

Abstract

Bacterial virus phi29 genomic DNA is packaged into a procapsid shell with the aid of a motor containing a 12-subunit connector channel and a hexameric pRNA (packaging RNA) ring. The wide end, or the C-terminus, of the cone-shaped connector is embedded within the procapsid shell, whereas the narrow end, or N-terminus, extends outside of the procapsid, providing a binding location for pRNA. Recently, we have reported the mechanism of in vivo assembly of an ellipsoid nanoparticle with seven connectors through an interaction among a peptide tag. Here we report the formation of a similar nanoparticle in vitrovia the addition of DNA or RNA oligos to connector proteins. Free connectors guided by one or two copies of oligonucleotides were assembled into a rosette structure containing 60 subunits of reengineered proteins. The number of oligonucleotides within the particle is length-dependent but sequence-independent. Reversible shifting between the 12- and 60-subunit nanoparticles (between individual connectors and rosette structures, respectively) was demonstrated by the alternative addition of oligonucleotides and the treatment of ribonuclease, suggesting a potential application as a switch or regulator in nanobiotechnology. This advancement allows for a simple method to produce multivalent nanoparticles that contain five 12-unit nanoparticles with defined structure and stoichiometry. That is, it will be possible to assemble nanoparticles in vitro with the combination of 60 assortments of ligands, tags, therapeutic drugs, and diagnostic moieties for multivalent delivery or enhancement of signal detection in nanotechnological and nanomedicinal applications.
Keywords: protein interaction, assembly of RNA/protein nanoparticles, multivalent protein nanoparticle, multivalent nanoparticle for diagnosis and detection, viral assembly
The dsDNA viruses of humans, animals, and bacteria, including herpes viruses,(1) adenovirus,(2) phi29,(3) λ,(4) SPP1,(5) P22,(6) T3,(7) T4,(8) T7,(9) and Epsilon 15,(10) package their genomic DNA inside a preformed protein shell, called a procapsid. This entropically and energetically unfavorable DNA condensation task is accomplished with the aid of a viral DNA-packaging motor involving two nonstructural components(11) and a connector core.12,13 The powerful motor converts the chemical energy from ATP hydrolysis into a physical force for motor motion.11,14−16 In bacteriophage phi29, the motor is geared by a hexameric pRNA ring17−19 and a DNA-packaging enzyme gp16.11,20,21 The in vitro phi29 DNA packaging(22) has been investigated by single molecule dual-view imaging3,23 and by using optical tweezers.(24)
The head−tail connector, also called a portal protein, plays an essential role in procapsid assembly and DNA packaging. Although the portal proteins from different viruses share little sequence identities, analysis of their structures reveals a significant similarity in morphology, displaying a truncated cone shape composed of 12 protein subunits, which accounts for the common function in DNA packaging. The detailed structure of the phi29 connector has been thoroughly studied and solved at the atomic level.25,26 The 12-fold symmetric dodecamer is 7.5 nm tall, with an external diameter of 13.8 nm at its wide end and 6.6 nm at its narrow end.(25) The diameter of the central channel at the wide end and narrow end is about 6.0 and 3.6 nm, respectively. The C-terminus of each protein is located at the wide end and is embedded in the procapsid, while the N-terminus is at the narrow end and is exposed to the solvent.
The phi29 pRNA contains two functional domains.(27) One is the gp16 binding domain, located at the 5′/3′ paired helical region.(20) The other domain, located at the central region,28,29 is responsible for inter-RNA interaction. This domain contains two left- and right-hand interlocking loops required for specifically assembling a hexameric pRNA ring with the appropriate size to encircle the connector.18,30−32 However, individual pRNA subunits may bind to the connector or procapsid nonspecifically, like that of nonspecific tRNA.(19) Therefore, specificity in connector−pRNA interaction is attributed to the formation of the static hexameric pRNA ring that surrounds the connector.
Previous studies on the phi29 DNA-packaging motor focused on the motor embedded in a procapsid (1) that included the capsid protein (gp8) and the scaffolding protein (gp7).26,33−35 Although the mechanism of the connector−pRNA (attached to the procapsid) interaction was previously illustrated,(19) for the construction of a biomimetic DNA-packaging motor, it is of interest to determine the mechanism of the pRNA−connector interaction using the isolated connector, free of the capsid protein. It has been reported that rosettes were produced when free connectors were mixed with pRNA.36,37 Such a finding is intriguing, unexpected, and puzzling. However, the mechanism of the rosette formation and the presence or absence of RNA molecules in the complex is unknown. Here, we elucidate the mechanism of the pRNA interaction with the free connector and demonstrate that the interaction process and the resulting product are very different from that obtained when the connector is embedded in the procapsid shell.
