

Abstract
Nephrolithiasis in the Slc26a6−/− mouse is accompanied by 50–75% reduction in intestinal oxalate secretion with unchanged intestinal oxalate absorption. The molecular identities of enterocyte pathways for oxalate absorption and for Slc26a6-independent oxalate secretion remain undefined. The reported intestinal expression of SO42− transporter SLC26A2 prompted us to characterize transport of oxalate and other anions by human SLC26A2 and mouse Slc26a2 expressed in Xenopus oocytes. We found that hSLC26A2-mediated [14C]oxalate uptake (K1/2 of 0.65 ± 0.08 mM) was cis-inhibited by external SO42− (K1/2 of 3.1 mM). hSLC26A2-mediated bidirectional oxalate/SO42− exchange exhibited extracellular SO42− K1/2 of 1.58 ± 0.44 mM for exchange with intracellular [14C]oxalate, and extracellular oxalate K1/2 of 0.14 ± 0.11 mM for exchange with intracellular 35SO42−. Influx rates and K1/2 values for mSlc26a2 were similar. hSLC26A2-mediated oxalate/Cl− exchange and bidirectional SO42−/Cl− exchange were not detectably electrogenic. Both SLC26A2 orthologs exhibited nonsaturable extracellular Cl− dependence for efflux of intracellular [14C]oxalate, 35SO42−, or 36Cl−. Rate constants for 36Cl− efflux into extracellular Cl−, SO42−, and oxalate were uniformly 10-fold lower than for oppositely directed exchange. Acidic extracellular pH (pHo) inhibited all modes of hSLC26A2-mediated anion exchange. In contrast, acidic intracellular pH (pHi) selectively activated exchange of extracellular Cl− for intracellular 35SO42− but not for intracellular 36Cl− or [14C]oxalate. Protein kinase C inhibited hSLC26A2 by reducing its surface abundance. Diastrophic dysplasia mutants R279W and A386V of hSLC26A2 exhibited similar reductions in uptake of both 35SO42− and [14C]oxalate. A386V surface abundance was reduced, but R279W surface abundance was at wild-type levels.
Keywords: chondrodysplasia, nephrolithiasis, oxalosis, anion exchange, Xenopus oocyte
the gene family SLC26/SulP encodes anion exchangers and channels (12) expressed throughout evolution. Mutations in several SLC26 genes have been associated with human disease, including chondrodysplasia [SLC26A2 (22)], diarrhea [SLC26A3; (24)], and deafness [SLC26A4 (13) and SLC26A5 (33)]. Additional pathological phenotypes have been reported in knockout mice deficient in expression of other SLC26 genes for which human mutations have not yet been associated with disease. These potential disease models include distal renal tubular acidosis [Slc26a9−/− (46)], gastric achlorhydria [Slc26a7−/− and Slc26a9−/− (45, 46)], male infertility [Slc26a8−/− (41)], and nephrolithiasis [Slc26a6−/− (27) and Slc26a1−/− (10a)].
Nephrolithiasis in the Slc26a6−/− mouse was associated with enteric absorptive hyperoxaluria secondary to selective reduction of intestinal oxalate secretion by 50–75%, with unchanged rates of oxalate absorption (27, 44). Elevated levels of plasma oxalate derived from endogenous (predominantly hepatic) biosynthesis and from dietary sources were filtered by the glomerulus, producing hyperoxaluria and urinary supersaturation with calcium oxalate, leading in turn to calcium oxalate precipitation, nephrocalcinosis, and nephrolithiasis.
Pharmacological reduction of hepatic oxalate biosynthesis should reduce hyperoxaluria, but such inhibitors are currently unavailable. Restriction of dietary oxalate has been proposed as an approach to reduce hyperoxaluria, but the proportion of urinary oxalate excretion attributable to the diet has been estimated to range between 5% and 40% (25), and lifetime compliance is difficult. Thus, attempts to stimulate intestinal oxalate secretion, and/or to inhibit intestinal oxalate reabsorption, continue to constitute important therapeutic approaches to divert plasma oxalate from the urine by increasing fecal oxalate excretion.
The molecular identities of enteric oxalate absorption pathways remain unknown, as do those of the apparently Slc26a6-independent pathways mediating oxalate secretion into the intestinal lumen. Intestinal SLC26A3/DRA, although primarily an apical Cl−/HCO3− exchanger, is capable of some oxalate transport (2, 7). mRNA encoding the chondrodysplasia gene SLC26A2/DTDST (diastrophic dysplasia sulfate transporter) is expressed not only in cartilage and chondrocytes but in the small intestine of rats (37) and in the colon and other tissues of humans (20, 22). SLC26A2/DTDST polypeptide was detected at substantial levels at the apical pole of human enterocytes (20), in rat renal proximal tubule brush border (5), as well as in chondrocytes and many other cell types (20, 31).
SLC26A2 was discovered as a SO42− uptake transporter in human chondrocytes (22) and is expressed in chondrocytes, fibroblasts, and osteoblasts of the mouse (14). 35SO42− efflux from rat chondrocytes was noted to require the presence of extracellular Cl− (37). Functional studies of recombinant SLC26A2 have been restricted to 35SO42− uptake assays in Xenopus oocytes (29, 37) and in human embryonic kidney (HEK)-293 cells (28). The oocyte studies have demonstrated cis-inhibition by Cl− and HCO3− (100 mM), by SO42−, thiosulfate, and oxalate (5 mM), and by DIDS (1 mM) (37). However, tests of anion exchange and oxalate transport by recombinant SLC26A2 have not yet been published. hSLC26A2 and mSlc26a2 polypeptides, although of identical length, share only 80% amino acid identity, a degree of divergence previously shown for SLC26A6 (at 78%) to encode substantial differences in function. We have therefore extended the functional characterization of both human SLC26A2 (hSLC26A2) and mouse Slc26a2 (mSlc26a2) and tested the hypothesis that they transport oxalate.
