locus, Civq1. None of these Civq loci exhibit epistatic interactions with other regions of the genome.
Chromosome Substitution Strains Between B6 and A/J Validate Civq1 and Civq2
To confirm the presence of an infarct volume locus mapping to mouse chromosome 7, we used the C57BL/6J-Chr7A/J/NaJ chromosome substitution strain (hereafter CSS7) in which strain A/J chromosome 7 has been substituted into the B6 background.22 We first examined infarct volume of the donor A/J strain. A/J mice exhibited an infarct volume ≈6-fold larger than that of B6 but not statistically different from that observed for BALB/c (Figure 4). Based on this finding, we next determined the phenotype of the CSS7 mice. The CSS7 mice displayed large infarct volumes identical to the A/J parental strain that provides chromosome 7. This confirms that chromosome 7 harbors a locus with a large effect on cerebral infarction, with the B6 allele providing a protective effect. Additionally, this suggests that the A/J and BALB/c alleles at the chromosome 7 locus may be identical because of ancestral relatedness.
Figure 4.
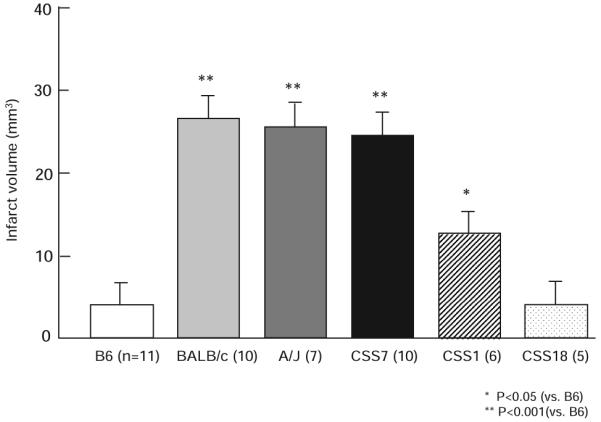
The B6 chromosome 7 confers a strong protective effect on ischemic infarct volume in a chromosome substitution strain. Similar to BALB/c, A/J mice display larger infarct volumes than B6 mice. The CSS C57BL/6J-Chr7A/J/NaJ (CSS7) mice which have an A/J chromosome 7 substituted into the B6 genomic background, show a similar infarct volume as A/J (the source of chromosome 7) and BALB/c, but significantly different than B6 mice (the genomic background). The CSS1 mice, where a minor locus was mapped, also exhibit larger infarct volumes than B6, but consistent with the lower effect size, smaller than BALB/c, A/J, and CSS7 mice. CSS18 mice, substituting a copy of A/J chromosome 18, where no infarct volume locus was mapped, show no difference with B6 mice, consistent with the QTL mapping data. Error bars indicate SD.
To confirm existence of the B6 protective allele on Civq2 responsible for 7% of phenotypic variance, we also measured infarct volumes of CSS1 mice. As predicted, CSS1 exhibited a significantly larger infarct volume than B6 (Figure 4). In the CSS1 line, the contribution of chromosome 1 to the phenotype seems larger than would be predicted by the effect size of Civq2 calculated from the F2 intercross. This was not unexpected, because a locus that is isolated from the effects of other loci across the genome by chromosome substitution can often show stronger effects than that predicted from a mapping cross.23 Because we did not map an infarct volume locus to chromosome 18, the CSS18 was used as a negative control for the CSS validation approach. The CSS18 mice showed infarct volumes identical to the B6 parental mice, confirming the negative mapping data and the use of CSS mice for locus validation for this phenotype.
An Intercross Between Strain B6 and SWR/J Reidentifies Civq1
We conducted a second intercross between inbred strains B6 and SWR/J, which also differ >6-fold in infarct volume (P<0.001; Figure 1B). Infarct volumes were measured in both sexes for a total of 78 F2 intercross progeny. F2 mice were genotyped for 215 SNP markers that were informative between the B6 and SWR/J strains. From this cross, we identified a highly significant locus (LOD=9.7) that mapped to the identical position (peak LOD at rs13479513) on chromosome 7 as that of Civq1 (Figure 5). Similar to Civq1 mapped in the original B6×BALB/c cross, Civq1 identified in this second cross also explains the majority of the effect (57%) of the total variance of infarct volume and shows the same genotype-phenotype correlation. These data further validate the importance of Civq1 in the determination of infarct volume in common inbred mouse strains.
