

Abstract
To develop novel bifunctional agents for tumor imaging (MR) and photodynamic therapy (PDT) certain tumor-avid photosensitizers derived from chlorophyll-a were conjugated with variable number of Gd(III)aminobenzyl DTPA moieties. All the conjugates containing three or six gadolinium units showed significant T1 and T2 relaxivities. However, as a bifunctional agent, the 3-(1′-hexyloxyethyl)pyropheophorbide-a (HPPH) containing 3Gd(III) aminophenyl DTPA was most promising with possible applications in tumor-imaging and PDT. Compared to HPPH, the corresponding 3- and 6Gd(III)aminobenzyl DTPA conjugates exhibited similar electronic absorption characteristics with a slightly decreased intensity of the absorption band at 660 nm. However, compared to HPPH, the excitation of the broad “Soret” band (near 400 nm) of the corresponding 3Gd(III)aminobenzyl-DTPA analogs showed a significant decrease in the fluorescence intensity at 667 nm, which was further diminished by increasing the number of Gd(III)units.
INTRODUCTION
Magnetic resonance imaging (MRI) is a medical imaging technique (1) commonly used in radiology to visualize the internal structure and function of the body. MRI provides greater contrast between the different soft tissues of the body than computed tomography (CT), making it especially useful in neurological (brain), musculoskeletal, cardiovascular, and oncological (cancer) imaging (2). However, MR imaging has some practical disadvantages. For example, benign tumors may be difficult to distinguish from malignant lesions. The fibrocystic disease of the breast, fibroadenomas, sclerosing adenosis, atypical hyperplasia, lobular carcinoma in situ (LCIS), and breast papillomas can all produce contrast enhancement patterns that are hard to distinguish from malignant processes. In addition, some benign processes such as proliferative dysplasia, inflammation, wounds, and benign tumors can enhance in a similar fashion to malignant tumors (3). These shortcoming of MRI in measuring tissue properties offer opportunities that may be satisfied with information provided by optical fluorescence imaging with near-infrared light (4). Optical techniques offer the potential to contribute greatly to the expansion of clinical multimodality techniques. Their ability to image structural, functional and molecular information at different spatial and temporal scales makes them very attractive. In this case, the multimodality approach can be used as the combination of multiple optical techniques in one instrument and/or the fusion of an optical technique with another well-established clinical modality, such as PET, CT or MRI. (5,6).
Clinically, optical imaging modality has been employed to enable or improve PDT (7). Most of the porphyrin-based photosensitizers generally fluoresce and the fluorescent nature of these porphyrins has been exploited for the detection of early-stage cancers in the lung, bladder and other sites (8). In addition, for treatment of early disease or for deep seated tumors the fluorescence can be used to guide the activated light. Most of the porphyrin-based compounds are not optimal fluorophores for tumor detection for several reasons, especially a small shift between the long-wavelength absorption and emission bands, which make it technically difficult to separate the fluorescence from excitation wavelength (9). However, certain pyropheophorbide-a analogs (e. g., HPPH) produce very high fluorescence quantum yields and exciting them at their long wavelength absorption (665 nm) produces two emission bands; a strong peak at 670 nm, and a weaker band at 720 nm. This characteristic of HPPH has been used for in vivo fluorescence imaging of canine and other tumors (10). In recent years, integrating the strengths of each imaging modality has become a major research area. The research in our laboratory has also been focused in designing certain porphyrin-based compounds as “Multifunctional Agents” for tumor imaging and photodynamic therapy (PDT) (11-15).
We have previously shown that HPPH (16-21) a tumor-avid photosensitizer (currently undergoing Phase I/II human clinical trials) on conjugating with Gd(III)aminobenzyl DTPA [Gd(III)ADTPA] can be used for both tumor MR imaging and PDT (22, 23). In our initial study, among the compounds investigated, the conjugates with two Gd(III)ADTPA moieties showed greater tumor-contrast than the corresponding mono- Gd(III)ADTPA derivative. However, poor solubility of these agents in various clinically acceptable formulations restricted their use for detailed in vivo studies, including toxicity. To investigate the effect of increased number of GD(III)ADTPA moieties in tumor imaging/PDT and also to improve water solubility, we decided to extend this approach further and a series of tumor-avid long-wavelength absorbing chlorophyll-a analogs (pyropheophorbide-a and purpurinimide) containing three- or six Gd(III)ADTPA moieties with variable lipophilicity were synthesized. For developing “bifunctional agents” with longer wavelength absorption than HPPH. (λ max: 660 nm) a related conjugate containing a highly tumor-avid purpurinimide (λ max: 700nm) (24) was also prepared. In this manuscript the synthesis, photophysical properties, T1/T2 relaxivity and in vitro photosensitizing efficacy of a series of novel photosensitizer-Gd(III)ADTPA conjugates are discussed. The second part of our study (see the following paper in this issue) is focused in selecting the optimal “Multifunctional Agent” for tumor-imaging (MRI, fluorescence imaging) and PDT. In vivo PDT response is discussed in terms of long-term cure rate and monitoring of the vascular response via immunohistochemistry. Finally, the biodistribution and organ specific toxicity of the lead compound (C-14 labeled) at variable time points is also discussed.
EXPERIMENTAL PROCEDURES
Chemistry
The synthetic intermediates and the final products were characterized by NMR (400 MHz), mass spectrometry (EIMS and HRMS) and elemental analyses. 1H NMR spectra were recorded on a Bruker AMX-400 spectrometer. Chemical shifts are expressed in δ ppm. All photo physical experiments were carried out using spectroscopic grade solvents. The reactions were monitored by TLC and/or spectrophotometrically. Column chromatography was performed either over Silica Gel 60 (70-230 mesh) or neutral Alumina (Brockmann grade III, 50 mesh). UV-visible spectrums were recorded on Varian Cary 50 Bio UV-visible spectrophotometer using dichloromethane as solvent unless otherwise specified. Fluorescence spectra were recorded on a Varian Cary Eclipse Fluorescence spectrophotometer with an excitation wavelength in the “Soret” band region between 410 – 425 nm.
Compound No. 1
HPPH (100.0 mg, 0.157 mmol), amine A (97.8 mg, 0.235 mmol), EDCI (1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride); (60.2 mg, 0.314 mmol) and DMAP (4-dimethylaminopyridine); (38.36 mg, 0.314 mmol) were taken in a dry round-bottom flask (100 ml). Dry dichloromethane (30 ml) was added and the reaction mixture was stirred at room temperature for 16 hr under N2 atm. The reaction mixture was diluted with dichloromethane (100 ml), washed with brine solution, organic layer separated, dried over sodium sulfate and concentrated. The crude mixture was chromatographed over silica gel using 1-3% methanol/dichloromethane mixture as eluent to give product 1. Yield: 130.0 mg (80.0%). UV-vis (λmax, dichloromethane): 318, 412, 506, 537, 604 & 660 nm. 1HNMR (400MHz, CDCl3): δ 9.78 (s, 1H, H-5), 9.41 (s, 1H, H-10), 8.54 (s, 1H, H-20), 5.96-5.91 (m, 2H, CH3CHOhexyl, NH), 5.33 (d, 1H, CH-151, J = 19.6 Hz), 5.14 (d, 1H, CH-151, J = 19.6 Hz), 4.53 (q, 1H, H-17, J = 7.2 Hz), 4.32 (m, 1H, H-18), 3.71-3.67 (m, 2H, −OCH2-hexyl), 3.63-3.60 (m, 2H, 8-CH2CH3), 3.52 (s, 3H, 7-CH3), 3.39 (s, 3H, 2-CH3), 3.27 (s, 3H, 12-CH3), 2.67 (m, 1H, CH-172), 2.37 (m, 1H, H-172), 2.26 (m, 1H, H-171), 2.15-2.13 (m, 9H, 3CH2-chain & CH3CHOhexyl), 1.97 (m, 1H, H-171), 1.92 (t, 6H, 3CH2-chain, J = 7.6 Hz), 1.80 (d, 3H, 18-CH3, J = 7.2 Hz), 1.73 (m, 2H, CH2hexyl), 1.66 (t, 3H, 8-CH2CH3, J = 7.6 Hz), 1.44 (m, 2H, CH2hexyl), 1.30 (s, 27H, 3CO2tBu), 1.24 (m, 4H, 2CH2hexyl), 0.78 (t, 3H, CH3hexyl, J = 6.8Hz), 0.04 (brs, 1H, NH), - 1.68 (brs, 1H, NH). EIMS: 1035 (MH+). HRMS: Calculated for C61H87N5O9: 1033.6503, found: 1034.6564 (MH+).