Figure 1.
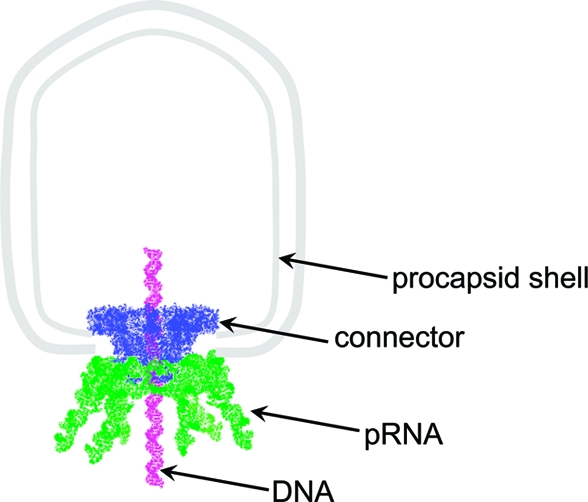
Schematic illustration of procapsid−pRNA interaction.
Results
RNA Nomenclature
To facilitate the description of the pRNA, uppercase and lowercase letters are included to represent the right-hand and left-hand interlocking loops, respectively.18,19 The same letter in upper and lower cases (e.g., C and c′, or A and a′) indicates their ability to complement each other, whereas different letters, such as C and d′, indicate noncomplementary base pairing.
Equal Binding of pRNA and Nonspecific RNA to Free Connector
In the gel shift assay, a final concentration of 0.5 μM purified connector was mixed with an increasing concentration of pRNA monomer Cd′, dimer (Cd′ + Dc′), or nonspecific tRNA. The complex was separated in a 0.8% agarose gel. The mobility patterns of different connector−RNA complexes were similar (2). Quantification of the dissociation constant (Kd) of the connector−RNA interaction revealed that the binding affinity of nonspecific tRNA to the connector was identical to that of the pRNA Aa′ (2). The Kd of connector/tRNA is 565.97 nM, while the Kd of connector/Aa′ is 588.66 nM.
Figure 2.

Gel shift assay to compare the binding capacity between different RNAs and the connector. (A) Connector with dimer Cd′−Dc′; (B) connector with monomer Cd′; (C) connector with tRNA. The triangle indicates a 2-fold increase in RNA concentration. The complex was separated by 0.8% agarose gel. The gel was stained by ethidium bromide to track the RNA bands first (left), and then the same gel was stained by Coomassie brilliant blue to detect protein bands (right). Connector/tRNA (D) and connector/Aa′ (E) were analyzed to determine the binding constant. Lane 1 indicates RNA only. Triangle above lanes 2−10 indicates a 2-fold increase in connector concentration.
The connector binding efficiency with different RNAs was also evaluated by sucrose gradient sedimentation (3), which showed that the efficiency of the connector−RNA complex, promoted by tRNA, was approximately that of monomeric pRNA Cd′ (3).
Figure 3.

5−20% sucrose gradient sedimentation to detect the binding efficiency of pRNA to connector. Connector with pRNA Aa′ (closed diamond), Cd′ (open square), Cd′−Dc′ (open triangle), and tRNA (dot) were analyzed by 5−20% sucrose gradient sedimentation. All RNAs were labeled by [3H]. [3H]-Aa′ without connector (closed triangle) was used as a control.
Comparable Binding of RNA and DNA to Free Connector
The ssDNA, dsDNA, and ssRNA, all of 55 nucleotides in length, were incubated with free connectors. At low concentrations of nucleic acid (4, lanes 2, 6, and 10), the migrations of the rosettes were nearly identical with all three kinds of nucleic acids. When the concentration of the nucleic acids was increased, dsDNA caused a more significant band shift than that of the ssDNA and ssRNA.
Figure 4.

Comparison of connector binding ability to DNA and RNA of the same length. A fixed amount of connector was mixed with 55 nt ssDNA, dsDNA, and ssRNA at different ratios. The mixtures were separated by 0.8% agarose gel. The gel was first stained with ethidium bromide to detect nucleic acids (A) and then stained by Coomassie brilliant blue to detect the proteins (B).
Rosette Formation Induced by RNA
The 117 nt pRNA Aa′ was mixed with connector at a ratio of 4:1 at room temperature for 10 min. The resulting products were separated by 5−20% sucrose gradient sedimentation. TEM imaging of the mixture before sedimentation revealed that the sample contained two kinds of structures. One was the connector with a diameter from 6.6 nm (the narrow end) to 13.8 nm (the wider end), and the other was a rosette structure with a diameter of 30 nm (5). After separation by sucrose gradient sedimentation, the peak centered at fraction 18 (3) contained both connector and nucleic acids, as identified by radioactive labeling. TEM imaging clearly revealed that fraction 18 contained homogeneous rosette particles (5).