Figure 5.

Civq1 is reidentified in a second intercross between B6 and SWR/J inbred mouse strains. The graph presents the results of a genome-wide linkage scan for infarct volumes in 78 (B6×SWR/J) F2 progeny. The y axis represents the LOD score. The significant (P<0.05) and suggestive (P<0.63) levels of linkage were determined by 1000 permutation tests. One region of the genome, distal chromosome 7, displays significant linkage to the trait, reaching a LOD score of 9.7. This linkage peak completely overlaps Civq1 originally mapped in the B6×BALB/c intercross.
Combined Cross Analysis and Interval-Specific SNP Haplotype Analysis Narrowed Civq1 to 12 Candidate Genes
The limited number of crossovers in a traditional mapping cross results in a large confidence interval for the typical QTL. Similarly, in our crosses the portion of the linkage peak above the significance threshold extends over 42 Mb of chromosome 7 in the B6×BALB/c and 32 Mb in the B6×SWR/J cross, implicating hundreds of genes as potential candidates. Recently, Churchill and coworkers24 have shown that by combining and analyzing data from multiple crosses, the number of crossovers is increased and the QTL interval can be reduced. Thus, we merged the genotype and phenotype data from the 2 intercrosses and performed genome-wide linkage analysis on the combined data. The combined animals from both intercrosses yielded an overall LOD score of 21.7 (Figure 6). This analysis also reduced the 1.5-LOD interval to 9.9 Mb encompassing 225 potential candidate genes.25
Figure 6.

Fine-mapping of the Civq1 interval using combined cross and interval-specific SNP haplotype analysis. The top graph presents the LOD score plots on chromosome 7 in crosses B6×BALB/c (dashed line), B6×SWR/J (dotted line), and combined cross (solid line). The combined cross approach narrowed 1.5-LOD interval of Civq1 to 9.9 Mb. Interval-specific haplotype sharing patterns among the strains further reduced the interval to 12 possible candidate genes.
The combined cross analysis provides a much tighter location for the causative gene, but still containing a larger number of candidates. Thus, to further dissect the interval we compared ancestral SNP haplotype patterns within the inbred mouse lineage. Interval-specific SNP haplotype analysis can reduce confidence intervals by identifying high-priority regions within a QTL interval that are likely to harbor the causal polymorphism.14 Approximately 97% of the genetic variation between the inbred laboratory mouse strains is found in ancestral haplotype DNA regions that seem to be identical by descent, with a minority of the sequence variation being unique to any individual strain.26,27 Civq1 was identified in 2 different genetic crosses and in the CSS series, each of which include B6 as one of the parental strains. Furthermore, multiple inbred strains exhibit small infarct volumes, suggesting that the protective B6 allele at Civq1 might not be unique to this strain. Allelic variation at Civq1 is therefore most likely attributable to a gene that maps within an ancestral haplotype block that is shared between BALB/c, A/J, and SWR/J, but that is different from B6. We examined all haplotypes throughout the 1.5-LOD interval of Civq1, defining a haplotype block to be 3 or more adjacent consecutive shared SNP alleles.14 Sixteen haplotype blocks, encompassing a combined total of 900 kb, exhibited the appropriate pattern of haplotype variation or relatedness. These 16 haplotype blocks contained only 12 possible candidate genes for Civq1 (Table 2).
Table 2. Candidate Genes Within the Shared SNP Haplotype Blocks for Civq1.