We found that SLC26A2 exchanges oxalate with trans-SO42−, Cl−, or oxalate itself, in addition to mediating SO42−/Cl− and Cl−/Cl− exchange. The two species orthologs exhibit similar anion transport affinities, share nonsaturating extracellular Cl− dependence, and exhibit low rates of 36Cl− efflux except as Cl−/Cl− self-exchange. hSLC26A2 is differentially regulated by extracellular pH (pHo) and intracellular pH (pHi). hSLC26A2 is inhibited by protein kinase C (PKC), likely through regulation of surface abundance. Two diastrophic dysplasia mutants of hSLC26A2 exhibit parallel reductions in transport of sulfate and oxalate but differ in impairment of surface expression.
METHODS
Materials.
Na36Cl, Na235SO42−, and [14C]butyrate were from ICN (Irvine, CA). [14C]oxalate originally from NEN-DuPont was the generous gift of C. Scheid and T. Honeyman (Univ. of Massachusetts Medical Center). Restriction enzymes and T4 DNA ligase were from New England Biolabs (Beverly, MA). The EXPAND High-fidelity PCR System was from Roche Diagnostics (Indianapolis, IN). 4,4′-Diisothiocyanostilbene-2,2′-disulfonic acid (DIDS) was from Calbiochem (La Jolla, CA). 4,4′-Dinitrostilbene-2,2′-disulfonic acid (DNDS) was from Pfalz and Bauer (Waterbury, CT). 4β-Phorbol-12-myristate-13-acetate (PMA) and 4α-phorbol-didecanoate (4α-PDD) were from LC Laboratories (Woburn, MA). All other chemical reagents were from Sigma (St. Louis, MO) or Fluka (Milwaukee, WI) and were of reagent grade.
Solutions.
MBS consisted of (in mM) 85 NaCl, 1 KCl, 2.4 NaHCO3, 0.82 Mg(SO4)2, 0.33 Ca(NO3)2, 0.41 CaCl2, and 10 HEPES (pH adjusted to 7.40 with NaOH). ND-96 (pH 7.40) consisted of (in mM) 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES. In Cl−-free ND-96 or partial Cl− substitution solutions, NaCl was replaced mole-for-mole with Na cyclamate or, in the case of two-electrode voltage-clamp experiments, Na gluconate. In some experiments, 96 mM NaCl was replaced with 64 mM Na2SO42−. As needed, the Cl− salts of K+, Ca2+, and Mg2+ were substituted on an equimolar basis with the corresponding gluconate salts. Na oxalate-containing bath solutions were nominally Ca2+ free. Addition to flux media of the weak acid salt sodium butyrate (40 mM) was in equimolar substitution for Na cyclamate. Bath addition of NH4Cl (20 mM) was in equimolar substitution for NaCl. pH 5.0 ND96 solutions were buffered with 5 mM MES. (Occasional use of 5 mM HEPES during the four 1-min exposures to pH 5.0 was without bath pH drift and without change in results.)
Construction and mutagenesis of cDNA expression plasmids.
Human SLC26A2/DTDST (hSCL26A2) cDNA in a modified pGEM vector as described (29) was subcloned into the Xenopus oocyte expression vector pXT7. Mouse Slc26a2 (mSlc26a2) cDNA in pCMV-SPORT6 (clone ID 4014785; BC028345) was obtained from Open Biosystems (Huntsville, AL). The cDNA plasmid encoding a COOH-terminal green fluorescent protein (GFP)-fusion hSLC26A2-GFP (gift of J. Schwartzbauer, Princeton University) was subcloned into pXT7.
hSLC26A2 mutations R297W and A386V were generated by four primer polymerase chain reaction (PCR) as described (8) with specific mutagenic oligonucleotides and flanking oligonucleotides hA2-F and hA2-R (see supplemental Table 1 online at the AJP-Cell Physiology website). All PCR products were sequenced in entirety in both strands to ensure absence of unintended mutations.
Expression of cRNAs in Xenopus oocytes.
Capped cRNA was transcribed from linearized cDNA templates with T7 or SP6 RNA polymerase (Ambion, Austin, TX) and purified with an RNeasy mini-kit (Qiagen). cRNA concentration (A260) was measured by Nanodrop spectrometer (ThermoFisher), and integrity was confirmed by formaldehyde agarose gel electrophoresis. Mature female Xenopus (Dept. of Systems Biology, Harvard Medical School; or NASCO, Madison, WI) were maintained and subjected to partial ovariectomy under hypothermic tricaine anesthesia following protocols approved by the Institutional Animal Care and Use Committee of Beth Israel Deaconess Medical Center. Stage V-VI oocytes were prepared by overnight incubation of ovarian fragments in MBS with 1.3 mg/ml collagenase B (Roche, Indianapolis, IN), followed by a 20-min rinse in Ca2+-free MBS with subsequent manual selection and defolliculation as needed. Oocytes were injected on the same day with cRNA (0.5–50 ng) or with water in a volume of 50 nl. Injected and uninjected oocytes were then maintained before use for 2–6 days at 19°C in MBS containing gentamicin. Injection of increasing amounts of hSLC26A2-GFP cDNA led to corresponding increases in GFP fluorescence intensity at or near the oocyte surface (see supplemental Fig. 1 online at the AJP-Cell Physiology website). Injection of increasing amounts of untagged hSLC26A2 also led to corresponding increases in transporter polypeptide at or near the oocyte surface, as detected by confocal immunofluorescence microscopy (see supplemental Fig. 2 at the AJP-Cell Physiol website).
Isotopic influx experiments.
Unidirectional 35SO42− influx studies were carried out for periods of 15 or 30 min in a bath containing (in mM) 1 Na2SO4, 94.5 Na cyclamate, 2 K glutamate, 1.8 Ca glutamate, 1 Mg glutamate, and 5 HEPES (2 μCi/150 μl in a microtiter plate well) (6). 35SO42− uptake increased as a function of the amount of cRNA previously injected (supplemental Fig. 1). 36Cl− influx studies were carried out for periods of 15 or 30 min in ND-96 as previously described (7). Total bath [Cl−] was 103.6 mM (0.5 μCi/well). [14C]oxalate influx studies were carried out for 30-min periods in nominally Ca2+- and Mg2+-free influx medium containing (in mM) 96 mM Na cyclamate, 2 K glutamate, 5 HEPES, pH 7.40, with added 1.0 mM sodium oxalate (0.375 μCi/well; 150 μl), or the indicated oxalate concentrations with balancing cyclamate.