Figure 5.

Negative stained electron micrograph images of connector and rosette. (A) Mixture of connector and pRNA; (B) purified connector−pRNA complex. Bar = 100 nm.
Nucleotide Length Dependence and Sequence Independence in Rosette Formation
The finding of nonspecific binding between the connector and nucleic acids led to the question of whether the interaction was purine- or pyrimidine-specific. It was found that all the 20 nt poly-dG, poly-dT, and poly-dC showed similar connector binding activity (6).
Figure 6.

(A) Connector binding to nucleic acids is not purine- or pyrimidine-dependent. Constant amount of connector was mixed with various amounts of 20 nt poly-d(purine) and poly-d(pyrimidine), and the mixtures were analyzed by 0.8% agarose gel shift assays, followed by Coomassie brilliant blue staining. (B) Rosette formation by connector−nucleic acid interaction is sequence-length-dependent. Different lengths of DNA oligos were mixed with connector and analyzed by agarose gel electrophoresis. The connector protein itself appears as a smeared band in the gel, but the band pattern becomes more defined with increasing length of ssDNA.
DNA oligos with different lengths were also tested. The results showed that sequences necessitated a length of greater than 21 nt in order to bind to the connector. We found that 31 nt was the minimal length to induce the rosette formation at low concentration (connector: DNA = 1:1) (6, lane 10), but the resulting band corresponding to the complex was not as sharp as was observed at higher concentration (6, lanes 11 and 12). The bands were heterogeneous, indicating that DNA with 31 nt was sufficient to form a stable rosette complex. The figure also illustrates that a greater concentration of DNA is needed to maintain the complex structure. The finding is in accord with the result of single molecule studies (see below) demonstrating that a 31 nt polynucleotide chain is required to promote the formation of the rosette while multiple short nucleotides are needed for a stable rosette formation.
Counting RNA Molecules within Each Rosette Particle by Single Molecule Photobleaching
Single molecule photobleaching analysis revealed that the connector/dsDNA, connector/ssDNA, and connector/ssRNA complexes all resulted in a comparable histogram of photobleaching steps (7). The majority of both types of complexes contained only one fluorescent 50 nt RNA molecule. The results indicate that one RNA molecule with 50 nt is long enough to bring the connectors together. However, we also noticed that the complex composed of connector and dimeric pRNA Cd′−Dc′ contained many two-step photobleaching events (7), which means that two Cy3-labeled Cd′ were involved in the rosette formation. Since pRNA Cd′−Dc′ forms a dimer in the solution spontaneously, two pRNA dimers Cd′−Dc′ were more prone to be involved in the rosette formation than the monomeric pRNA Cd′.
Figure 7.

Single molecule photobleaching analysis on rosette particle formed by connector−RNA interaction. (A−F) Histogram showing the frequency of photobleaching steps of Cy3-RNA in connector−RNA complex excited by a 532 nm laser beam. Each step in photobleaching represents the presence of one Cy3-RNA molecule. In the complex of connector−pRNA (Cd′−Dc′), the Cy3 molecule was only attached to the pRNA Cd′.
Alternative Switching between Rosette and Free Connector by the Addition and Removal of Nucleic Acids
When the rosette (connector/ssDNA complex) was treated with Mung Bean Nuclease, the rosette (8, lane 2) dissociated into individual connectors (8, lane 3). However, after adding pRNA (8, lane 4) to the above solution, the rosette was re-formed and the complex migrated to the same location as the control of the connector/pRNA complex (8, lane 6). Digesting the mixture with RNase A resulted in the dissociation of the rosette into the individual connectors again (8, lane 5). Lanes 7−10 represent the corresponding controls in the absence of connector.
Figure 8.

Reversible structural shift between the connector and the rosette as induced by nucleic acids and ribonuclease. Connector (lane 1) was transformed into a rosette by ssDNA (lane 2). After treating the connector−ssDNA complex by Mung Bean Nuclease (lane 3), ssDNA was degraded and the rosette reverted into free connector protein. The rosette was formed again when pRNA was added into the solution (lane 4). Rosette was dissociated into the connector when the connector−pRNA complex was treated by RNase A (lane 5). Lane 6, control of connector−pRNA complex only. Lanes 7−10, negative control without connector. The gel was first stained with ethidium bromide to detect nucleic acids (A) and then stained by Coomassie brilliant blue to detect the proteins (B).