| Position (Build 37) | Gene | Description |
|---|---|---|
| 132473157–132493286 | 4933440M02Rik | RIKEN cDNA 4933440M02 gene |
| 132849588–133018791 | D430042O09Rik | RIKEN cDNA D430042O09 gene |
| 133020221–133226358 | Gsg1l | Germ cell–specific gene 1-like |
| 133941182–133974050 | 1500016O10Rik | RIKEN cDNA 1500016O10 gene |
| 134250176–134266022 | Qprt | Quinolinate phosphoribosyltransferase |
| 134438374–134478651 | Itgal | Integrin alpha L |
| 135515178–135561686 | Rgs10 | Regulator of G-protein signaling 10 |
| 135665334–135690923 | Bag3 | Bcl2-associated athanogene |
| 135754564–135839950 | Inpp5f | Inositol polyphosphate-5-phosphatase F |
| 140176874–140315166 | Ctbp2 | C-terminal binding protein 2 |
| 141614861–141691072 | LOC100043248 | Hypothelical protein LOC100043248 |
| 141860759–142365780 | Dock1 | Dedicator of cytokinesis 1 |
Discussion
Although significant progress has been made in the identification of genetic risk factors for human ischemic stroke susceptibility,28 identification of genetic risk factors for stroke outcomes has been severely limited. Because variation in the anatomic location of the occluded artery, extent/duration of occlusion, time until treatment, and other contributing factors cannot be controlled in patients, few genetic factors have been identified that contribute to the severity of tissue damage in human ischemic stroke. By contrast, these same factors are readily controlled using experimental animal model systems.
Different inbred mouse strains show robust differences in stroke outcomes after MCA occlusion, providing evidence that the innate response to focal cerebral ischemia is under strong genetic control.7-10 Hsu and coworkers8 examined the size of cortical infarct after permanent distal MCA occlusion in 3 different inbred strains; B6, BALB/c, and 129X1/SvJ. BALB/c mice had significantly larger infarcts than B6 and 129X1/SvJ strains. The authors postulated that the greater susceptibility of BALB/c to ischemic damage was not associated with a greater reduction in flow to the MCA territory, because blood flow before and after occlusion was nearly identical in all 3 strains. Similarly, although some strains had a poorly developed posterior segment of the Circle of Willis, this anatomic difference did not strictly correlate with either distal blood flow or infarct volume. Thus, Majid et al8 suggested that the differential ischemic outcomes after permanent MCA occlusion are not caused by interstrain differences in cerebrovascular anatomy but may instead be related to intrinsic differences in ischemic tolerance or protection pathways in neural tissue.
However, more recently, Faber and coworkers17 have postulated that the differential tissue damage after MCA ligation is attributable to differences in preexisting vascular anatomy between B6 and BALB/c strains. B6 mice have a well-developed collateral vessel network in the pial area of the brain leading to robust perfusion after MCA ligation, whereas BALB/c showed almost no collateral vessels, and greatly impaired perfusion after ligation. Their data suggest that the observed differences in infarct volume between strains are due to sequence variation in genes regulating collateral vessel formation.
These and other studies8,17,29-33 indicate that the patho-physiological mechanisms responsible for the differential stroke outcomes in inbred strains remain uncertain. Despite the ongoing debate on mechanism, experimental proof is lacking for any of the hypotheses, because current data supporting each view derives primarily from correlative studies of a limited number of inbred strains, studies by their very nature which lack any genetic support. The observed phenotypic differences among strains may be attributable to multiple mechanisms, each driven by different genetic loci.
Our study confirms that the differences in the severity of ischemic tissue damage after distal MCA occlusion in the common inbred strains are highly heritable because of natural genetic variation. Through the use of different inbred strains and genetic crosses, we present evidence of 3 distinct QTL that modulate the volume of cerebral infarction. These Civq loci do not map to regions of conserved synteny for any of the loci mapped for human stroke susceptibility, illustrating distinct genetic contributions to stroke susceptibility versus stroke outcomes. In particular, a single locus (Civq1) mapping to distal mouse chromosome 7, accounts for the major portion of variation in infarct volume in multiple inbred lines. These data demonstrate that natural variation in infarct volume in this species is predominantly attributable to the effects caused by sequence variation at a single locus, and possibly at a single gene. These data further suggest the presence of an ancestral allele of the gene determining the outcome of focal cerebral ischemia. The identification of the same QTL in different crosses facilitated the use of ancestral haplotype analysis to greatly reduce the number of potential candidate genes at this locus.