Compound No. 2
Compound 1 (73.0 mg, 0.07 mmol) was stirred with 70% Trifluoroacetic acid (TFA) / dichloromethane (DCM, 5.0 ml) at room temperature for 3 hr. The resultant mixture was concentrated and dried under high vacuum to remove trace of TFA. To this crude preparation were added, amino-benzyl-DTPA-penta-tert-butyl ester (219.0 mg, 0.282 mmol), EDCI (67.0 mg, 0.352 mmol) and DMAP (43.0 mg, 0.352 mmol). Dry dichloromethane (30 ml) was added and the reaction mixture was stirred at RT for 16 hr under N2 atm. The reaction mixture was diluted with dichloromethane (100 ml), washed with brine solution, organic layer separated, dried over sodium sulfate and concentrated. The crude mixture was chromatographed over alumina G (III) using 1-3% methanol/dichloromethane mixture as eluent to give product 2. Yield: 130.0 mg (58.47%). UV-vis (λmax, dichloromethane): 319, 411, 506, 537, 604, 661. 1HNMR (400MHz, CDCl3): δ 9.74 (splitted s, 1H, H-5), 9.36 (splitted s, 1H, H-10), 8.54 (splitted s, 1H, H-20), 8.17 (brs, 1H, NH), 7.74 (m, 1H, Ph-DTPA), 7.61-7.56 (m, 2H, Ph-DTPA), 7.43 (m, 4H, Ph-DTPA), 7.12 (m, 5H, Ph-DTPA), 6.96 (brs, 1H, NH), 5.96 (m, 1H, CH3CHOhexyl), 5.29 (d, 1H, CH-151, J = 19.2 Hz), 5.11 (d, 1H, CH-151, J = 19.2 Hz), 4.47 (m, 1H, H-17), 4.26 (m, 1H, H-18), 3.64 (m, 2H, & OCH2hexyl), 3.54 (m, 2H, 8-CH2CH3), 3.45-3.38 (m, 30H, 15CH2-DTPA), 3.36 (s, 3H, 7-CH3), 3.32 (s, 3H, 2-CH3), 3.22 (s, 3H, 12-CH3), 3.12 (m, 3H, CH-DTPA), 2.84-2.70 (m, 19H, 9CH2-DTPA & CH-172), 2.59 (m, 6H, 6CH2-benzyl), 2.47-2.41 (m, 8H, 3CH2-chain, CH-172 & CH-171), 2.19-2.15 (m, 9H, 3CH2-chain, CH3CHOhexyl), 2.06 (d, 3H, 18-CH3, J = 7.6 Hz) 2.00 (m, 1H, CH-171), 1.74 (m, 4H, 2CH2-hexyl), 1.66 (t, 3H, 8-CH2CH3, J = 7.2 Hz), 1.60 (m, 135H for 15 CO2tBu), 1.26 (m, 4H, 2CH2-hexyl), 0.77 (m, 3H, CH3-Ohexyl), 0.55 (brs, 1H, NH), −0.24 (brs, 1H, NH). HRMS: Calculated for C172H267N17O36: 3149.053, found: 3150.10 (MH+).
Compound No. 3
Compound 2 (110.0 mg, 0.034 mmol) was stirred with 70% TFA/DCM (5.0 ml) at room temperature for 3 hr. The resultant mixture was concentrated and dried under high vacuum to remove trace TFA. The crude preparation thus obtained was dissolved in pyridine (10 ml) and while stirring, GdCl3.6H2O (77.9 mg, 0.21 mmol) in 1 ml of water was added slowly and the resultant mixture was stirred for 16 hr. The reaction mixture was concentrated to dryness under high vacuum. The residue was washed with water (10 ml × 3), acetone (10 ml × 3) and finally dried under high vacuum using P2O5 as drying agent. Yield: 75.0 mg (77.55%). UV-vis (λmax, MeOH): 620, 408, 504, 537, 604 & 660 nm. Elemental analysis: Calculated for C113H151Gd3N17O36: C, 48.55; H, 5.44; Gd, 16.88; N, 8.52; O, 20.61, found: C, 48.68; H, 5.49; N, 8.57.
Compound No. 5
Acid 4 (100.0 mg, 0.143 mmol), amine A (89.1 mg, 0.214 mmol), EDCI (54.9 mg, 0.28 mmol) and DMAP (34.9 mg, 0.28 mmol) were taken in a dry round bottom flask (RBF, 100 ml). Dry dichloromethane (30 ml) was added to it and the reaction mixture was stirred at RT for 16 hr under N2 atm. The reaction mixture was diluted with dichloromethane (100 ml), washed with brine solution, the organic layer separated, dried over sodium sulfate and concentrated. The crude mixture was chromatographed over silica gel using 1-2% methanol/dichloromethane mixture as eluent to give product 5. Yield: 134.0 mg (85.3%). UV-vis (λmax nm, dichloromethane): 318, 411, 506, 536, 604 & 661. 1HNMR (400MHz, CDCl3): δ 9.76 (splitted s, 1H, H-5), 9.38 (splitted s, 1H, H-10), 8.55 (s, 1H, H-20), 6.02 (d, 1H, CH3CHOOTEG, J = 6.4 Hz), 5.99 (brs, 1H, NH), 5.34 (d, 1H, CH-151, J = 20.0 Hz), 5.15 (d, 1H, CH-151, J = 20.0 Hz), 4.58 (q, 1H, H-17, J = 6.8 Hz), 4.33 (m, 1H, H-18), 3.88-3.75 (m, 4H, 2CH2-O-TEG), 3.70-3.62 (m, 6H, 3CH2-O-TEG), 3.55 (m, 2H, 8-CH2CH3), 3.47-3.44 (m, 2H, CH2-O-TEG), 3.40 (s, 3H, 7-CH3), 3.39 (s, 3H, 2-CH3), 3.29 (s, 3H, 12-CH3), 3.27 (s, 3H, CH3-O-TEG), 2.69 (m, 1H, CH-172), 2.39 (m, 1H, CH-172), 2.31 (m, 1H, CH-171), 2.14 (m, 8H, 4CH2-chain), 2.00 (m, 1H, CH-171), 1.92 (m, 7H, 2CH2-chain, CH3CHOTEG), 1.82 (d, 3H, 18-CH3, J = 7.2 Hz), 1.68 (t, 3H, 8-CH2CH3, J = 7.6 Hz), 1.31 (s, 27H, 3CO2tBu), 0.42 (brs, 1H, NH), −1.69 (brs, 1H, NH). EIMS: 1097 (MH+). Calculated for C62H89N5O12: 1095.6507, found: 1096.6571 (MH+).
Compound No. 6
Compound 5 (100.0 mg, 0.091 mmol) was stirred with 80% TFA/DCM (5.0 ml) at room temperature for 3 hr. The resultant mixture was concentrated and dried under high vacuum to remove trace TFA. To this crude preparation were added, amino-benzyl-DTPA-penta-tert-butyl ester (285.0 mg, 0.365 mmol), EDCI (105.0 mg, 0.54 mmol) and DMAP (66.8 mg, 0.54 mmol). Dry dichloromethane (30 ml) was added and the reaction mixture was stirred at RT for 16 hr under N2 atm. The reaction mixture was diluted with dichloromethane (100 ml), washed with brine solution, the organic layer was separated, dried over sodium sulfate and concentrated. The crude mixture was chromatographed over alumina Gr(III) using 1-3% methanol/dichloromethane mixture as eluent to give product 6. Yield: 250.0 mg (85.3%). UV-vis (λmax nm, dichloromethane): 319, 411, 506, 537, 606, 661. 1HNMR (400MHz, CDCl3): δ 9.70 (splitted s, 1H, H-5), 9.44 (splitted s, 1H, H-10), 9.37 (brs, 1H, NH), 9.20 (brs, 1H, NH), 8.56 (m, 1H, NH), 8.47 (s, 1H, H-20), 7.77 (m, 1H, Ph-DTPA), 7.59 (m, 2H, Ph-DTPA), 7.44 (m, 2H, Ph-DTPA), 7.10 (m, 6H, Ph-DTPA), 6.81 (m, 1H, Ph-DTPA), 5.97 (m, 1H, CH3CHOTEG), 5.20 (m, 2H, CH2-151), 4.60 (m, 1H, H-17), 4.22 (m, 1H, H-18), 3.81-3.64 (m, 4H, 2CH2-OTEG), 3.57-3.50 (m, 4H, 8-CH2CH3, CH2-OTEG), 3.60 (m, 6H, 3CH2OTEG), 3.38 (s, 30H, 15CH2-DTPA), 3.35 (s, 3H, 7-CH3), 3.23 (m, 6H, 12-CH3, OCH3-TEG), 3.04 (m, 3H, 3CH-DTPA), 2.70 (m, 19H, 9CH2-DTPA, CH-172), 2.55 (m, 7H, 3CH2-benzyl & CH-172), 2.32 (t, 6H, 3CH2-chain, J = 6.8 Hz), 2.23 (m, 1H, CH-171), 2.10 (d, 3H, CH3CH-OTEG, J = 6.4 Hz), 2.01 (m, 1H, CH-171), 1.77 (d, 3H, 18-CH3, J = 7.2 Hz), 1.68 (t, 3H, 8-CH2CH3, J = 7.6 Hz), 1.59 (t, 6H, 3CH2-chain, J = 6.4 Hz), 1.44 (m, 135H, 15CO2tBu), −1.75 (brs, 1H, NH). HRMS: Calculated for C173H269 N17O39: 3211.077, found: 3212.20 (MH+).