Influx experiments were terminated with four washes in Cl−-free ND96, followed by oocyte lysis in 150 μl of 2% sodium dodecyl sulfate (SDS). Duplicate 10-μl aliquots of influx solution were used to calculate specific activity of radiolabeled substrate anions. Oocyte anion uptake was calculated from oocyte-associated counts per minute (cpm) and bath specific activity.
Isotopic efflux experiments.
For unidirectional 36Cl− efflux studies individual oocytes in Cl−-free ND-96 were injected with 50 nl of 260 mM Na36Cl (20,000–24,000 cpm). After a 5- to 10-min recovery period in Cl−-free ND-96, the efflux assay was initiated by transfer of individual oocytes to 6-ml borosilicate glass tubes, each containing 1 ml efflux solution. At intervals of 1 or 3 min, 0.95 ml of this efflux solution was removed for scintillation counting and replaced with an equal volume of fresh efflux solution. After completion of the assay with a final efflux period either in Cl−-free cyclamate solution or in the presence of the inhibitor DIDS (100 or 200 μM), each oocyte was lysed in 150 μl of 2% SDS. Samples were counted for 3–5 min such that the magnitude of 2SD was <5% of the sample mean.
For [14C]oxalate efflux assays, oocytes were injected with 50 nl of 50 mM Na [14C]oxalate (6,000–8,000 cpm, with final estimated intracellular concentration 5 mM). After a recovery period of at least 20 min, efflux was measured in baths containing up to 103.6 mM NaCl or 64 mM Na2SO4, or as indicated. In baths of reduced [Cl−] or [SO42−] the respective anions were replaced with equimolar cyclamate. Bath anion concentration-response curves were generated from individual oocytes subjected sequentially to increasing concentrations of the bath anion under study. Single oocytes were exposed to a maximum of six conditions.
To vary pHi, oocytes were preexposed to 40 mM Na butyrate (substituting for Na cyclamate) for 30 min before initiation of an efflux experiment to produce intracellular acidification to pHi 6.8. Upon removal of bath butyrate (with substitution by Na cyclamate) during the efflux experiment, pHi rapidly alkalinized back toward initial pHi while pHo remained constant. Variation of pHo was achieved at near-constant pHi (39). Some oocyte groups were exposed to 20 mM NH4Cl during the efflux experiments, acidifying pHi to 6.9 (26). Drugs were added to the bath or were injected into oocytes either before or together with an isotope as indicated.
Efflux data were plotted as the natural logarithm (ln) of the quantity (%cpm remaining in the oocyte) versus time. Efflux rate constants for 35SO42−, 36Cl−, and [14C]oxalate were measured from linear fits to data from the last three time points sampled within each experimental period. For each experiment, water-injected or uninjected oocytes from the same frog were subjected to parallel measurements with cRNA-injected oocytes. Each experimental condition was tested in oocytes from at least two frogs. Two exclusion criteria were defined to categorize efflux experiments as reflecting “nonspecific” efflux or “leaky” oocytes. One criterion was <50% reduction in efflux rate constant upon final addition of the inhibitor DIDS or upon final bath substitution of exchangeable substrate anion for the impermeant anion, cyclamate. The second criterion was loss of >85% of injected isotope before completion of the efflux assay.
Application of these a priori criteria resulted in exclusion from analysis of 11% of 176 36Cl−-injected oocytes, 12% of 330 35SO42−-injected oocytes, and 15% of 683 [14C]oxalate-injected oocytes.
Two-electrode voltage-clamp measurements.
Microelectrodes from borosilicate glass made with a Sutter P-87 puller were filled with 3 M KCl and had resistances of 2–3 MΩ. Oocytes previously injected with water or with 10 ng of the indicated cRNA were placed in a 1-ml chamber (model RC-11, Warner Instruments, Hamden, CT) on the stage of a dissecting microscope and impaled with microelectrodes under direct view. Steady-state currents achieved within 2–5 min following bath change or drug addition were measured with a Geneclamp 500 amplifier (Axon Instruments, Burlingame, CA) interfaced to an Dell computer with a Digidata 1322A digitizer (Axon). Standard recording bath solution was (in mM) 93.5 NaCl, 2 KCl, 5 HEPES, 2.8 MgCl2, pH 7.40, or occasionally ND-96. In anion substitution experiments NaCl was replaced with Na cyclamate, with addition of 5 mM sulfate or oxalate. In some experiments, 96 mM NaCl was replaced with 64 mM Na2SO42−. Bath addition of 5 mM Na oxalate was performed in the presence of nominally Ca2+-free cyclamate solution.
Data acquisition and analysis utilized pCLAMP 8.0 software (Axon). The voltage pulse protocol generated with the Clampex subroutine consisted of 20-mV steps between −100 mV and +40 mV, with durations of 738 ms separated by 30 ms at the holding potential of −30 mV. Bath resistance was minimized by the use of agar bridges filled with 3 M KCl, and a virtual ground circuit clamped bath potential to zero during voltage-clamp experiments.
Confocal immunofluorescence microscopy.
Two days after injection with H2O or with cRNA encoding COOH terminally GFP-tagged hSLC26A2, 10–12 oocytes were fixed in 3% paraformaldehyde (PFA) in phosphate-buffered saline (PBS) for 30 min at room temperature, washed three times in PBS supplemented with 0.002% Na azide (PBS-azide), and saved at 4°C for direct imaging of GFP. Oocytes were aligned in uniform orientation along a Plexiglas groove and sequentially imaged through the ×10 objective of a Zeiss LSM510 laser scanning confocal microscope using the 488-nm laser line at 512 × 512 resolution at a uniform setting of 80% intensity, pinhole 61 μm (1.0 Airy units), detector gain 650, Amp gain 1, 0 amp offset.