In a MCA occlusion study using the spontaneous hypertensive stroke-prone rat (SHRSP), a QTL modulating infarct volume has been mapped to rat chromosome 5.34 The major locus we have mapped in the mouse, Civq1, is not the region of conserved synteny for the rat locus on chromosome 5. Furthermore, we did not uncover a significant locus on mouse chromosome 4, the genomic region of conserved synteny for rat chromosome 5. Therefore, the same gene cannot be responsible at these 2 loci. In an intercross between SHRSP with WKR rats, blood pressure was influenced by 5 different loci across the genome. One of these, mapping to rat chromosome 1, also influenced infarct volume after distal MCA occlusion.35 Intriguingly, congenic animals for this locus also showed increased cerebral blood flow, providing genetic evidence for the importance of the regulation of cerebral blood flow in the modulation of infarct volume in this species. We note that a portion of Civq1 on mouse chromosome 7 is partially overlapping with the region of conserved synteny for the rat chromosome 1 linkage peak. The width of the linkage peaks in both species precludes further conjecture on the relationship between the 2 loci.
Intriguingly, Civq1 maps in the identical genomic region as a locus that was mapped in a mouse model of hindlimb ischemia.21 Similar to the focal cerebral ischemia model, different mouse strains show differences in limb salvage in surgically induced hindlimb ischemia.17,21,36-39 In a cross between B6 and BALB/c, a very strong locus influencing limb necrosis and recovery of perfusion (LSq1) maps to the identical position as Civq1 on chromosome 7.21 Because Civq1 was identified in 2 different intercrosses, and the SNP databases have recently increased in density and accuracy, interval-specific SNP haplotype analysis for Civq1 provided increased resolution resulting in fewer candidate genes than that determined for LSq1,21 which did not share these same advantages. Intriguingly, 6 new genes now appear for Civq1 that were not originally described for LSq1. More complete and accurate SNP data sharpened and sometimes extended poorly defined borders of certain haplotype blocks, and also uncovered new, smaller blocks in regions that previously did not exhibit haplotype structure. Assuming that Civq1 and LSq1 represent the identical allelic variant, these additional 6 candidate genes for Civq1 should be added to the list of those under consideration for LSq1.
Given the physiological similarities between the 2 ischemic models and the identical map position, we propose that the observed effects of Civq1 and LSq1 do in fact represent different phenotypic manifestations of the same gene. Civq1/LSq1 may confer sensitivity to the effects of ischemia in multiple tissues through the same physiological mechanism. Identification of the underlying genes will provide a better understanding of the innate processes modulating ischemic tissue damage in the brain, peripheral limbs, and possibly other tissues and organs. This gene and the others underlying the other Civq loci may provide novel targets for therapeutic intervention in human vascular occlusive diseases.
CLINICAL PERSPECTIVE.
Although significant progress has been made in the identification of genetic risk factors for human ischemic stroke susceptibility, identification of genetic risk factors for stroke outcomes has proven more challenging. Because variation in the anatomic location of the occluded artery, extent/duration of occlusion, time until treatment, and other contributing factors are not easily controlled for in patients, few genetic variants have been identified that contribute to the severity of tissue damage in human ischemic stroke. By contrast, these same confounding factors are readily controlled using experimental animal model systems. Different inbred mouse strains exhibit robust differences in infarct volume after distal middle cerebral artery occlusion, providing evidence that the response to focal cerebral ischemia is under strong genetic control. We have extended these observations to show that in 16 different inbred strains of mice, infarct volume can vary by >30-fold. Using a genetic mapping approach in this experimental model, we have exploited these innate differences to identify regions of the mouse genome that modulate infarct volume. Three different genomic regions significantly altered infarct volume, with a small region of mouse chromosome 7 accounting for more than half of the observed variation. Molecular genetic analysis reduced the list of candidate genes for this region of chromosome 7 to only 12 genes. The identification of this and other genes in this experimental model may uncover novel genetic and physiological pathways that modulate cerebral infarction, and provide new targets for therapeutic intervention in ischemic stroke and other vascular occlusive diseases.
Supplementary Material
Acknowledgments
We thank Drs Lan Mao and Howard Rockman for instruction on MCAO surgery and Christopher Clayton for animal husbandry.
Sources of Funding
This work was supported by National Institutes of Health grant 1R01 HL097281 to D.A.M. and American Heart Association Predoctoral Fellowship 0815132E to S.K.
Footnotes
The online-only Data Supplement is available at http://circgenetics.ahajournals.org/cgi/content/full/CIRCGENETICS.109.883231/DC1.