Compound No. 7
Compound 6 (226.0 mg, 0.07 mmol) was stirred with 80% TFA/DCM (5.0 ml) at room temperature for 3 hr. The resultant mixture was concentrated and dried under high vacuum to remove trace TFA. The crude mixture thus obtained was dissolved in pyridine (10 ml) and while stirring, GdCl3.6H2O (156.9 mg, 0.422 mmol) in 1 ml of water was added slowly and the resultant mixture was stirred for 16 hr. The reaction mixture was concentrated to dryness under high vacuum. The residue was washed with water (10 ml × 3), acetone (10 ml × 3) and finally dried under high vacuum using P2O5 as drying agent. Yield: 165.0 mg (82.5%). UV-vis (λmax nm, MeOH): 320, 408, 505, 537, 605 and 660 nm. Elemental analysis: Calculated for C113H149Gd3N17O39: C, 47.77; H, 5.29; Gd, 16.60; N, 8.38; O, 21.96, found: C, 47.85; H, 5.30; N, 8.43.
Compound No. 9
Acid 8 (82.0 mg, 0.118 mmol), amine A (73.6 mg, 0.177 mmol), EDCI (45.3 mg, 0.236 mmol) and DMAP (28.8 mg, 0.236 mmol) were taken in a dry RBF (100 ml). Dry dichloromethane (30 ml) was added and the reaction mixture was stirred at RT for 16 hr under N2 atm. The reaction mixture was diluted with dichloromethane (100 ml), washed with brine solution, the organic layer separated, dried over sodium sulfate and concentrated. The crude mixture was chromatographed over silica gel using 1-2% methanol/dichloromethane mixture as eluent to give product 9. Yield: 90.0 mg (69.8%). UV-vis (λmax, dichloromethane): 365, 418, 509, 545 and 699 nm. 1HNMR (400MHz, CDCl3): δ 9.75 (splitted s, 1H, meso-H), 9.64 (s, 1H, meso-H), 8.55 (s, 1H, meso-H), 6.25 (m, 1H, CONH), 5.79 (q, 1H, CH3CHObutyl, J = 6.4 Hz), 5.34 (m, 1H, H-17), 4.52 (t, 2H, −NCH2butyl, J = 7.2 Hz), 4.42 (m, 1H, H-18), 3.84 (s, 3H, ring-CH3), 3.70-3.59 (m, 4H, −OCH2-butyl, 8-CH2CH3), 3.32 (s, 3H, ring-CH3), 3.17 (s, 3H, ring-CH3), 2.61 (m, 1H, CH-172), 2.43 (m, 1H, H-172), 2.27 (m, 1H, H-171), 2.20 (t, 6H, 3CH2-chain, J = 7.2 Hz), 2.06 (m, 3H, CH3CHObutyl), 2.01 (t, 6H, 3CH2-chain, J = 7.6 Hz), 1.82 (m, 1H, H-171), 1.75 (d, 3H, 18-CH3, J = 6.0 Hz), 1.68 (t, 3H, 8-CH2CH3, J = 7.6 Hz), 1.62 (m, 8H, 4CH2butyl), 1.34 (s, 27H, 3CO2tBu), 1.10 (t, 3H, CH3-obutyl, J = 7.2 Hz), 0.87 (t, 3H, CH3-Nbutyl, J = 7.2 Hz), 0.40 (brs, 1H, NH), −0.06 (brs, 1H, NH). EIMS: 1092 (MH+). HRMS: Calculated for C63H90N6O10: 1090.6718, found: 1091.6823 (MH+).
Compound No. 10
Compound 9 (80.0 mg, 0.073 mmol) was stirred with 70% TFA/DCM (5.0 ml) at room temperature for 3 hr. The resultant mixture was concentrated and dried under high vacuum to remove trace TFA. To this crude mixture were added, amino-benzyl-DTPA-penta-tert-butyl ester (286.0 mg, 0.366 mmol), EDCI (84.4 mg, 0.44 mmol) and DMAP (53.7 mg, 0.44 mmol). Dry dichloromethane (30 ml) was added and the reaction mixture was stirred at RT for 16 hr under N2 atm. The reaction mixture was diluted with dichloromethane (100 ml), washed with brine solution, the organic layer separated, dried over sodium sulfate and concentrated. The crude mixture was chromatographed over alumina G (III) using 1-3% methanol/dichloromethane mixture as eluent to give product 10. Yield: 140.0 mg (59.57%). UV-vis (λmax, dichloromethane): 365, 418, 509, 546, 699 nm. 1HNMR (400MHz, CDCl3): δ 9.75 (splitted s, 1H, meso-H), 9.63 (splitted s, 1H, meso-H), 9.33 (brs, 1H, NH), 8.60 (splitted s, 1H, meso-H), 7.61 (m, 2H, Ph-DTPA), 7.58 (m, 1H, Ph-DTPA), 7.37 (m, 3H, Ph-DTPA), 7.12 (m, 5H, Ph-DTPA), 6.84 (m, 1H, Ph-DTPA), 5.76 (m, 1H, CH3CHObutyl), 5.39 (m, 1H, H-17), 4.45 (m, 3H, H-18 & NCH2butyl), 3.82 (s, 3H, ring-CH3), 3.65 (m, 4H, 8-CH2CH3 & OCH2butyl), 3.38 (m, 22H, 11CH2-DTPA), 3.31 (m, 11H, 4CH2-DTPA & ring-CH3), 3.17 (s, 3H, ring-CH3), 3.03 (m, 3H, CH-DTPA), 2.84-2.61 (m, 19H, 9CH2-DTPA & CH-172), 2.47 (m, 8H, 6CH2-benzyl & CH-172 & CH-171), 2.20 (m, 6H, 3CH2-chain), 2.04 (d, 3H, CH3CHObutyl, J = 6.8 Hz), 1.96 (m, 6H, 3CH2-chain), 1.84 (m, 1H, CH-171), 1.73 (s, 3H, 17-CH3), 1.67 (t, 3H, 8-CH2CH3, J = 7.2 Hz), 1.60 (m, 4H, 2CH2-Obutyl), 1.41 (m, 135H for 15 CO2tBu), 1.37 (m, 4H, 2CH2-N-butyl), 1.03 (t, 3H, CH3-Obutyl, J = 6.8 Hz), 0.86 (t, 3H, CH3-N-butyl, J = 6.8 Hz), −0.09 (brs, 1H, NH). HRMS: Calculated for C174H270N18O37: 3206.104, found: 3207.250 (MH+).
Compound No. 11
Compound 10 (130.0 mg, 0.04 mmol) was stirred with 70% TFA/DCM (5.0 ml) at room temperature for 3 hr. The resultant mixture was concentrated and dried under high vacuum to remove trace TFA. The crude preparation thus obtained was dissolved in pyridine (10 ml) and while stirring, GdCl3.6H2O (90.3 mg, 0.243 mmol) in 1 ml of water was added slowly and the resultant mixture was stirred for 16 hr. The reaction mixture was concentrated to dryness under high vacuum. The residue was washed with water (10 ml × 3), acetone (10 ml × 3) and finally dried under high vacuum using P2O5 as drying agent. Yield: 80.0 mg (69.9%). UV-vis (λmax, MeOH): 364, 415, 546 and 700 nm. Elemental analysis: Calculated for C114H150Gd3N18O37: C, 48.28; H, 5.33; Gd, 16.63; N, 8.89; O, 20.87, found: C, 48.14; H, 5.40; N, 8.93.