Two or three days after injection with H2O or with cRNA encoding wild-type or mutant untagged hSLC26A2, groups of 10–12 oocytes were similarly fixed with 3% PFA and washed with PBS-azide. Oocytes were then placed in PBS containing 1% SDS to permeabilize the surface membranes and unmask epitope. Fixed, permeabilized oocytes were blocked in PBS with 1% bovine serum albumin (PBS-BSA) containing 0.05% saponin for 1 h at 4°C and then washed three times with PBS-BSA. Oocytes were incubated overnight at 4°C with mouse monoclonal anti-hSLC26A2 antibody I-G2 at 1:400 dilution in PBS-BSA (28) and then washed three times in cold PBS-BSA. Antibody-labeled oocytes were then incubated 2 h with Cy3-conjugated secondary goat anti-mouse Ig (dilution 1:500, Jackson Immunochemicals), again thoroughly washed in PBS-BSA, and stored at 4°C until imaging.
Cy3-labeled oocytes were aligned in uniform orientation along a Plexiglas groove and sequentially imaged through the ×10 objective of a Zeiss LSM510 laser scanning confocal microscope by using the 543-nm laser line at 512 × 512 resolution at a uniform setting of 80% intensity, pinhole 54 (1.0 Airy units), detector gain 650, Amp gain 1, 0 amp offset.
Polypeptide abundance at or near each oocyte surface was estimated by quantitation of specific fluorescence intensity (FI) at the circumference of one quadrant of an equatorial focal plane (Image J v. 1.38, National Institutes of Health). Mean background-corrected FI for quadrants of oocytes previously injected with water was subtracted from the background-corrected FI for quadrants of individual cRNA-injected oocytes to yield intensity values for surface-associated specific FI for each oocyte.
Statistics.
Data are reported as means ± SE. Flux data were compared by Student's paired or unpaired two-tailed t tests (Microsoft Excel), or by ANOVA with Bonferroni post hoc analysis (SigmaPlot). Concentration-dependence data were fit to a four-parameter Hill equation in SigmaPlot 8.0. Two-electrode voltage-clamp data were analyzed by ANOVA followed by Bonferroni's T-test for multiple samples (SigmaPlot 8.0). Image intensity data were compared by ANOVA. P < 0.05 was interpreted as significant.
RESULTS
SO42− uptake by SLC26A2 is cis-inhibited by Cl− and oxalate.
Previous isotopic flux studies of recombinant rat and human SLC26A2 have been limited to 35SO42−. hSLC26A2-mediated 35SO42− influx exhibited a K1/2 of 67 μM in Xenopus oocytes (29), was cis-inhibited by 100–150 mM Cl− or HCO3−, and by 5 mM oxalate, SO42−, or thiosulfate (37). We confirmed that 35SO42− influx mediated by hSLC26A2 was cis-inhibited by extracellular Cl− with K1/2 of 24.1 ± 3.8 mM (Fig. 1A). 35SO42− influx mediated by mSlc26a2 was inhibited by extracellular Cl− with a similar K1/2 of 18.4 ± 3.4 mM (Fig. 1C). hSLC26A2-mediated 35SO42− influx was cis-inhibited by extracellular oxalate with K1/2 of 5.1 ± 1.7 mM (Fig. 1B), and mSlc26a2 showed similar results (Fig. 1D). Cis-inhibition by 24 mM HCO3− was 20% for hSLC26A2 and 45% by mSlc26a2. 35SO42− influx by both orthologs was completely inhibited in the presence of 200 μM DIDS (Fig. 1D and data not shown), consistent with previous reports (29, 37). Inhibition by DIDS (12 min exposure) was only partially reversible, whereas the lower affinity inhibition by DNDS was completely reversible (supplemental Fig. 3).
Fig. 1.
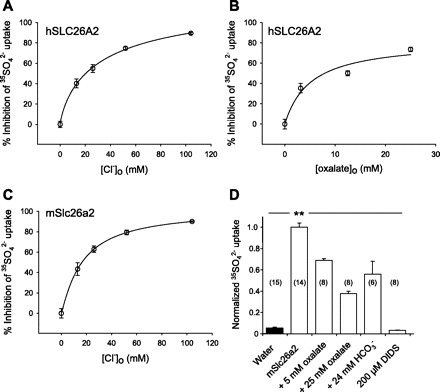
Cis-inhibition of SLC26A2-mediated SO42− uptake by extracellular anions. A: extracellular [Cl−] dependence for cis-inhibition of 35SO42− uptake by hSLC26A2. K1/2 = 24.1 ± 3.8 mM Cl− (R2 = 0.90, n = 12–14). B: extracellular [oxalate] dependence for cis-inhibition of 35SO42− uptake by hSLC26A2. K1/2 = 5.1 ± 1.7 mM oxalate (R2 = 0.86, n = 7–9). C: extracellular [Cl−] dependence for cis-inhibition of 35SO42− uptake by mSlc26a2. K1/2 = 18.4 ± 3.4 mM Cl− (R2 = 0.93, n = 6–7). D: cis-inhibition of mSlc26a2-mediated 35SO42− uptake by oxalate or HCO3− at the indicated concentrations for (n) oocytes compared with complete inhibition by DIDS (200 μM). Oocytes previously injected with 50 ng cRNA or water were subjected to 30 min uptake in the presence of 1 mM extracellular SO42−. Uptake data were normalized to same-day experiments carried out in cyclamate. Solid bar indicates water-injected oocytes. Normalized uninhibited SO42− uptake was (in nmol/oocyte−1·h−1) 1.42 ± 0.05 (A), 1.48 ± 0.07 (B), 1.13 ± 0.05 (C), and 1.03 ± 0.19 (D). Values are means ± SE. **P < 0.01.
SLC26A2 mediates bidirectional oxalate/SO42− exchange.