Discussion
Extensive studies have revealed that the connector, whether incorporated into the procapsid or in free form, interacts with individual pRNA or other nucleic acids nonspecifically. This is reasonable as nucleic acids contain a negatively charged phosphate backbone and the N-terminus of the connector contains a stretch of positively charged amino acids.38,39 However, the resulting connector−nucleic acid complexes are very different in observed shape and conformation depending upon whether or not the connector is associated with the procapsid. When pRNA interacts with the connector embedded in the procapsid, the pRNA forms a hexameric ring around the connector. Yet, when pRNA interacts with purified, isolated connector in the absence of the capsid protein, the resulting complex is a rosette containing one or two copies of the pRNA. The studies in this report elucidate the mechanism of the latter connector−nucleic acid reaction.
The difference in particle size between the procapsid and the connector leads to a difference in nucleation during the protein−pRNA complex formation. The size of the procapsid is approximately 20 million Da, while the connector is approximately 0.4 million Da. The larger size of the procapsid makes it relatively difficult to move or turn, whereas the smaller connector can turn quickly. Subsequently, during the interaction of connector/procapsid with pRNA, the connector serves as the nuclear core for the assembly, which results in the binding of six copies of pRNA to the connector. However, as to the interaction of free connector with pRNA (or DNA), the pRNA serves as the nuclear core and is surrounded by five connectors instead.
The connector possesses a truncated cone structure composed of three distinct surface layers: hydrophilic regions at both terminal ends and a hydrophobic region within the central domain (8). The polar nature of this dynamic protein enables it to self-assemble into nanoparticles.36,40−42 Thus, the number of connectors in the rosette is predominantly driven and controlled by the size and shape of the connector. The extension of both sides of the connector results in the formation of an angle smaller than 72° (8). Although six- or four-subunit particles might occasionally form, the pentameric rosette is the dominant configuration with a central angle of 72°. Each rosette contains a central portal with dimensions of 11.2 nm in diameter and 35 nm in circumference. If only one oligonucleotide is used to link the five connectors, it must be at least 28 nm (4/5 of the circumference length) to promote the formation of the rosette, corresponding to about 70 nt (0.4 nm/nt(43)). The N-terminal RKR sequence of gp10 is responsible for nucleotide binding;38,39,44 however, the rosette can be formed by either (1) long oligos linking all five connectors together at once (8), or (2) short oligos linking two neighboring connectors at a time (8). Considering the nonspecific, random, and overlapping linkage between any nucleotides in the assembly of the rosette, shorter nucleotides would suffice (9), but higher concentrations of the nucleotide would be needed because each rosette formation involves the random incorporation of multiple nucleotides with overlapping regions within the connector contact regions (6).
Figure 9.

Illustration of mechanism in rosette formation. (A) Five connectors arranged into a pentagonal structure via side-to-side interactions with its external hydrophilic−hydrophobic−hydrophilic property. (B−D) Interaction of oligonucleotides with the narrow end of connectors containing “RKR” at the N-terminus of each gp10 subunit. Formation of rosette can be promoted by only one long oligo (DNA/RNA) (B), by short oligos to link the neighboring connectors (C), or by multiple overlapping oligos (D).
Sedimentation velocity (SV) experiments of RNA−connector complexes were used to estimate the size and shape of multiple solutes in a mixture of macromolecules. These experiments showed the presence of multiple species with higher sedimentation coefficients, consistent with higher order association states of connectors forming rosette species (10). These experiments further show that the connector proteins appear fully saturated at a 1:1 molar ratio, with increasing RNA amounts remaining mostly free in solution. It appears that an excess of RNA may possibly be disrupting the rosette formation, and that the formation of the rosette may be mass-action driven. The model of assembly shown in 10 is further consistent with the observed frictional ratio, which shows increasing globularity for the larger species observed in the mixture. Taken together, these experiments are consistent with the model of one RNA molecule interacting with one connector molecule, and then another connector joining the interaction. Gradually, more connectors are linked, with an apparent maximum of five connectors linked together by RNA molecules. The number of connectors within each rosette is determined by the shape and size of the connector. The number of RNA or DNA oligomers in each rosette also portrays the mechanism of rosette assembly (7). Single molecule analysis revealed that each rosette contained one or two DNA or pRNA monomers or dimers (7). The data agrees with the mechanism as proposed above.
Figure 10.

(A−C) Genetic algorithm−Monte Carlo analysis of sedimentation velocity data: frictional ratio vs sedimentation coefficients for three different ratios of connector to RNA. The color gradient legend in each panel indicates the relative concentrations based on color. (D) Cartoons suggesting a mechanism of rosette formation. In the beginning, one RNA molecule interacts with one connector molecule, and then another connector joins the interaction. A stoichiometry of one RNA for each connector appears likely because a molar excess of RNA only increases the partial concentration of the free RNA (∼5 S).