Compound No. 12
HPPH (100.0 mg, 0.157 mmol), Di-tert-butyl iminodiacetate (77.0 mg, 0.314 mmol), EDCI (60.2 mg, 0.314 mmol) and DMAP (38.36 mg, 0.314 mmol) were taken in a dry RBF (100 ml). Dry dichloromethane (30 ml) was added and the reaction mixture was stirred at room temperature for 16 hr under N2 atm. The reaction mixture was diluted with dichloromethane (100 ml), washed with brine solution, the organic layer separated, dried over sodium sulfate and concentrated. The crude mixture was chromatographed over silica gel using 1-1.5% methanol/dichloromethane mixture as eluent to give product 12. Yield: 120.0 mg (88.3%). UV-vis (λmax nm, dichloromethane): 317, 411, 506, 538, 605 and 660 nm. 1HNMR (400MHz, CDCl3): δ 9.80 (s, 1H, H-5), 9.51 (s, 1H, H-10), 8.55 (s, 1H, H-20), 5.94 (q, 1H, CH3CHOhexyl, J = 6.4 Hz), 5.33 (d, 1H, CH-151, J = 20.0 Hz), 5.17 (d, 1H, CH-151, J = 20.0 Hz), 4.52 (q, 1H, H-17, J = 7.6 Hz), 4.41 (m, 1H, H-18), 4.04 (m, 2H, CH2chain), 3.75 (m, 2H, −OCH2-hexyl), 3.67 (s, 3H, 7-CH3), 3.62 (m, 2H, 8-CH2CH3), 3.42 (s, 3H, 2-CH3), 3.37 (m, 2H, CH2chain), 3.29 (s, 3H, 12-CH3), 2.77 (m, 1H, CH-172), 2.46 (m, 1H, CH-172), 2.16 (m, 1H, CH-171), 2.13 (m, 3H, & CH3CHOhexyl), 1.97 (m, 1H, CH-171), 1.84 (d, 3H, 18-CH3, J = 7.2 Hz), 1.78 (m, 2H, CH2hexyl), 1.72 (t, 3H, 8-CH2CH3, J = 7.6 Hz), 1.49 (s, 9H, CO2tBu), 1.46 (m, 2H, CH2hexyl), 1.45 (s, 9H, CO2tBu), 1.25 (m, 4H, 2CH2hexyl), 0.8 (t, 3H, CH3hexyl, J = 6.8Hz), 0.42 (brs, 1H, NH), −1.7 (brs, 1H, NH). EIMS: 865 (MH+) (25).
Compound No. 13
Compound 12 (120.0 mg, 0.139 mmol) was stirred with 70% TFA/DCM (5.0 ml) at room temperature for 3 hr. The resultant mixture was concentrated and dried under high vacuum to remove trace TFA. To this crude preparation were added, amine A (144.2 mg, 0.34 mmol), EDCI (106.6 mg, 0.556 mmol) and DMAP (67.8 mg, 0.556 mmol). Dry dichloromethane (30 ml) was added and the reaction mixture was stirred at RT for 16 hr under N2 atm. The reaction mixture was diluted with dichloromethane (100 ml), washed with brine solution, organic layer separated, dried over sodium sulfate and concentrated. The cude mixture was chromatographed over silica gel using 2-6% methanol/dichloromethane mixture as eluent to give product 13. Yield: 190.0 mg (88.3%). UV-vis (λmax, dichloromethane): 318, 411, 506, 537, 605 and 660 nm. 1HNMR (400MHz, CDCl3): δ 9.78 (s, 1H, H-5), 9.52 (s, 1H, H-10), 8.3 (s, 1H, H-20), 8.27 (brs, 1H, NH), 8.17 (brs, 1H, NH), 6.59 (brs, 1H, NH), 5.90 (m, 1H, CH3CHOhexyl), 5.34 (d, 1H, CH-151, J = 20.0 Hz), 5.15 (d, 1H, CH-151, J =20.0 Hz), 4.53 (q, 1H, H-17, J = 6.0 Hz), 4.36 (m, 1H, H-18), 3.77 (m, 2H, -OCH2-hexyl), 3.69 (m, 2H, 8-CH2CH3), 3.67 (s, 3H, 7-CH3), 3.61 (m, 4H, 2CH2chain), 3.36 (s, 3H, 2-CH3), 3.26 (s, 3H, 12-CH3), 2.77 (m, 1H, CH-172), 2.66 (m, 1H, CH-172), 2.52 (m, 1H, CH-171), 2.23 (m, 12H, 6CH2-chain), 2.11 (d, 3H, CH3CHOhexyl, J = 6.4 Hz), 2.07 (m, 6H, 3CH2-chain), 1.95 (m, 6H, 3CH2-chain), 1.93 (m, 1H, CH-171), 1.81 (d, 3H, 18-CH3, J = 7.2 Hz), 1.75 (m, 2H, CH2hexyl), 1.71 (t, 3H, 8-CH2CH3, J = 8.0 Hz), 1.43 (m, 2H, CH2hexyl), 1.41 (s, 27H, 3CO2tBu), 1.32 (s, 27H, 3CO2tBu), 1.24 (m, 4H, 2CH2hexyl), 0.77 (t, 3H, CH3hexyl, J = 6.8Hz). EIMS: 1548 (MH+). HRMS: Calculated for C87H131N7O17: 1545.9601, found: 1545.9574 (M+).
Compound No. 14
Compound 13 (100.0 mg, 0.064 mmol) was stirred with 80% TFA/DCM (5.0 ml) at room temperature for 3 hr. The resultant mixture was concentrated and dried under high vacuum to remove trace TFA. To this crude preparation were added, amino-benzyl-DTPA-penta-tert-butyl ester (503.5 mg, 0.64 mmol), EDCI (123.9 mg, 0.64 mmol) and DMAP (78.8 mg, 0.64 mmol). Dry N, N-dimethylformamide (15 ml) was added and reaction mixture was stirred at RT for 16 hr under N2 atm. The reaction mixture was concentrated under vacuum, dichloromethane added (100 ml), washed with brine solution, the organic layer separated, dried over sodium sulfate and concentrated. The crude mixture was chromatographed over alumina G (III) using 1-3% methanol/dichloromethane mixture as eluent to give product 14. Yield: 250.0 mg (70.0%). UV-vis (λmax, dichloromethane): 318, 413, 507, 539, 606 & 660. Elemental analysis: Calculated for C309H491N31O71: C, 64.25; H, 8.57; N, 7.52; O, 19.67, found: C, 64.30; H, 8.59; N, 7.56.
Compound No. 15
Compound 14 (225.0 mg, 0.038 mmol) was stirred with 70% TFA/DCM (5.0 ml) at room temperature for 3 hr. The resultant mixture was concentrated and dried under high vacuum to remove trace TFA. The crude preparation thus obtained was dissolved in pyridine (10 ml) and while stirring, GdCl3.6H2O (173.7 mg, 0.46 mmol) in 1 ml of water was added slowly and the resultant mixture was stirred for 16 hr. The reaction mixture was concentrated to dryness under high vacuum. The residue was washed with water (10 ml × 3), acetone (10 ml × 3) and finally dried under high vacuum using P2O5 as drying agent. Yield: 170.0 mg (86.7%). UV-vis (λmax, MeOH): 320, 411, 507, 539, 606 and 660 nm. Elemental analysis: Calculated for C189H251Gd6N31O71: C, 45.07; H, 5.02; Gd, 18.73; N, 8.62; O, 22.55, found: C, 45.15; H, 5.10; N, 8.58.
Compound No. 16
Acid 4 (150.0 mg, 0.214 mmol), Di-tert-butyl iminodiacetate (105.0 mg, 0.429 mmol), EDCI (82.3 mg, 0.429 mmol) and DMAP (52.0 mg, 0.429 mmol) were taken into a dry RBF (100 ml). Dry dichloromethane (30 ml) was added and the reaction mixture was stirred at RT for 16 hr under N2 atm. The reaction mixture was diluted with dichloromethane (100 ml), washed with brine solution, the organic layer was separated, dried over sodium sulfate and concentrated. The crude mixture was chromatographed over silica gel using 1-1.5% methanol/dichloromethane mixture as eluent to give product 16. Yield: 165.0 mg (82.9%). UV-vis (λmax, dichloromethane): 318, 411, 506, 536, 605 and 661 nm. 1HNMR (400MHz, CDCl3): δ 9.75 (splitted s, 1H, H-5), 9.52 (splitted s, 1H, H-10), 8.53 (s, 1H, H-20), 6.01 (m, 1H, CH3CHOTEG), 5.31 (d, 1H, CH-151, J = 20.0 Hz), 5.13 (d, 1H, CH-151, J = 20.0 Hz), 4.50 (q, 1H, H-17, J = 7.2 Hz), 4.36 (m, 1H, H-18), 4.02 (m, 2H, CH2chain), 3.85 (m, 2H, CH2-O-TEG), 3.79 (m, 2H, CH2-O-TEG), 3.73 (m, 4H, 3CH2-O-TEG), 3.68 (s, 3H, 7-CH3), 3.66 (m, 2H, 8-CH2CH3), 3.55 (m, 2H, CH2-O-TEG), 3.39 (s, 3H, 2-CH3), 3.27 (s, 6H, 12-CH3 & OCH3-TEG), 2.75 (m, 1H, CH-172), 2.44 (m, 1H, CH-172), 2.41 (m, 1H, CH-171), 2.16 (m, 1H, CH-171), 2.14 (d, 3H, CH3CHOTEG, J = 6.4 Hz), 1.81 (d, 3H, 18-CH3, J = 7.6 Hz), 1.71 (t, 3H, 8-CH2CH3, J = 7.6 Hz), 1.44 (splitted s, 9H, CO2tBu), 1.06 (splitted s, 9H, CO2tBu), 0.39 (brs, 1H, NH), −1.80 (brs, 1H, NH). EIMS: 927 (MH+). HRMS: Calculated for C52H71N5O10: 925.5201, found: 926.5288 (MH+).