Although oxalate had been shown to cis-inhibit uptake of SO42− by SLC26A2, SLC26A2-mediated transport of oxalate had not been reported. We found that hSLC26A2 transported oxalate with a K1/2 for [14C]oxalate uptake of 0.65 ± 0.08 mM (Fig. 2A). [14C]oxalate uptake was cis-inhibited by extracellular SO42− with K1/2 of 3.1 ± 0.7 mM. Bidirectional oxalate/SO42− exchange by hSLC26A2 was demonstrated by 35SO42− efflux dependent on extracellular oxalate (Fig. 2C, SO42−i/oxalateo exchange) and by [14C]oxalate efflux dependent on extracellular SO42− (Fig. 2D, oxalatei/SO42−o exchange). hSLC26A2-mediated oxalate/SO42− exchange measured as 35SO42− efflux was characterized by K1/2 for extracellular oxalate of 1.6 ± 0.4 mM, and a K1/2 for extracellular SO42− of 0.14 ± 0.05 mM measured as [14C]oxalate efflux. The corresponding extracellular K1/2 values for mSlc26a2 were 3.7 ± 0.7 mM for oxalate (Fig. 3C) and 0.20 ± 0.08 mM for SO42− (Fig. 3D). These K1/2 values for extracellular SO42− in [14C]oxalatei/SO42−o exchange were within two- to threefold of the K1/2 for SO42− influx of 0.067 mM reported by Karniski for hSLC26A2 (29).
Fig. 2.

hSLC26A2 mediates bidirectional oxalate/SO42− exchange. A: bath [oxalate] dependence of 30 min oxalate uptake by oocytes expressing hSLC26A2. K1/2 = 0.65 ± 0.08 mM oxalate, Hill coefficient = 2.01 ± 0.41 (R2 = 0.66, n = 19–20). Normalized oxalate uptake = 1.0 represented 0.15 nmol/h. B: bath [SO42−] dependence for cis-inhibition of 1 mM bath oxalate uptake by oocytes expressing hSLC26A2. K1/2 = 3.07 ± 0.74 mM sulfate (R2 = 0.72, n = 7–17). C: traces of 35SO42− efflux from individual oocytes previously injected with water (open squares) or hSLC26A2 (filled circles) during sequential exposure to Cl−-free cyclamate baths in the initial presence and subsequent absence of 25 mM oxalate. D: traces of [14C]oxalate efflux from individual oocytes previously injected with water (open squares) or hSLC26A2 (filled circles) during sequential exposure to Cl−-free baths in the initial presence and subsequent absence of 64 mM SO42−. Oocytes previously injected with 50 ng cRNA or water were assayed 4 days postinjection. Values are means ± SE.
Fig. 3.

[Substrate] dependence of SLC26A2-mediated oxalate/SO42− exchange. A: bath [oxalate] dependence of hSLC26A2-mediated 35SO42− efflux. K1/2 = 1.58 ± 0.44 mM oxalate (n = 4–5). B: bath [SO42−] dependence of hSLC26A2-mediated oxalate efflux. K1/2 = 0.14 ± 0.05 mM SO42− (n = 5–22). C: bath [oxalate] dependence of mSlc26a2-mediated 35SO42− efflux. K1/2 = 3.74 ± 0.73 mM oxalate (n = 3–15). D: bath [SO42−] dependence of mSlc26a2-mediated oxalate efflux. K1/2 = 0.20 ± 0.08 mM SO42− (n = 4–20). Oocytes previously injected with 50 ng cRNA were assayed 3–4 days postinjection. Values are means ± SE. Curve fits in B and D were unchanged by inclusion of additional [14C]oxalate efflux rate constants measured at higher bath [SO42−] values of 8 and 9 mM (n = 5–6, data not shown). There was no sulfate self-inhibition at still higher bath [SO42−] values of 16, 32, or 64 mM (not shown).
SLC26A2-mediated exchange of intracellular SO42−, [14C]oxalate, and 36Cl− for extracellular Cl− is nonsaturable with respect to extracellular Cl−.
Efflux of 35SO42− from oocytes expressing hSLC26A2 or mSlc26a2 into bath Cl− (SO42−i/Clo− exchange) exhibited a quasilinear, nonsaturable extracellular [Cl−] dependence of low affinity (Fig. 4). hSLC26A2-mediated [14C]oxalate efflux into bath Cl− (oxalatei/Clo− exchange, supplemental Fig. 3) exhibited similar quasi-linear [Cl−] dependence of low affinity (Fig. 5, A and B). The low affinity for extracellular Cl− was also evident in measurements of hSLC26A2-mediated 36Cl− efflux into the Cl− bath (Cli−/Clo− exchange, Fig. 5, C and D). In oocytes expressing mSlc26a2 or hSLC26A2, 36Cl− influx in exchange for intracellular anion (nominal Cl−) was also linear and nonsaturable out to 104 mM (supplemental Fig. 4, A and C). Thus inward translocation of extracellular Cl− was of low affinity in exchange for each of the three intracellular anions tested. This property of SLC26A2 differed from its higher-affinity, saturable transport of extracellular SO42− and oxalate. The nonsaturable extracellular [Cl−] dependence for SLC26A2-mediated anion exchange also contrasted markedly with the saturable cis-inhibition of SO42− uptake by extracellular Cl− (Fig. 1).
Fig. 4.

SLC26A2-mediated exchange of intracellular 35SO42− for bath Cl− exhibits very low Cl− affinity. A: traces of 35SO42− efflux from individual oocytes previously injected with water (open squares, n = 2) or hSLC26A2 cRNA (filled circles, n = 6) during sequential increases in bath [Cl−], followed by exposure to 200 μM DIDS. B: extracellular Cl− concentration ([Cl−]o) dependence of hSLC26A2-mediated [35SO42−]i/[Cl−]o exchange determined from A. C: traces of 35SO42− efflux from individual oocytes previously injected with water (open squares, n = 2) or mSlc26a2 cRNA (filled circles, n = 6) during sequential increases in bath [Cl−], followed by addition of 200 μM DIDS. D: [Cl−]o dependence of mSlc26a2-mediated [35SO42−]i/[Cl−]o exchange determined from C. Values in B and D are means ± SE measured 3 days after oocyte injection with cRNA encoding hSLC26A2 (1 ng) or mSlc26a2 (5 ng). Data are representative of 2–3 similar experiments.
Fig. 5.