Guest Editor for this article was Donna K. Arnett.
Disclosures
None.
References
- 1.Mohr JP, Choi D, Grotta J, Wolf P. Stroke: Pathophysiology, Diagnosis, and Management. IV ed. Churchill Livingstone; New York, NY: 2004. [Google Scholar]
- 2.Hossmann KA. Genetically modified animals in molecular stroke research. Acta Neurochir Suppl. 2004;89:37–45. doi: 10.1007/978-3-7091-0603-7_5. [DOI] [PubMed] [Google Scholar]
- 3.Hossmann KA. Pathophysiology and therapy of experimental stroke. Cell Mol Neurobiol. 2006;26:1057–1083. doi: 10.1007/s10571-006-9008-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dirnagl U, Simon RP, Hallenbeck JM. Ischemic tolerance and endogenous neuroprotection. Trends Neurosci. 2003;26:248–254. doi: 10.1016/S0166-2236(03)00071-7. [DOI] [PubMed] [Google Scholar]
- 5.Mergenthaler P, Dirnagl U, Meisel A. Pathophysiology of stroke: lessons from animal models. Metab Brain Dis. 2004;19:151–167. doi: 10.1023/b:mebr.0000043966.46964.e6. [DOI] [PubMed] [Google Scholar]
- 6.Mehta SL, Manhas N, Raghubir R. Molecular targets in cerebral ischemia for developing novel therapeutics. Brain Res Rev. 2007;54:34–66. doi: 10.1016/j.brainresrev.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 7.Barone FC, Knudsen DJ, Nelson AH, Feuerstein GZ, Willette RN. Mouse strain differences in susceptibility to cerebral ischemia are related to cerebral vascular anatomy. J Cereb Blood Flow Metab. 1993;13:683–692. doi: 10.1038/jcbfm.1993.87. [DOI] [PubMed] [Google Scholar]
- 8.Majid A, He YY, Gidday JM, Kaplan SS, Gonzales ER, Park TS, Fenstermacher JD, Wei L, Choi DW, Hsu CY. Differences in vulnerability to permanent focal cerebral ischemia among 3 common mouse strains. Stroke. 2000;31:2707–2714. doi: 10.1161/01.str.31.11.2707. [DOI] [PubMed] [Google Scholar]
- 9.Lambertsen KL, Gregersen R, Finsen B. Microglial-macrophage synthesis of tumor necrosis factor after focal cerebral ischemia in mice is strain dependent. J Cereb Blood Flow Metab. 2002;22:785–797. doi: 10.1097/00004647-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 10.Sugimori H, Yao H, Ooboshi H, Ibayashi S, Iida M. Krypton laser-induced photothrombotic distal middle cerebral artery occlusion without craniectomy in mice. Brain Res Brain Res Protoc. 2004;13:189–196. doi: 10.1016/j.brainresprot.2004.06.001. [DOI] [PubMed] [Google Scholar]
- 11.Goldlust EJ, Paczynski RP, He YY, Hsu CY, Goldberg MP. Automated measurement of infarct size with scanned images of triphenyltetrazolium chloride-stained rat brains. Stroke. 1996;27:1657–1662. doi: 10.1161/01.str.27.9.1657. [DOI] [PubMed] [Google Scholar]
- 12.Wexler EJ, Peters EE, Gonzales A, Gonzales ML, Slee AM, Kerr JS. An objective procedure for ischemic area evaluation of the stroke intraluminal thread model in the mouse and rat. J Neurosci Methods. 2002;113:51–58. doi: 10.1016/s0165-0270(01)00476-9. [DOI] [PubMed] [Google Scholar]
- 13.Lander E, Kruglyak L. Genetic dissection of complex traits: guidelines for interpreting and reporting linkage results. Nat Genet. 1995;11:241–247. doi: 10.1038/ng1195-241. [DOI] [PubMed] [Google Scholar]
- 14.Burgess-Herbert SL, Cox A, Tsaih SW, Paigen B. Practical applications of the bioinformatics toolbox for narrowing quantitative trait loci. Genetics. 2008;180:2227–2235. doi: 10.1534/genetics.108.090175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Virley D. Choice, methodology, and characterization of focal ischemic stroke models: the search for clinical relevance. Methods Mol Med. 2005;104:19–48. doi: 10.1385/1-59259-836-6:019. [DOI] [PubMed] [Google Scholar]