Compound No. 17
Compound 16 (140.0 mg, 0.151 mmol) was stirred with 70% TFA/DCM (5.0 ml) at room temperature for 3 hr. The resultant mixture was concentrated and dried under high vacuum to remove trace TFA. To this crude preparation were added, amine A (188.2 mg, 0.453 mmol), EDCI (115.9 mg, 0.604 mmol) and DMAP (73.7 mg, 0.604 mmol). Dry dichloromethane (30 ml) was added and the reaction mixture was stirred at RT for 16 hr under N2 atm. The reaction mixture was diluted with dichloromethane (100 ml), washed with brine solution, the organic layer separated, dried over sodium sulfate and concentrated. The crude mixture was chromatographed over silica gel using 3-7% methanol/dichloromethane mixture as eluent to give product 17. Yield: 210.0 mg (86.4%). UV-vis (λmax, dichloromethane): 318, 411, 506, 536, 604 and 661 nm. 1HNMR (400MHz, CDCl3): δ 9.76 (splitted s, 1H, H-5), 9.53 (s, 1H, H-10), 8.63 (s, 1H, H-20), 8.31 (splitted s, 1H, CONH), 6.56 (splitted s, 1H, CONH), 6.00 (m, 1H, CH3CHOTEG), 5.35 (d, 1H, CH-151, J = 20.0 Hz), 5.16 (d, 1H, CH-151, J = 20.0 Hz), 4.53 (q, 1H, H-17, J = 7.6 Hz), 4.36 (d, 1H, H-18, J = 10.4 Hz), 3.85-3.80 (m, 4H, 2CH2-O-TEG), 3.74-3.71 (m, 6H, 2CH2chain, CH2-O-TEG), 3.67 (s, 3H, 7-CH3), 3.66 (m, 4H, 8-CH2CH3, CH2-O-TEG), 3.53 (m, 2H, CH2-O-TEG), 3.42-3.39 (m, 5H, CH2-O-TEG, 2-CH3), 3.27-3.26 (m, 6H, 12-CH3, OCH3TEG), 2.75 (m, 1H, CH-172), 2.67 (m, 1H, CH-172), 2.52 (m, 1H, CH-171), 2.24-2.22 (m, 13H, 6CH2-chain, CH-171), 2.14 (d, 3H, CH3CHOTEG, J = 6.8 Hz), 2.09-2.04 (m, 6H, 3CH2-chain), 1.96 (m, 6H, 3CH2-chain), 1.81 (d, 3H, 18-CH3, J = 7.2 Hz), 1.71 (t, 3H, 8-CH2CH3, J = 8.0 Hz), 1.41 (s, 27H, 3CO2tBu), 1.33 (s, 27H, 3CO2tBu), 0.39 (brs, 1H, NH), −1.79 (brs, 1H, NH). EIMS: 1610 (MH+). HRMS: Calculated for C88H133N7O20: 1607.9605, found: 1607.9650 (M+).
Compound No. 18
Compound 17 (100.0 mg, 0.06 mmol) was stirred with 80% TFA/DCM (5.0 ml) at room temperature for 3 hr. The resultant mixture was concentrated and dried under high vacuum to remove trace TFA. To this crude preparation were added, amino-benzyl-DTPA-penta-tert-butyl ester (471.0 mg, 0.60 mmol), EDCI (115.9 mg, 0.60 mmol) and DMAP (73.8 mg, 0.60 mmol). Dry N, N-dimethylformamide (15 ml) was added and the reaction mixture was stirred at RT for 16 hr under N2 atm. The reaction mixture was concentrated under vacuum, dichloromethane added (100 ml), washed with brine solution, the organic layer separated, dried over sodium sulfate and concentrated. The crude mixture was chromatographed over alumina G (III) using 1-3% methanol/dichloromethane mixture as eluent to give product 18. Yield: 250.0 mg (69.0%). UV-vis (λmax, dichloromethane): 319, 412, 508, 538, 606 and 661 nm. Elemental analysis: Calculated for C309H491N31O74: C, 63.72; H, 8.50; N, 7.46; O, 20.33, found: C, 63.67; H, 8.57; N, 7.46.
Compound No. 19
Compound 18 (215.0 mg, 0.0369 mmol) was stirred with 80% TFA/DCM (5.0 ml) at room temperature for 3 hr. The resultant mixture was concentrated and dried under high vacuum to remove trace TFA. The crude preparation thus obtained was dissolved in pyridine (10 ml) and while stirring, GdCl3.6H2O (164.6 mg, 0.44 mmol) in 1 ml of water was added slowly and resultant mixture was stirred for 16 hr. The reaction mixture was concentrated to dryness under high vacuum. The residue was washed with water (10 ml × 3), acetone (10 ml × 3) and finally dried under high vacuum using P2O5 as drying agent. Yield: 160.0 mg (85.0%). UV-vis (λmax, MeOH): 320, 410, 507, 539, 606 and 661 nm. Elemental analysis: Calculated for C190H253Gd6N31O74: C, 44.76; H, 5.00; Gd, 18.50; N, 8.52; O. found: C, 44.80; H, 5.07; N, 8.51.
Calculation of In Vitro Relaxivities
MRI acquisitions were performed using a General Electric 4.7T / 33 cm horizontal bore magnet (GE NMR instruments, Fremont, CA) incorporating AVANCE™ digital electronics (Bruker BioSpec platform with ParaVision® Version 3.0.2 acquisition software, Bruker Medical, Billerica, MA).
Each conjugate was diluted with phosphate-buffered saline (pH 7.4) to a concentration ranging from 0.02 mM to 0.10 mM and imaged at 25°C. T1 relaxation rates (R1) were acquired utilizing a saturation recovery, spin-echo (SE) sequence with a fixed echo time (TE) = 10 ms and repetition times (TR) ranging from 75 to 8000 ms. Additional MR acquisition parameters were as follows: field of view (FOV) 32 × 32 mm, slice thickness = 1 mm, slices = 3, interslice gap = 2 mm, matrix = 192 × 192, number of averages (NEX) = 1. Signal intensities at each repetition time were sampled by taking the mean intensity within regions of interest (ROI’s) using commercially available image processing software (Analyze 7.0, AnalyzeDirect, Overland, KS), and R1 and SMAX were calculated by nonlinear fitting of the equation:
using Matlab’s Curve Fitting Toolbox (Matlab 7.0, MathWorks Inc., Natick, MA). The T1 relaxivity for each conjugate was then determined by obtaining the slope of the compound’s molar concentration vs. R1 via linear regression fitting of the data. Similarly, T2 relaxation rates (R2) were acquired using a multi-echo, Carr-Purcell-Meiboom Gill (CPMG) SE sequence with a fixed TR of 2500 ms and TE times ranging from 15 to 300 ms, NEX = 2. R2 and SMAX were calculated as described above using the equation:
As before, the T2 relaxivity was determined by obtaining the slope of concentration vs. R2 via linear regression fitting of the data.
Determination of in vitro photobleaching
In vitro photobleaching (26) of each compound was performed using a continuous light source at 665 nm at a power of 500 mW and a dose rate of 75 mW/cm2. Samples were prepared in 17% bovine calf serum. Individual, 2.5 mL samples in 17% bovine calf serum were irradiated in quartz cuvettes and monitored via a UV-Vis spectroscopy (Varian Cary Bio 50 spectrophotometer, Palo Alto, CA) at 665 nanometers until the peak had been reduced to a minimum of 70%.
Evaluation of phototocitiy viability
The compounds were tested for photodynamic efficacy at 4 μM and variable light doses (at 665 nm for HPPH analogues and 705 nm for purpurinimides) after incubating for 24 h with (Colon26) cells seeded in 96 well plates. Viability was measured 48 h later using the 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazoliumbromide (MTT) assay (26). The results were plotted as percent survival compared with the untreated dark control (no drug, no light) at each light dose (in J/cm2).