The effect of the capsid protein on the pRNA orientation is an important factor that governs the formation of the pRNA hexameric ring. If the capsid protein is present, it serves as a barrier, and the entire gp16 binding domain (the 5′/3′ helix tail) will extend in one direction away from the wider end of the connector. The appropriate orientation facilitates the interaction between the interlocking loops for the assembly of the hexamer. However, in the absence of the capsid protein, the gp16 binding domain will be disordered and cannot support contact between the interlocking loops. Therefore, the formation of a pRNA hexamer is not possible.
Methods
Preparation of RNA, DNA, and Connector
The preparation of DNA oligos and RNA18,31 and connector41,42 has been reported previously. Poly-d(N) refers to homopolymeric single-stranded DNA.
Gel Shift Assay and Kd Determination of Connector Binding to Different RNAs
A constant amount (0.5 μM final) of purified connector was mixed with different pRNA, ssDNA, dsDNA, and tRNA molecules from 2 to 0.0625 μM by 2-fold dilution and incubated for 10 min at room temperature. The final products were resolved on a 0.8% agarose gel. The gel was first stained by ethidium bromide to visualize nucleic acids and then by Coomassie blue to visualize the protein.
The dissociation constant (Kd) determination was performed in gel shift assays. The pRNA Aa′ and tRNA were labeled at the 5′-end with radioactive 32P. In this experiment, the RNA concentration was constant (20 nM), while the connector concentration varied from 1280 to 5 nM by 2-fold dilution. After 10 min incubation, the complex was then autoradiographed. The Kd was calculated as the concentration of the RNA when 50% of the RNA formed the connector/RNA complex. The data points of the binding curves were fit to the equation F = P/(Kd + P) to determine the dissociation constants by Origin 7.5 (Microcal Software Inc.), where F is the fraction of RNA bound, P is the concentration of protein, and Kd is the dissociation constant.
Sucrose Gradient Sedimentation
Sucrose gradient sedimentation was performed to detect the binding of pRNA to connector and procapsid. [3H]-labeled RNA was mixed with the connector at a molar ratio of 1:1 at room temperature for 30 min. Linear 5−20% sucrose gradient sedimentation was performed to separate the connector/[3H]-RNA complex from the free [3H]-RNAs.
Purification of Connector/Cy3-RNA Complexes by 5−20% Sucrose Gradient Sedimentation
Sucrose gradient sedimentation was performed to separate the procapsid/5′Cy3-RNA. The complexes were prepared as described before and were loaded on top of a linear 5−20% sucrose gradient in TMS. After spinning in a Beckman L-80 ultracentrifuge at 35 000 rpm for 1 h at 20 °C in a SW55 rotor, fractions were collected from the bottom of the tube.
Single Molecule Imaging of Connector/Nucleic Acids Complex
The purified connector/Cy3-nucleic acids complex from sucrose gradient sedimentation was immobilized in a flow chamber on a quartz slide by means of an anti-phi29 procapsid antibody. Photobleaching in the presence of an oxygen scavenging system (5% β-d-glucose, 10 mM β-mercaptoethanol, and 1% GODCAT solution, a mixture of glucose oxidase and catalase) was carried out with a single molecule total internal reflection microscopy setup3,23 excited by a laser beam. The concentration of the samples was adjusted to show discrete fluorescing spots in the images. Laser power was adjusted to approximately 15 mW to photobleach Cy3 fluorophores at a reasonable speed in order that the photobleaching steps might be easily distinguished. Sequential images were taken with an exposure time of 200 ms. The recorded movie, with more than 1000 frames, was analyzed by Kinetic Imaging (Andor Technology). Each step of decreasing fluorescence intensity represents one single Cy3 fluorophore that is attached to a single RNA. The number of photobleaching steps reveals the copy number of Cy3-RNA bound to each procapsid or connector. A histogram of photobleaching steps was obtained for each sample.
Analytical Ultracentrifugation (AUC) To Determine the Sedimentation Coefficient as well as the Shapes and Sizes of the Particles
All analytical ultracentrifugation (AUC) experiments were performed at the Center for Analytical Ultracentrifugation of Macromolecular Assemblies at the University Texas Health Science Center at San Antonio. Purified proteins, C-strep connector together with pRNA Cd′ in different molar ratios (1:1, 2:1, and 4:1) were studied by sedimentation velocity (SV) at 25 000 rpm and 4 °C. All experiments were performed in a Beckman XLA using absorbance optics in intensity mode by scanning at 230, 260, and 280 nm. All data were analyzed by the UltraScan software version 9.9.(45) Time invariant noise was eliminated from the data during analysis by two-dimensional spectrum analysis.(46) The resulting shape and molecular weight spectrum was further refined using genetic algorithm analysis.(47) Monte Carlo analysis was performed on the resulting parsimoniously regularized distributions,(48) and the results at 280 nm were plotted as pseudo-3D graphs with color depth indicating partial concentration (10). All calculations were performed at the Texas Advanced Computing Center using the Lonestar supercomputer.