- 16.Kuraoka M, Furuta T, Matsuwaki T, Omatsu T, Ishii Y, Kyuwa S, Yoshikawa Y. Direct experimental occlusion of the distal middle cerebral artery induces high reproducibility of brain ischemia in mice. Exp Anim. 2009;58:19–29. doi: 10.1538/expanim.58.19. [DOI] [PubMed] [Google Scholar]
- 17.Chalothorn D, Clayton JA, Zhang H, Pomp D, Faber JE. Collateral density, remodeling, and VEGF-A expression differ widely between mouse strains. Physiol Genomics. 2007;30:179–191. doi: 10.1152/physiolgenomics.00047.2007. [DOI] [PubMed] [Google Scholar]
- 18.Frazer KA, Eskin E, Kang HM, Bogue MA, Hinds DA, Beilharz EJ, Gupta RV, Montgomery J, Morenzoni MM, Nilsen GB, Pethiyagoda CL, Stuve LL, Johnson FM, Daly MJ, Wade CM, Cox DR. A sequence-based variation map of 8.27 million SNPs in inbred mouse strains. Nature. 2007;448:1050–1053. doi: 10.1038/nature06067. [DOI] [PubMed] [Google Scholar]
- 19.Yang H, Bell TA, Churchill GA, Pardo-Manuel de Villena F. On the subspecific origin of the laboratory mouse. Nat Genet. 2007;39:1100–1107. doi: 10.1038/ng2087. [DOI] [PubMed] [Google Scholar]
- 20.McCullough LD, Hurn PD. Estrogen and ischemic neuroprotection: an integrated view. Trends Endocrinol Metab. 2003;14:228–235. doi: 10.1016/s1043-2760(03)00076-6. [DOI] [PubMed] [Google Scholar]
- 21.Dokun AO, Keum S, Hazarika S, Li Y, Lamonte GM, Wheeler F, Marchuk DA, Annex BH. A QTL (LSq-1) on mouse chromosome 7 is linked to the absence of tissue loss following surgical hind-limb ischemia. Circulation. 2008;117:1207–1215. doi: 10.1161/CIRCULATIONAHA.107.736447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Singer JB, Hill AE, Burrage LC, Olszens KR, Song J, Justice M, O’Brien WE, Conti DV, Witte JS, Lander ES, Nadeau JH. Genetic dissection of complex traits with chromosome substitution strains of mice. Science. 2004;16(304):445–448. doi: 10.1126/science.1093139. [DOI] [PubMed] [Google Scholar]
- 23.Belknap JK. Chromosome substitution strains: some quantitative considerations for genome scans and fine mapping. Mamm Genome. 2003;14:723–732. doi: 10.1007/s00335-003-2264-1. [DOI] [PubMed] [Google Scholar]
- 24.Li R, Lyons MA, Wittenburg H, Paigen B, Churchill GA. Combining data from multiple inbred line crosses improves the power and resolution of quantitative trait loci mapping. Genetics. 2005;169:1699–1709. doi: 10.1534/genetics.104.033993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Manichaikul A, Dupuis J, Sen S, Broman KW. Poor performance of bootstrap confidence intervals for the location of a quantitative trait locus. Genetics. 2006;174:481–489. doi: 10.1534/genetics.106.061549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wade CM, Kulbokas EJ, III, Kirby AW, Zody MC, Mullikin JC, Lander ES, Lindblad-Toh K, Daly MJ. The mosaic structure of variation in the laboratory mouse genome. Nature. 2002;420:574–578. doi: 10.1038/nature01252. [DOI] [PubMed] [Google Scholar]
- 27.Wiltshire T, Pletcher MT, Batalov S, Barnes SW, Tarantino LM, Cooke MP, Wu H, Smylie K, Santrosyan A, Copeland NG, Jenkins NA, Kalush F, Mural RJ, Glynne RJ, Kay SA, Adams MD, Fletcher CF. Genome-wide single-nucleotide polymorphism analysis defines haplotype patterns in mouse. Proc Natl Acad Sci USA. 2003;100:3380–3385. doi: 10.1073/pnas.0130101100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dichgans M. Genetics of ischaemic stroke. Lancet Neurol. 2007;6:149–161. doi: 10.1016/S1474-4422(07)70028-5. [DOI] [PubMed] [Google Scholar]