Determination of in vitro intracellular localization
Colon26 cells were seeded at 1 × 105 cells in 2 ml growth medium on poly-L-lysine coated glass coverslips in 6-well plates and cultured for 48 h to allow attachment and spreading. The photosensitizer (1.0 μM) was incubated with the cells in the dark at 37°C for 4 h or 24 h. The cells were co-incubated with organelle specific dyes MitoTracker Green FM for 30 minutes at 0.5 μM; Lysotracker Green at 1 μM for 2 h and Bodipy Ceramide C5-albumin complexes at 5 μM (27) according to manufacturer’s instructions (Invitrogen). Prior to microscopy the cells were rinsed with PBS and mounted with PBS containing 1 μM DAPI. Live cells were examined with a Zeiss Axiovert 200 inverted fluorescence with a Fluoarc mercury vapor shortarc lamp as light source. Fluorescent images from the same fields were captured successively in detector channels using suitable filter sets (Chromatechnology) for the PS and the organelle probes with an AxioCam MR-MRGrab frame grabber and a monochromatic CCD camera (Zeiss AxioCam MRm). Filter cubes contained 410/40 nm excitation, beamsplitter 505 and bandpass emission 675/50 nm for the PS; excitation bandpass 470/40 nm, beamsplitter 495, and bandpass emission 525/50 nm for MitoTracker Green and Lysotracker Green; excitation bandpass 565/30 nm, beamsplitter 585 and bandpass emission 620/60 nm for the Bodipy C5 ceramide and excitation 390/22 nm, beamsplitter 420 nm and bandpass emission 460/50 nm. Images were digitally processed with the Zeiss AxioVision LE v. 4.1 software.
Statistical Evaluation
In vitro PDT of the compounds was presented as the mean and standard deviation of three replicate experiments. In vivo PDT of the compounds was compared in Kaplan-Meier plots. GraphPad Prism 5.0 was utilized to compare tumor response curves
RESULTS AND DISCUSSION
Chemistry
For the preparation of photosensitizer-Gd(III) ADTPA conjugates, 3-(1′-hexyloxy ethyl)-3-devinylpyropheophorbide-a (HPPH) HPPH and 3-(1′-butyloxyethyl)3-devinyl-purpurin-18-N-butylbutylimide 8 were synthesized by following the methodologies developed in our laboratory (28). For the synthesis of HPPH-3Gd(III)ADTPA 3, HPPH was reacted with aminotriester 1 which on treating with trifluoroacetic acid (TFA) and further reaction with aminobenzylDTPAbutyl ester gave the desired conjugate. To investigate the effect of lipophilicity of the conjugate in tumor-imaging (MRI) and PDT, we synthesized a series of compounds with increasing/decreasing lipophilicity/hydrophilicity by modifying the position-17 of pyropheophorbide with alkyl ether (butyl or hexyl) or polyethylene glycol moieties. We started with compound 4 containing an ethylene glycol group at position-3, which on a sequence of reactions illustrated in Scheme-2 produced the corresponding 3Gd(III)aminobenzyl DTPA conjugate 7 in modest yield. For the synthesis of a bifunctional agent with longer wavelength absorption pyropheophorbide-a analogs (HPPH or compound 4) was replaced with a highly effective photosensitizer 8 (developed on the basis of structure-activity relationships studies) exhibiting longer wavelength absorption near 700 nm, and the corresponding 3Gd(III)ADTPA conjugate 11 was synthesized. To further investigate the effect of increased Gd(III)units in MR imaging ability and the effect of overall lipophilicity in pharmacokinetic/pharmacodynamic characteristics pyropheophorbide-6Gd(III)aminobenzyl conjugates 15 and 19 containing a hexyl ether or an ethylene glycol group at position-3 were synthesized by following the methodology as depicted in Schemes 4 and 5 respectively. The structures of all the intermediates and the final products were confirmed by NMR, mass spectrometry and elemental analyses.
Scheme-2.

Scheme-4.

Scheme-5.

Effect of gadolinium chelates in absorption and fluorescence properties of HPPH
To investigate the effect of the number of Gd(III) units in absorption and fluorescence properties of the conjugates, HPPH and the corresponding conjugates 3 and 15 containing three- and six gadolinium chelates were dissolved in methanol, the electronic absorption spectra and fluorescence spectra were measured at equimolar concentrations. As can be seen from Figure 1 (A), subtle deviations were observed between the UV-Vis spectra of HPPH, 3 (HPPH-3Gd(III) chelate and 15 (HPPH-6Gd(III)chelate). The Soret band region in compound 15 was much broader. The long wavelength absorption intensity at 660 nm for conjugates 3 and 15 was slightly lower (ε = 35000, 33000 respectively) than that observed for HPPH (ε = 45000).
Figure 1.

Comparative absorption (A) and fluorescence spectra (B) of HPPH and the corresponding 3 Gd(III)aminobenzyl DTPA 3 and 6Gd(III)aminobenzyl DTPA conjugate 15 in methanol (conc. 19.6 μM).
Compared to HPPH, the emission spectra (Figure 1B) of the metallated analogs 3 and 15 showed a significant decrease in fluorescence intensity. Excitations of these compounds in the “Soret” band region (410 – 425 nm) exhibited an emission peak at 667 nm and as can be seen from Figure 1 (B), compared to HPPH the corresponding 3Gd(DTPA) analog 3 produced a log decrease in fluorescence intensity, which further decreased by increasing the gadolinium units in conjugate 15. A sharp decrease in fluorescence intensity indicates a possible interaction between the gadolinium chelates and the HPPH moiety and detailed photophysical studies of this phenomenon are currently in progress.
Effect of Gd(III)aminobenzyl DTPA moiety in photobleaching of HPPH
The singlet oxygen produced on exposing photosensitizer within a tumor to light is responsible for the destruction of tumor and also the photosensitizer (29,30). In order to achieve an optimal PDT response, it is extremely important to make the maximum use of the tumor oxygen supply and also to understand the photobleaching characteristics of the photosensitizer by the single oxygen produced in situ. In this study, HPPH and the corresponding gadolinium analogs 3 and 15 with 3 and 6 Gd(III)aminobenzyl DTPA units respectively were dissolved in 17% bovine calf serum (BCS) solution and irradiated with laser light at 665 nm. The absorption spectra were measured at regular intervals and the observed peak intensity values at λmax 665 nm were plotted against the light exposure time.
As can be seen from the results summarized in Figure 2, the presence of Gd(III)aminobenzl DTPA moieties in both conjugates 3 and 15 significantly reduced the rate of photobleaching of the HPPH. The longer stability of the Gd(III) containing photosensitizers provides an opportunity for an additional light treatment to those cancer patients, which did not produce satisfactory response after initial treatment without injecting more PDT agent.
Figure 2.

Comparative photobleaching characteristics of HPPH and the corresponding 3 and 6Gd(III) aminobenzyl DTPA 3 and 15 respectively at (9.8 M) in 17% BCS. The solutions were exposed with a laser light (500 mW, 665 nm, 75 mW/cm2) and the absorption peak at 665 nm were recorded at variable time points.
T1 and T2 relaxivity of HPPH-Gd(III)aminobenzylDTPA conjugates
The T1 and T2 relaxivity values of the photosensitizer-Gd(III)ADTPA conjugates are summarized in Table 1.
Table 1.
Comparative T1, T2 relaxivity values of the conjugates and Magnevist•
| (mM • s)-1 | ||
|---|---|---|
| Compound | T1 | T2 |
| 3 | 13.50 | 81.37 |
| 15 | 24.72 | 66.54 |
| 11 | 14.14 | 85.32 |
| 7 | 13.58 | 40.11 |
| Magnevist ® | 4.33 | 5.10 |
| 19 | 25.09 | 69.19 |
Compared to Magnevist®, the relaxivity values were higher for compounds containing 3-or 6Gd-ADTPA moieties. However, the increase in relaxivity was non-linear with the increase in the number of Gd (III) units. The reason for the lack of linearity is attributed to the relative close proximity of Gd-ADTPA groups, which may be reducing the degree of relaxation enhancement by limiting the hydration of the molecule.
In selection of a lead candidate, T1 relaxivity was favored over T2, as an increase in T1 relaxation rate reduces saturation of signal, leading to brighter intensity in tumors, while an increase in T2 relaxation rate causes loss of signal intensity, producing poorer contrast. Although all compounds exhibited adequate T1/T2 ratios, compounds 15 and 19 displayed the highest T1/T2 ratios, but limited photosensitizing efficacy.