Acknowledgments
We thank H. Zhang, D. Shu, A. Vonderheide, C. Schwartz, and F. Haque for their technical assistance and valuable comments. The research was mainly supported by NIH Grant R01-GM59944 (P.G.). The development of the UltraScan software is supported by NIH Grant R01-RR0222000 (B.D.).
Funding Statement
National Institutes of Health, United States
References
- Heymann J. B.; Cheng N. Q.; Newcomb W. W.; Trus B. L.; Brown J. C.; Steven A. C. Dynamics of Herpes Simplex Virus Capsid Maturation Visualized by Time-Lapse Cryo-Electron Microscopy. Nat. Struct. Biol. 2003, 10, 334–341. [DOI] [PubMed] [Google Scholar]
- Cusack S. Adenovirus Complex Structures. Curr. Opin. Struct. Biol. 2005, 15, 237–243. [DOI] [PubMed] [Google Scholar]
- Shu D.; Zhang H.; Jin J.; Guo P. Counting of Six pRNAs of Phi29 DNA-Packaging Motor with Customized Single Molecule Dual-View System. EMBO J. 2007, 26, 527–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catalano C. E.; Cue D.; Feiss M. Virus DNA Packaging: The Strategy Used by Phage Lambda. Mol. Microbiol. 1995, 16, 1075–1086. [DOI] [PubMed] [Google Scholar]
- Camacho A. G.; Gual A.; Lurz R.; Tavares P.; Alonso J. C. Bacillus subtilis Bacteriophage SPP1 DNA Packaging Motor Requires Terminase and Portal Proteins. J. Biol. Chem. 2003, 278, 23251–23259. [DOI] [PubMed] [Google Scholar]
- Lander G. C.; Tang L.; Casjens S. R.; Gilcrease E. B.; Prevelige P.; Poliakov A.; Potter C. S.; Carragher B.; Johnson J. E. The Structure of an Infectious P22 Virion Shows the Signal for Headful DNA Packaging. Science 2006, 312, 1791–1795. [DOI] [PubMed] [Google Scholar]
- Valpuesta J. M.; Sousa N.; Barthelemy I.; Fernandez J. J.; Fujisawa H.; Ibarra B.; Carrascosa J. L. Structural Analysis of the Bacteriophage T3 Head-to-Tail Connector. J. Struct. Biol. 2000, 131, 146–155. [DOI] [PubMed] [Google Scholar]
- Kanamaru S.; Leiman P. G.; Kostyuchenko V. A.; Chipman P. R.; Mesyanzhinov V. V.; Arisaka F.; Rossmann M. G. Structure of the Cell-Puncturing Device of Bacteriophage T4. Nature 2002, 415, 553–557. [DOI] [PubMed] [Google Scholar]
- Agirrezabala X.; Martin-Benito J.; Valle M.; Gonzalez J. M.; Valencia A.; Valpuesta J. M.; Carrascosa J. L. Structure of the Connector Of Bacteriophage T7 at 8Å Resolution: Structural Homologies of a Basic Component of a DNA Translocating Machinery. J. Mol. Biol. 2005, 347, 895–902. [DOI] [PubMed] [Google Scholar]
- Jiang W.; Chang J.; Jakana J.; Weigele P.; King J.; Chiu W. Structure of Epsilon15 Bacteriophage Reveals Genome Organization and DNA Packaging/Injection Apparatus. Nature 2006, 439, 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P.; Peterson C.; Anderson D. Prohead and DNA-Gp3-Dependent ATPase Activity of the DNA Packaging Protein Gp16 of Bacteriophage Φ29. J. Mol. Biol. 1987, 197, 229–236. [DOI] [PubMed] [Google Scholar]
- Guo P. X.; Lee T. J. Viral Nanomotors for Packaging of dsDNA and dsRNA. Mol. Microbiol. 2007, 64, 886–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov A. S.; Harvey S. C. Structural and Thermodynamic Principles of Viral Packaging. Structure 2007, 15, 21–27. [DOI] [PubMed] [Google Scholar]
- Morita M.; Tasaka M.; Fujisawa H. DNA Packaging ATPase of Bacteriophage T3. Virology 1993, 193, 748–752. [DOI] [PubMed] [Google Scholar]
- Yang K.; Baines J. D. The Putative Terminase Subunit of Herpes Simplex Virus 1 Encoded by UL28 Is Necessary and Sufficient To Mediate Interaction between Pul15 and pUL33. J. Virol. 2006, 80, 5733–5739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S. Y.; Kondabagil K.; Gentz P. M.; Rossmann M. G.; Rao V. B. The Structure of the ATPase That Powers DNA Packaging into Bacteriophage T4 Procapsids. Mol. Cell 2007, 25, 943–949. [DOI] [PubMed] [Google Scholar]