- 29.Connolly ES, Jr, Winfree CJ, Stern DM, Solomon RA, Pinsky DJ. Procedural and strain-related variables significantly affect outcome in a murine model of focal cerebral ischemia. Neurosurgery. 1996;38:523–531. doi: 10.1097/00006123-199603000-00021. [DOI] [PubMed] [Google Scholar]
- 30.Yang G, Kitagawa K, Matsushita K, Mabuchi T, Yagita Y, Yanagihara T, Matsumoto M. C57BL/6 strain is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: selective neuronal death in the murine transient forebrain ischemia. Brain Res. 1997;752:209–218. doi: 10.1016/s0006-8993(96)01453-9. [DOI] [PubMed] [Google Scholar]
- 31.Schauwecker PE, Steward O. Genetic determinants of susceptibility to excitotoxic cell death: implications for gene targeting approaches. Proc Natl Acad Sci USA. 1997;94:4103–4108. doi: 10.1073/pnas.94.8.4103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maeda K, Hata R, Hossmann KA. Regional metabolic disturbances and cerebrovascular anatomy after permanent middle cerebral artery occlusion in C57black/6 and SV129 mice. Neurobiol Dis. 1999;6:101–118. doi: 10.1006/nbdi.1998.0235. [DOI] [PubMed] [Google Scholar]
- 33.Wellons JC, III, Sheng H, Laskowitz DT, Burkhard Mackensen G, Pearlstein RD, Warner DS. A comparison of strain-related susceptibility in two murine recovery models of global cerebral ischemia. Brain Res. 2000;868:14–21. doi: 10.1016/s0006-8993(00)02216-2. [DOI] [PubMed] [Google Scholar]
- 34.Jeffs B, Clark JS, Anderson NH, Gratton J, Brosnan MJ, Gauguier D, Reid JL, Macrae IM, Dominiczak AF. Sensitivity to cerebral ischaemic insult in a rat model of stroke is determined by a single genetic locus. Nat Genet. 1997;16:364–367. doi: 10.1038/ng0897-364. [DOI] [PubMed] [Google Scholar]
- 35.Yao H, Cui ZH, Masuda J, Nabika T. Congenic removal of a QTL for blood pressure attenuates infarct size produced by middle cerebral artery occlusion in hypertensive rats. Physiol Genomics. 2007;30:69–73. doi: 10.1152/physiolgenomics.00149.2006. [DOI] [PubMed] [Google Scholar]
- 36.Fukino K, Sata M, Seko Y, Hirata Y, Nagai R. Genetic background influences therapeutic effectiveness of VEGF. Biochem Biophys Res Commun. 2003;310:143–147. doi: 10.1016/j.bbrc.2003.08.134. [DOI] [PubMed] [Google Scholar]
- 37.Scholz D, Ziegelhoeffer T, Helisch A, Wagner S, Friedrich C, Podzuweit T, Schaper W. Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J Mol Cell Cardiol. 2002;34:775–787. doi: 10.1006/jmcc.2002.2013. [DOI] [PubMed] [Google Scholar]
- 38.Helisch A, Wagner S, Khan N, Drinane M, Wolfram S, Heil M, Ziegelhoeffer T, Brandt U, Pearlman JD, Swartz HM, Schaper W. Impact of mouse strain differences in innate hindlimb collateral vasculature. Arterioscler Thromb Vasc Biol. 2006;26:520–526. doi: 10.1161/01.ATV.0000202677.55012.a0. [DOI] [PubMed] [Google Scholar]
- 39.van Weel V, Toes RE, Seghers L, Deckers MM, de Vries MR, Eilers PH, Sipkens J, Schepers A, Eefting D, van Hinsbergh VW, van Bockel JH, Quax PH. Natural killer cells and CD4+ T-cells modulate collateral artery development. Arterioscler Thromb Vasc Biol. 2007;27:2310–2318. doi: 10.1161/ATVBAHA.107.151407. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.