In vitro Photosensitizing Activity
To determine the effect of the nature of photosensitizer and the number of Gd(III)ADTPA moieties on photosensitizing efficacy, conjugates 3, 7, 11, 15 and 19 were evaluated in RIF cells at various concentrations and light doses.
The results obtained at a fixed concentration of the conjugates (4.0 μM), but at variable light doses are shown in Figure 3. As can be seen, among all the derivatives both HPPH and purpurinimide linked with 3Gd(III)ADTPA moieties (3 and 11 respectively) were more effective than those containing 6Gd(III)ADTPA moieties (15 and 19 respectively). Interestingly, replacing the hexyl ether functionality at position-3 with PEG (7 and 19) reduced the in vitro efficacy.
Figure 3.
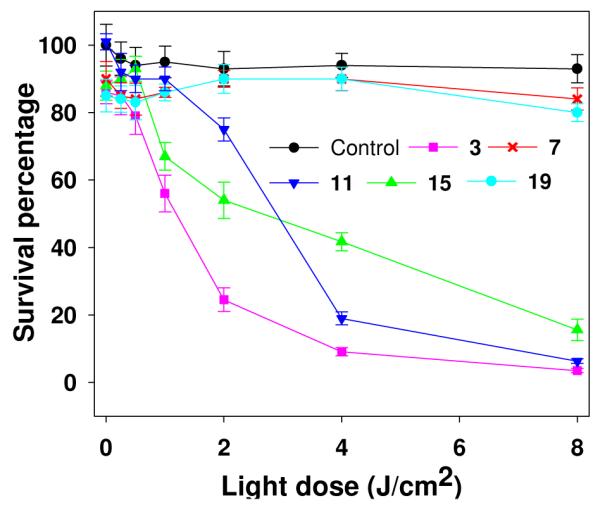
In vitro photosensitizing activity of photosensitizer-Gd(III)ADTPA conjugates (4.0 μM) in RIF cells. The cells were incubated for 24h and then exposed to light at a dose of 3.2 mW/cm2. Control cells were exposed to light only.
Intracellular localization of HPPH vs. HPPH-3Gd(III)ADTPA Conjugate
Most of the porphyrin-based compounds can be observed intracellularly with a fluorescence microscope. It has been shown that photosensitizers localize to cytosolic targets such as Golgi, endoplasmic reticulum, mitochondria, lysosomes and membranes (31, 32). Efforts have been made to investigate a correlation between the site(s) of localization and PDT activity of photosensitizers. Moan et. al. (33) suggested that lysosomes are a primary localization site but not the photodynamic target. However, it has not been determined whether the lysosomes repture after PDT damage and release the compound or whether some other cellular damage, caused by unobservable functions of the photosensitizer elsewhere in the cell, cause the lysosome to rupture and subsequently release the photosensitizer. We have previously shown that most of the effective PDT agents, including HPPH developed in our laboratory localize either in mitochondria or in lisosomes (34). We have also observed that besides the overall lipophilicity, the nature of the substituents introduced in photosensitizers also makes a remarkable difference in intracellular localization characteristics (35).
To determine the impact of Gd(III)ADTPA moieties in site of localization of HPPH, the intracellular localization of both the compounds was investigated by using various site-specific probes. Although some of the conjugates were also found in the mitochondra and lysosomes (see Figure 4 and the Supplementary Information), localization of the lead Gd-containing compound 3 appeared to be primarily in the Golgi apparatus as determined by its colocalization with a probe for that organelle. Addition of Gd alone may not account for the shift from mitochondria observed with HPPH to the Golgi, but may be partly due to the position at which the additional Moieties are attached, and a significant change in overall lipophilicity of the molecule. Primarily, photosensitizers which localize to the mitochondria are more effective at evoking cell death (34), however photosensitizers which localize to alternative cellular organelles also exhibit the ability to induce cell mortality upon light irradiation (36). Further studies to establish a correlation between the site(s) of localization and cell-death (apoptosis vs. necrosis) are currently in progress.
Figure 4.

False-color images showing subcellular localization of HPPH and compound 3 (red) in Colon26 cells after 4h (B) or 24 h incubations (A and C) respectively. Localization was determined by fluorescence microscopy by co-incubation with fluorescent organelle probes (green). Subcellular areas of co-localization in the overlay of compound 3 or HPPH with the probes for are yellowish orange. A, HPPH; B and C, compound 3; D, MitoTracker Green; E and F, Bodipy ceramide C5-albumin complex; G, overlay of HPPH and MitoTracker Green; H and I overlays of compound 3 and Bodipy ceramide C5-albumin complex
CONCLUSION
Our studies indicate that tumor-avid photosensitizers can be used as vehicles for delivering the MR imaging agents to tumors. Compared to Magnevist, a clinical standard, most of the conjugates showed enhanced tumor-imaging potential (T1/T2 relaxivity) which increased by increasing the number of Gd(III)ADTPA units. However, as a “Multifunctional Agent” for tumor-imaging (MR and fluorescence) and PDT, HPPH containing 3Gd(III)ADTPA showed the best PDT efficacy (in vivo results are presented in the following paper of this journal). Our approach has a great potential in developing dual-function agents for MR imaging and phototherapy with increased sensitivity by reducing false-positive and false-negative results in screening, diagnosis, treatment monitoring or image-guided intervention.
Supplementary Material
Scheme-1.

Scheme-3.

ACKNOWLEDGMENT
The financial support from the NIH (R21/R33 Grant CA109914) and the shared resources of the RPCI support grant (P30CA16056) is highly appreciated.
Footnotes
Supporting Information Available: The 1H NMR spectra of selected compounds and the intracellular localization profiles of the conjugates. This material is available free of charge via the Internet at http:/pubs.acs.org.
LITERACTURE CITED
- (1).Caravan P, Ellison JJ, McMurry TJ, Lauffer RB. Gadolinium(III) chelates as MRI contrast agents: Structure, dynamics and applications. Chem. Rev. 1999;99:2293–2352. doi: 10.1021/cr980440x. [DOI] [PubMed] [Google Scholar]
- (2).Kolokythas O, Shibata DK, Dubinsky TJ. In: Medical Imaging in Image-Guided Therapy Systems. Vaezy S, Zderic V, editors. Artech House; Boston: 2009. pp. 17–57. [Google Scholar]
- (3).Hamblin MR, Mroz P, editors. Advances in Photodynamic Therapy. Artech House; Norwood MA: 2008. [Google Scholar]
- (4).Carpenter CM, Pogue BW. In: Difuse Optical Spectroscopy with Magnetic Resonance Imaging in Translational Multimodality Optical Imaging. Azar FS, Intes X, editors. Artech House; Boston: 2008. pp. 125–162. [Google Scholar]
- (5).Povoski SP, Hall NC, Marin EW, Walker MJ. Multimodality approach of perioperative 18F-FDG PET/CT imaging, intraoperative 18F-FDG handheld gamma probe detection, and intraoperative ultrasound for tumor localization and verification of resection of all sites of hypermetabolic activity in case of occult recurrent metastatic melanoma. World J. Surg. Oncol. 2008;6:1. doi: 10.1186/1477-7819-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Shah NJ, Gibbs J, Wolverton D, Cerussi A, Hylton N, Tromberg BJ. Combined diffuse optical spectroscopy and contrast-enhanced magnetic resonance imaging for monitoring breast cancer neoadjuvant chemotherapy: A case study. J. Biomed. Opt. 2005;10:051503. doi: 10.1117/1.2070147. [DOI] [PubMed] [Google Scholar]
- (7).Ruck A, Dolp F, Hulshoff C, Hauser C, Scaffi-Happ C. Fluorescence lifetime in PDT. An overview. Medical Laser Application. 2005;20:125–129. [Google Scholar]
- (8).Van den Bergh H. Early detection of lung cancer and the role of endoscopic fluorescence imaging. Medical Laser Application. 2003;18:20–26. [Google Scholar]
- (9).Smith KM, editor. Porphyrins and metalloporphyrins. Elsevier Scientific Publishing Company; Amsterdam: 1975. [Google Scholar]