hSLC26A2-mediated exchange of intracellular [14C]oxalate or 36Cl− for bath Cl− exhibits low bath Cl− affinity. A: traces of [14C]oxalate efflux from individual oocytes previously injected with water (open squares, n = 3) or hSLC26A2 cRNA (closed circles, n = 6) during sequential increases in bath [Cl−], followed by addition of 200 μM DIDS. B: [Cl−]o dependence of hSLC26A2-mediated [14C]oxalatei/Clo− exchange determined from A. C: traces of 36Cl− efflux from individual oocytes previously injected with water (open squares, n = 3) or hSLC26A2 cRNA (filled circles, n = 6) during sequential increases in bath [Cl−], followed by exposure to 200 μM DIDS. D: [Cl−]o dependence of hSLC26A2-mediated 36Cli−/Clo−exchange determined from C. Values in B and D are means ± SE measured 3 days after oocyte injection with 1 ng hSLC26A2 cRNA and are representative of 2–3 similar experiments.
SLC26A2 exhibits very low rates of exchange of intracellular 36Cl− for extracellular SO42− or oxalate.
The unusual interaction of extracellular Cl− with SLC26A2 led us to examine exchange of intracellular Cl−. The substantial uptake rates of SO42− and Cl−, and the moderate uptake of oxalate, appeared likely to reflect exchange with intracellular Cl−. However, rates of SLC26A2-mediated exchange of intracellular 36Cl− into saturating extracellular [SO42−] (supplemental Fig. 4, B and D) or saturating extracellular [oxalate] (data not shown) were 10- to 30-fold lower (supplemental Fig. 4, C–F) than that of 36Cl− efflux into 104 mM extracellular Cl− (Fig. 5, C and D). Nonetheless, the K1/2 values for extracellular SO42− and oxalate at these low exchange rates were similar to those measured at the higher transport rates of the respective isotopically labeled anions. Thus the intracellular Cl− binding site of SLC26A2 exhibits highly specific trans-anion dependence. Occupancy of the intracellular anion binding site of SLC26A2 by 36Cl− allows robust 36Cli−/Clo− exchange. Rates of Cli−/SO42−o and Cli−/oxalateo exchange are much lower, despite the preservation of K1/2 values for these extracellular anion substrates.
SLC26A2-mediated exchange of extracellular oxalate or sulfate with intracelluar anion is not detectably electrogenic.
The above data shows that SLC26A2/DTDST is capable of exchanging Cl− for another intracellular anion of oocytes for divalent SO42− and oxalate. Bidirectional, electrogenic exchange of divalent oxalate with Cl− has been demonstrated with mSlc26a6 and with nonmammalian Slc26a5/prestin (8, 38). However, SO42−/HCO3− exchange and oxalate/HCO3− exchange by SLC26A1/SAT1 was observed to be electroneutral (30). Figure 6A shows that addition of 5 mM oxalate (a saturating concentration) to hSLC26A2-expressing oocytes in a Cl−-free gluconate bath produces no detectable increase of outward current (Fig. 6A). Figure 6B shows that addition of 5 mM SO42− [also a saturating concentration (see Ref. 29 and Figs. 2B, 3B)] to hSLC26A2-expressing oocytes in a Cl−-free cyclamate bath, or complete replacement of cyclamate with 64 mM SO42−, also fail to enhance detectable oocyte outward current. Similar lack of outward current enhancement by bath addition of oxalate (Fig. 6C) or SO42− (Fig. 6D) was noted for oocytes expressing mSlc26a2. In addition, substitution of 96 mM bath cyclamate with 96 mM Cl− failed to elicit currents in oocytes containing nominal [SO42−]i of 1 or 14 mM (n = 6 for each group, data not shown). Oocytes from the same frogs mediated robust isotopic influx of 36Cl− (not shown). Thus SLC26A2-mediated exchange of extracellular oxalate or SO42− with intracellular anion (nominally Cl−) and exchange of intracellular anion with extracellular Cl− were unaccompanied by detectable currents in voltage-clamped oocytes. We conclude that all tested modes of SLC26A2-mediated anion exchange in Xenopus oocytes are electroneutral within our limits of detection.
Fig. 6.

SLC26A2 mediates electroneutral exchange of bath oxalate or bath sulfate for nominal intracellular Cl− (Cli−). A: steady-state current-voltage (I-V) relationships of hSLC26A2-expressing oocytes measured first in Na gluconate bath (open circles) and again after subsequent bath addition of 5 mM oxalate (filled circles; n = 5). B: steady-state I-V relationships of hSLC26A2-expressing oocytes during sequential exposure to baths containing Na cyclamate (open circles), cyclamate with 5 mM SO42− (filled circles), and 64 mM SO42− (open triangles), with subsequent addition of 200 μM DIDS (filled triangles; n = 10). C: steady-state I-V relationships of mSlc26a2-expressing oocytes measured first in Na gluconate bath (open circles) and again after subsequent bath addition of 5 mM oxalate (filled circles; n = 4). D: steady-state I-V relationships of mSlc26a2-expressing oocytes during sequential exposure to baths containing Na cyclamate (open circles), cyclamate with 5 mM SO42− (filled circles), and 64 mM SO42− (open triangles), with subsequent addition of 200 μM DIDS (filled triangles; n = 9). Values are means ± SE.
Acidic pHo inhibits SLC26A2-mediated anion exchange.
hSLC26A2/DTDST was highly sensitive to pHo. hSLC26A2-expressing oocytes exposed to ND96 at pH 5.0 exhibited little efflux of 35SO42−, but efflux rapidly accelerated more than 10-fold upon bath pH elevation to 8.0 (SO42−i/Clo− exchange). The faster efflux did not represent nonspecific leak, since it remained sensitive to inhibition by DIDS (Fig. 7, A and B). Inhibition of hSLC26A2 by acidic pHo also applied to efflux of 36Cl− into ND96 (Cli−/Clo−exchange, Fig. 7C) and efflux of [14C]oxalate into ND96 (oxalatei/Clo− exchange, Fig. 7D). The strong inhibition of SLC26A2/DTDST by acidic pHo is paralog-specific. It contrasts with the modest inhibition by extracellular protons of hSLC26A6-mediated [14C]oxalatei/Clo− exchange (supplemental Fig. 5) and [14C]oxalate uptake (8), and with pHo-insensitive 36Cli−/Clo− exchange by hSLC26A3 (7).