- Guo P.; Erickson S.; Anderson D. A Small Viral RNA Is Required for In Vitro Packaging of Bacteriophage Phi29 DNA. Science 1987, 236, 690–694. [DOI] [PubMed] [Google Scholar]
- Guo P.; Zhang C.; Chen C.; Trottier M.; Garver K. Inter-RNA Interaction of Phage Phi29 pRNA To Form a Hexameric Complex for Viral DNA Transportation. Mol. Cell 1998, 2, 149–155. [DOI] [PubMed] [Google Scholar]
- Xiao F.; Zhang H.; Guo P. Novel Mechanism of Hexamer Ring Assembly in Protein/RNA Interactions Revealed by Single Molecule Imaging. Nucleic Acids Res. 2008, 36, 6620–6632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T. J.; Guo P. Interaction of Gp16 with pRNA and DNA for Genome Packaging by the Motor of Bacterial Virus Phi29. J. Mol. Biol. 2006, 356, 589–599. [DOI] [PubMed] [Google Scholar]
- Lee T. J.; Zhang H.; Liang D.; Guo P. Strand and Nucleotide-Dependent ATPase Activity of Gp16 of Bacterial Virus Phi29 DNA Packaging Motor. Virology 2008, 380, 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P.; Grimes S.; Anderson D. A Defined System for In Vitro Packaging of DNA-gp3 of the Bacillus subtilis Bacteriophage Phi29. Proc. Natl. Acad. Sci. U.S.A. 1986, 83, 3505–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Shu D.; Huang F.; Guo P. Instrumentation and Metrology for Single RNA Counting in Biological Complexes or Nanoparticles by a Single Molecule Dual-View System. RNA 2007, 13, 1793–1802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. E.; Tans S. J.; Smith S. B.; Grimes S.; Anderson D. L.; Bustamante C. The Bacteriophage Phi29 Portal Motor Can Package DNA against a Large Internal Force. Nature 2001, 413, 748–752. [DOI] [PubMed] [Google Scholar]
- Simpson A. A.; Leiman P. G.; Tao Y.; He Y.; Badasso M. O.; Jardine P. J.; Anderson D. L.; Rossman M. G. Structure Determination of the Head−Tail Connector of Bacteriophage Phi29. Acta Crystallogr. 2001, D57, 1260–1269. [DOI] [PubMed] [Google Scholar]
- Guasch A.; Pous J.; Ibarra B.; Gomis-Ruth F. X.; Valpuesta J. M.; Sousa N.; Carrascosa J. L.; Coll M. Detailed Architecture of a DNA Translocating Machine: The High-Resolution Structure of the Bacteriophage Phi29 Connector Particle. J. Mol. Biol. 2002, 315, 663–676. [DOI] [PubMed] [Google Scholar]
- Guo P. Structure and Function of Phi29 Hexameric RNA that Drive Viral DNA Packaging Motor: Review. Prog. Nucl. Acid. Res. Mol. Biol. 2002, 72, 415–472. [DOI] [PubMed] [Google Scholar]
- Reid R. J. D.; Bodley J. W.; Anderson D. Characterization of the Prohead−pRNA Interaction of Bacteriophage Phi29. J. Biol. Chem. 1994, 269, 5157–5162. [PubMed] [Google Scholar]
- Hoeprich S.; Guo P. Computer Modeling of Three-Dimensional Structure of DNA-Packaging RNA(pRNA) Monomer, Dimer, and Hexamer of Phi29 DNA Packaging Motor. J. Biol. Chem. 2002, 277, 20794–20803. [DOI] [PubMed] [Google Scholar]
- Chen C.; Zhang C.; Guo P. Sequence Requirement for Hand-in-Hand Interaction in Formation of pRNA Dimers and Hexamers to Gear Phi29 DNA Translocation Motor. RNA 1999, 5, 805–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F.; Lemieux S.; Wu X.; St.-Arnaud S.; McMurray C. T.; Major F.; Anderson D. Function of Hexameric RNA in Packaging of Bacteriophage Phi29 DNA In Vitro. Mol. Cell 1998, 2, 141–147. [DOI] [PubMed] [Google Scholar]
- Reid R. J. D.; Bodley J. W.; Anderson D. Identification of Bacteriophage phi29 Prohead RNA (pRNA) Domains Necessary for In Vitro DNA-gp3 Packaging. J. Biol. Chem. 1994, 269, 9084–9089. [PubMed] [Google Scholar]