- (10).Gurfinkel M, Thompson AB, Ralston W, Troy TL, Reynolds SR, Muggenberger B, Nikula K, Pandey RK, Mayer R, Hawrysz DJ, Sevick-Muraca EM. Pharmacokinetics of indocyanine green and carotene-HPPH-conjugate for detection of normal and tumor tissue using fluorescence near infrared continuous wave imaging. Photochem. Photobiol. 2000;72:94–102. doi: 10.1562/0031-8655(2000)072<0094:poiahc>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- (11).Li G, Slansky A, Dobhal MP, Goswami LN, Graham A, Chen Y, Kanter P, Aleberico RA, Spernyak J, Morgan J, Mazurchuk R, Oseroff A, Grossman Z, Pandey RK. Chlorophyll-a analogues conjugated with aminobenzyl DTPA as potential bifunctional agents for magnetic resonance imaging and photodynamic therapy. Bioconjugate Chem. 2005;16:32–42. doi: 10.1021/bc049807x. [DOI] [PubMed] [Google Scholar]
- (12).Chen Y, Gryshuk A, Achilefu S, Ohulchanskyy T, Potter W, Zhong T, Morgan J, Chance B, Prasad PN, Henderson BW, Oseroff A, Pandey RK. A novel approach to a bifunctional photosensitizer for tumor imaging and phototherapy. Bioconjugate Chem. 2005;16:1264–1274. doi: 10.1021/bc050177o. [DOI] [PubMed] [Google Scholar]
- (13).Pandey SK, Gryshuk AL, Sajjad M, Zgeng X, Chen Y, Abouzeid MM, Morgan J, Nabi HA, Oseroff A, Pandey RK. Multimodality agents for tumor imaging (PET, fluorescence) and PDT. A possible “see and treat” approach. J. Med. Chem. 2005;48:6286–6295. doi: 10.1021/jm050427m. [DOI] [PubMed] [Google Scholar]
- (14).Pandey SK, Chen Y, Zawada RH, Oseroff A, Pandey RK. Utility of tumor-avid photosensitizers in developing bifunctional agents for tumor-imaging and phototherapy. Proc. SPIE. 2006;6139:6139051–57. [Google Scholar]
- (15).Chen Y, Ohkubo K, Zhang M, Liu W, Pandey SK, Ciesielski M, Baumann H, Fukuzumi S, Kadish KM, Fenstermaker R, Oseroff A, Pandey RK. Photophysical, electrochemical characteristics and cross linking of STAT-3 proteins by an efficient bifunctional agent for fluorescence image-guided photodynamic therapy. Photochem. Photobiol. Sci. 2007;6:1257–1267. doi: 10.1039/b710395f. [DOI] [PubMed] [Google Scholar]
- (16).Pandey RK, Sumlin AB, Potter WR, Bellnier DA, Henderson BW, Constantine S, Aoudia M, Rodgers MA, Smith KM, Dougherty TJ. Structure and photodynamic efficacy among alkyl ether analogues of chlorophyll-a derivatives. Photochem. Photobiol. 1996;63:194–205. doi: 10.1111/j.1751-1097.1996.tb02442.x. [DOI] [PubMed] [Google Scholar]
- (17).Henderson BW, Bellnier DA, Graco WR, Sharma A, Pandey RK, Vaughan L, Weishaupt KR, Dougherty TJ. A quantitative structure-activity relationship for a congeneric series of pyropheophorbide derivatives as photosensitizers for photodynamic therapy. Cancer Res. 1997;57:4000–4007. [PubMed] [Google Scholar]
- (18).Dougherty TJ, Pandey RK, Nava HR, Smith JA, Douglass HO, Edge SB, Bellnier DA, O’Malley L, Cooper M. Preliminary clinical data on a new photodynamic therapy photosensitizer, HPPH for treatment of obstructive esophageal cancer. Proc. SPIE. 2000;3909:25–27. [Google Scholar]
- (19).Bellnier DA, Greco WR, Loewen GM, Nava V, Oseroff A, Pandey RK, Tsuchida T, Dougherty TJ. Population pharmacokinetics of the photodynamic therapy agent HPPH in cancer patients. Cancer Res. 2003;63:1806–1813. [PubMed] [Google Scholar]
- (20).Loewen GM, Pandey RK, Bellnier DA, Henderson BW, Pandey RK. Endobronchial photodynamic therapy for lung cancer. Lasers in Surgery and Medicine. 2006;38:364–370. doi: 10.1002/lsm.20354. [DOI] [PubMed] [Google Scholar]
- (21).Bellnier DA, Greco WR, Nava H, Loewen GM, Oseroff AR, Dougherty TJ. Mild skin phototoxicity in cancer patients following injection of Photochlor (HPPH) for photodynamic therapy. Cancer Chemother. Pharmacol. 2005;57:40–45. doi: 10.1007/s00280-005-0015-6. [DOI] [PubMed] [Google Scholar]
- (22).Pandey RK, Goswami LN, Chen Y, Gryshuk A, Missert JR, Oseroff A, Dougherty TJ. Nature: A rich source for developing multifunctional agents. Tumor-imaging and photodynamic therapy. Lasers in Surgery and Medicine. 2006;38:445–467. doi: 10.1002/lsm.20352. [DOI] [PubMed] [Google Scholar]
- (23).Spernyak J, White WH, Ethirajan M, Patel N, Goswami L, Chen Y, Turowski S, Missert JR, Batt C, Mazurchuk R, Pandey RK. Hexylether Derivative of Pyropheophorbide-a (HPPH) on Conjugating with 3Gadolinium(III) Aminobenzyl diethylenetriaminopentaacetic Acid Shows Potential for In vivo Tumor-Imaging (MR, Fluorescence) and Photodynamic Therapy. Bioconjugate Chem. 2009 doi: 10.1021/bc9005317. Submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zheng G, Camacho S, Potter W, Bellnier DA, Henderson BW, Dougherty TJ, Pandey RK. Synthesis, tumor-uptake and in vivo photosensitizing efficacy of a homologues series of 3-(1’-alkoxyethyl)purpurin-18-N-alkylimides. J. Med. Chem. 2001;44:1540–1559. doi: 10.1021/jm0005510. [DOI] [PubMed] [Google Scholar]
- (25).Pandey RK. Unpublished results.
- (26).Merlin JL, Azzi S, Lignon D, Ramacci C, Zeghari N, Guillemin F. MTT assays allow quick and reliable measurement of the response of human tumour cells to photodynamic therapy. Eur. J. Can. 1992;28A:1452–1458. doi: 10.1016/0959-8049(92)90542-a. [DOI] [PubMed] [Google Scholar]
- (27).Kessel D, Castelli M. Evidence that bcl-2 is the target of three photosensitizers that induce a rapid apoptotic response. Photochem. Photobiol. 2001;74:318–322. doi: 10.1562/0031-8655(2001)074<0318:etbitt>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- (28).Pandey RK. Unpublished results.
- (29).Finlay JC, Mitra S, Foster TH. In vivo mTHPC photobleaching in normal rat skin exhibits irradiance dependent features. Photochem. Photobiol. 2002;75:282–288. doi: 10.1562/0031-8655(2002)075<0282:ivmpin>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- (30).Sharman WM, Allen CM, Van Lier JE. Role of activated oxygen species in photodynamic therapy. Enzymology. 2000;319:57. doi: 10.1016/s0076-6879(00)19037-8. [DOI] [PubMed] [Google Scholar]
- (31).Kessel D, Woodburn K. Biodistribution of photosensitizing agents. Int. J. Biochem. 1993;10:1377–1383. doi: 10.1016/0020-711x(93)90685-8. [DOI] [PubMed] [Google Scholar]
- (32).Malik Z, Amit I, Rothmann C. Subcellular localization of sulfonated tetraphenyl porphines in colon carcinoma cells by spectrally resolved imaging. Photochem. Photobiol. 1997;65:389–396. doi: 10.1111/j.1751-1097.1997.tb08576.x. [DOI] [PubMed] [Google Scholar]
- (33).Moan J, Berg K, Anholt A, Madslien K. Sulfonated aluminum phthalocyanines as sensitizers for photochemotherapy. Effects of small doses on localization, dye fluorescence and photosensitivity in V79 cells. Int. J. Cancer. 1994;58:865–870. doi: 10.1002/ijc.2910580620. [DOI] [PubMed] [Google Scholar]
- (34).Morgan J, Oseroff AO, Torchilin VP. Mitochondrial based anti-cancer photodynamic therapy. Adv. Drug Delivery Rev. 2001;49:71–86. doi: 10.1016/s0169-409x(01)00126-0. [DOI] [PubMed] [Google Scholar]
- (35).Zheng X, Morgan J, Pandey SK, Chen Y, Tracy E, Baumann H, Missert JR, Batt C, Jackson J, Bellnier DA, Henderson BW, Pandey RK. Conjugation of HPPH to carbohydrates changes its subcellular distribution and enhances photodynamic activity in vivo. J. Med. Chem. 2009;52:4306–4318. doi: 10.1021/jm9001617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sibrian-Vazquez M, Ortiz J, Nesterova IV, Fernandez-Lazaro F, Sastre-Sanstos A, Soper SA, Vicente GH. Synthesis and properties of cell-targeted Zn(II)-phthalocyanine-peptide conjugates. Bioconjugate Chem. 2007;18:410–420. doi: 10.1021/bc060297b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.