Fig. 7.

Regulation of hSLC26A2-mediated anion exchange by extracellular pH. A: 35SO42− efflux traces from individual oocytes previously injected with water (open squares, n = 3) or with 0.5 ng hSLC26A2 (filled circles, n = 5) during sequential exposure to ND96 bath at pH 5.0, pH 8.0, and subsequent addition of 200 μM DIDS. B: 35SO42− efflux rate constants of oocytes expressing hSLC26A2 during sequential exposure to ND96 baths of pH 5.0, pH 8.0, and after addition of DIDS (open bars, n = 16). Solid bars show water-injected oocytes (n = 5). These data include those of A. C: 36Cl− efflux rate constants of oocytes expressing hSLC26A2 (10 ng cRNA) during sequential exposure to ND96 baths of pH 5.0, pH 8.0, and after addition of DIDS (open bars, n = 12). Solid bars show water-injected oocytes (n = 6). D: [14C]oxalate efflux rate constants of oocytes expressing hSLC26A2 (50 ng cRNA) during sequential exposure to ND96 baths of pH 5.0, pH 8.0, and after addition of DIDS (open bars, n = 11). Solid bars show water-injected oocytes (n = 4). Values in B–D are means ± SE measured 3–4 days post-cRNA injection.
Acidic pHi activates SLC26A2-mediated exchange of bath Cl− for intracellular 35SO42− but not with intracellular 36Cl− or [14C]oxalate.
hSLC26A2 pHi sensitivity was tested by addition and removal of 40 mM butyrate, which reversibly decreases oocyte pHi 0.5 pH units (39). hSLC26A2-mediated 35SO42−i/Clo− exchange was more active at acidic pHi (in the presence of butyrate) than at resting pHi (in the absence of butyrate), as shown in Fig. 8, A and B. However, hSLC26A2 was nearly or completely pHi insensitive when it mediated efflux of 36Cli− (Fig. 8C) or of [14C]oxalate (Fig. 8D) in exchange for extracellular Cl− (Cli−/Clo− exchange or oxalatei/Clo− exchange).
Fig. 8.

Regulation of hSLC26A2-mediated anion exchange by intracellular pH. A: 35SO42− efflux traces from individual uninjected oocytes (open squares, n = 2) or from oocytes previously injected with 0.5 ng hSLC26A2 cRNA (closed circles, n = 4) during sequential exposure to baths containing 64 mM Cl− with 40 mM Na butyrate, 64 mM Cl− with 40 mM sodium cyclamate, and Cl−-free cyclamate. B: effect of butyrate removal (intracellular alkalinization) on 35SO42−i/Clo− exchange by oocytes previously injected with 0.5 ng hSLC26A2 (open bars, n = 10) or from uninjected oocytes (solid bars, n = 2) determined as in A. C: effect of butyrate removal on Cli−/Clo− exchange by oocytes previously injected with 10 ng hSLC26A2 (open bars, n = 11) and by uninjected (solid bars, n = 5). D: effect of butyrate removal on [14C]oxalatei/Clo− exchange from oocytes previously injected with 50 ng hSLC26A2 (open bars, n = 11) and by uninjected oocytes (solid bars, n = 6). Values in B–D are means ± SE measured 3–4 days post-cRNA injection. All oocytes were preincubated 30 min in Cl−-free 40 mM Na butyrate before initiation of efflux experiments.
The apparent pHi sensitivity of SO42− efflux might also be explained by SCL26A2-mediated butyrate/SO42− exchange. However, Fig. 9 demonstrates that extracellular butyrate did not itself serve as a substrate for hSLC26A2-mediated exchange with 35SO42−, whereas subsequent introduction of bath Cl− elicited robust 35SO42− efflux activity sensitive to DIDS.
Fig. 9.
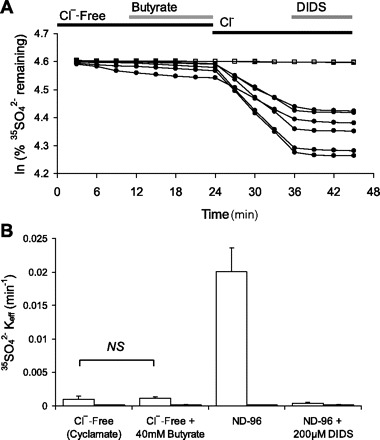
hSLC26A2 does not mediate 35SO42−i/butyrateo exchange. A. 35SO42− efflux traces from individual uninjected oocytes (open squares, n = 2) or from oocytes previously injected with 0.5 ng hSLC26A2 cRNA (closed circles, n = 7) during sequential exposure to baths containing Cl−-free cyclamate, cyclamate with 40 mM Na butyrate, butyrate-free ND96, and ND96 containing 200 μM DIDS. B: 35SO42− efflux rate constants (means ± SE) of oocytes shown in A. Values are measured 3 days post-cRNA injection.
Exposure of oocytes to 20 mM NH4Cl also acidifies pHi (∼0.4 pH units) (4, 26, 36), likely reflecting a native ratio of NH4+/NH3 permeabilities higher than that of mammalian tissue culture cells. This route of intracellular acidification, in contrast to butyrate, inhibited hSLC26A2-mediated efflux of 35SO42− into bath Cl− (SO42−i/Clo− exchange; Supplemental Fig. 6, A and B). Moreover, 20 mM NH4+ stimulated 36Cl− efflux into bath Cl− (Cli−/Clo− exchange; supplemental Fig. 6C), and had minimal effect on [14C]oxalate efflux (oxalatei/Clo− exchange; supplemental Fig. 6D). Thus, as observed previously for other anion exchangers (26), NH4+ regulates SLC26A2 by a mechanism likely distinct from that mediating regulation by pHi.
The anion substrate-selective pHi-sensitivity phenotype observed for hSLC26A2 has not been previously described among SLC26 transporters. It also contrasts remarkably with the pH sensitivity of anion exchange mediated by Slc4a2/Ae2, which is inhibited by both intracellular and extracellular protons acting through distinct structural elements of the polypeptide (39).