- Guo P.; Erickson S.; Xu W.; Olson N.; Baker T. S.; Anderson D. Regulation of the Phage Φ29 Prohead Shape and Size by the Portal Vertex. Virology 1991, 183, 366–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao Y.; Olson N. H.; Xu W.; Anderson D. L.; Rossmann M. G.; Baker T. S. Assembly of a Tailed Bacterial Virus and Its Genome Release Studied in Three Dimensions. Cell 1998, 95, 431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morais M. C.; Kanamaru S.; Badasso M. O.; Koti J. S.; Owen B. A.; McMurray C. T.; Anderson D. L.; Rossmann M. G. Bacteriophage Phi29 Scaffolding Protein gp7 before and after Prohead Assembly. Nat. Struct. Biol. 2003, 10, 572–576. [DOI] [PubMed] [Google Scholar]
- Tsuprun V.; Anderson D.; Egelman E. H. The Bacteriophage Φ29 Head−Tail Connector Shows 13-Fold Symmetry in Both Hexagonally Packed Arrays and as Single Particles. Biophys. J. 1994, 66, 2139–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garver K.; Guo P. Boundary of pRNA Functional Domains and Minimum pRNA Sequence Requirement For Specific Connector Binding and DNA Packaging of Phage Phi29. RNA 1997, 3, 1068–1079. [PMC free article] [PubMed] [Google Scholar]
- Xiao F.; Moll D.; Guo S.; Guo P. Binding of pRNA to the N-Terminal 14 Amino Acids of Connector Protein of Bacterial Phage Phi29. Nucleic Acids Res. 2005, 33, 2640–2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atz R.; Ma S.; Gao J.; Anderson D. L.; Grimes S. Alanine Scanning and Fe-BABE Probing of the Bacteriophage phi29 Prohead RNA−Connector Interaction. J. Mol. Biol. 2007, 369, 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guasch A.; Pous J.; Parraga A.; Valpuesta J. M.; Carrascosa J. L.; Coll M. Crystallographic Analysis Reveals the 12-Fold Symmetry of the Bacteriophage phi29 Connector Particle. J. Mol. Biol. 1998, 281, 219–225. [DOI] [PubMed] [Google Scholar]
- Guo Y.; Blocker F.; Xiao F.; Guo P. Construction and 3-D Computer Modeling of Connector Arrays with Tetragonal to Decagonal Transition Induced by pRNA of Phi29 DNA-Packaging Motor. J. Nanosci. Nanotechnol. 2005, 5, 856–863. [DOI] [PubMed] [Google Scholar]
- Wendell D.; Jing P.; Geng J.; Subramaniam V.; Lee T. J.; Montemagno C.; Guo P. Translocation of Double-Stranded DNA through Membrane-Adapted Phi29 Motor Protein Nanopores. Nat. Nano 2009, 4, 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolley A. T.; Kelly R. T. Deposition and Characterization of Extended Single-Stranded DNA Molecules on Surfaces. Nano Lett. 2001, 1, 345–348. [Google Scholar]
- Donate L. E.; Valpuesta J. M.; Rocher A.; Mendez E.; Rojo F.; Salas M.; Carrascosa J. L. Role of the Amino-Terminal Domain of Bacteriophage Phi 29 Connector in DNA Binding and Packaging. J. Biol. Chem. 1992, 267, 10919–10924. [PubMed] [Google Scholar]
- Demeler B.UltraScan—A Comprehensive Software Package for the Analysis of Sedimentation Velocity and Sedimentation Equilibrium Data; http://www.ultrascan.uthscasa.edu, 2009.
- Brookes E.; Cao W.; Demeler B. A Two-Dimensional Spectrum Analysis for Sedimentation Velocity Experiments of Mixtures with Heterogeneity in Molecular Weight and Shape. Eur. Biophys. J. 2010, 39, 405–414. [DOI] [PubMed] [Google Scholar]
- Brookes E.; Demeler B.. Parsimonious Regularization Using Genetic Algorithms Applied to the Analysis of Analytical Ultracentrifugation Experiments. In GECCO Proceedings ACM 978-1-59593-697-1/07/0007, 2007.
- Brookes E.; Demeler B. Parallel Computational Techniques for the Analysis of Sedimentation Velocity Experiments in Ultrascan. Colloid Polym. Sci. 2008, 286, 139–148. [Google Scholar]