PKC inhibits hSLC26A2 through reduction of surface expression.
Phorbol ester-sensitive PKC inhibits mSlc26a6 activity, largely through reduction in oocyte surface expression, whereas hSLC26A4/pendrin lacks this response to PKC (21). We reproduced this pattern of hSLC26A6 regulation by PKC (data not shown) and then tested the response of hSLC26A2 to PKC. As shown in Fig. 10, A and B, exposure to the classical PKC activator PMA in the absence of bath Cl− dramatically reduced subsequent 35SO42− efflux after reintroduction of bath Cl−, whereas exposure to the inactive PMA analog 4α-PDD was without effect. PKC activation was associated with reduced abundance of hSLC26A2-GFP at or near the oocyte surface, as shown in Fig. 10, C and D. Again, 4α-PDD was without effect. Thus PKC inhibits hSLC26A2/DTDST largely by reducing its abundance at the cell surface, similar to its regulation of mSlc26a6.
Fig. 10.
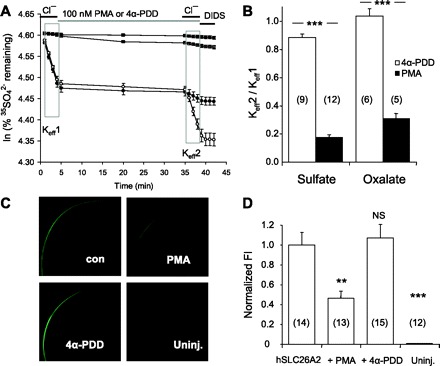
Protein kinase C inhibits hSLC26A2-mediated efflux of SO42− and of oxalate by reducing surface expression. A: 35SO42− efflux traces averaged from representative individual uninjected oocytes (two top traces, each n = 3) or from oocytes previously injected with 0.5 ng hSLC26A2 cRNA (two bottom traces, each n = 6) during sequential exposure to baths containing ND96, Cl−-free cyclamate containing PMA (closed symbols) or 4α-phorbol-didecanoate (4α-PDD) (open symbols), then ND96 in the continued presence of PMA or 4α-PDD, and finally, ND96 with 200 μM DIDS. Efflux trace slopes from the first and second bath Cl− periods (boxed areas) yield Keff1 and Keff2 as indicated. B: ratios of mean rate constants for 35SO42− efflux (Keff2/Keff1) into ND96 in the presence of 4α-PDD (open bars) or PMA (solid bars). C: confocal fluorescence micrographs of representative oocytes, uninjected or previously injected with 10 ng hSLC26A2-GFP cRNA, then incubated 30 min in the absence (con) or presence of PMA or 4α-PDD as indicated. D: normalized hSLC26A6-GFP fluorescence intensity (FI, means ± SE) at the periphery of (n) oocytes treated with PMA or 4α-PDD. Untreated hSLC26A2-GFP-expressing oocytes were assigned a mean FI value of 1.0. **P < 0.01; ***P < 0.001.
DTDST mutations inhibit hSLC26A2 activity at the cell surface or by regulating surface abundance.
Mutations in hSLC26A2/DTDST cause several related clinical phenotypes of chondrodysplasia (22, 35). A related murine phenotype was generated by genetic knock-in of a mutation homologous to a human mutation associated with disease of moderate severity (10, 14). The several hSLC26A2 disease mutations that have been subjected to functional study exhibited decreased SO42− uptake into oocytes (29) or HEK-293 cells (28). We therefore compared the effects of chondrodysplasia mutations on oxalate and SO42− transport. The severe hSLC26A2 mutation R279W impaired uptake of 35SO42− (Fig. 11A), as described earlier (28, 29). Uptake of [14C]oxalate was reduced to a comparable degree (Fig. 11B). The mutation A386V, analogous to that in the Slc26 mutant mouse (14) but not yet tested functionally, was associated with less severely reduced uptake of both 35SO42− and [14C]oxalate (Fig. 11, A and B). The impaired function of mutant A386V was largely secondary to reduced surface abundance of mutant polypeptide, whereas that of mutant R279W exhibited near wild-type surface abundance despite impaired SO42− uptake activity (Fig. 11, C and D).
Fig. 11.

Two diastrophic dysplasia sulfate transporter (DTDST) mutants of hSLC26A2 exhibit impaired efflux of 35SO42− and of oxalate but differ in surface expression. A: normalized 35SO42− uptake by (n) uninjected oocytes or oocytes expressing wild-type (WT) hSLC26A2 or the indicated hSLC26A2 mutants (0.5 ng cRNA). WT uptake 0.17 ± 0.02 nmol/h. B: normalized [14C]oxalate uptake by (n) uninjected oocytes or oocytes expressing WT or mutant hSLC26A2 (50 ng cRNA). WT uptake was 0.20 ± 0.02 nmol/h. C: confocal immunofluorescence micrographs of representative oocytes expressing WT hSLC26A2 or the indicated mutants. D: normalized mean fluorescence intensity (FI) measured at the periphery of (n) oocytes expressing WT or mutant SLC26A2. WT hSLC26A2 intensity was assigned a mean FI of 1.0. ***P < 0.001.
DISCUSSION
SLC26A2 was the first human member of the SulP gene superfamily linked to human genetic disease. Its homology with the previously identified SO42− uptake transporters hSAT-1 and sulfate permease II of Neurospora crassa led to the demonstration of loss of SO42− uptake by primary fibroblasts from a patient with diastrophic dysplasia (DTD) (22). Subsequent studies of recombinant SLC26A2 documented SO42− transport, defined cis-inhibitor anions (37) and K1/2 for extracellular SO42− in Xenopus oocytes, and documented reduced SO42− uptake by most SLC26A2 mutants of DTD patients in oocytes and HEK-293 cells (28, 29).
In the current paper, we have extended the functional characterization of SLC26A2. We establish that hSLC26A2 and mSlc26a2 mediate bidirectional anion exchange with similar characteristics. We find that SLC26A2 transports oxalate and Cl− (in addition to SO42−) by an apparently electroneutral mechanism. We show that SLC26A2 is regulated independently by pHo, pHi, NH4+, and PKC, and that disease mutations alter oxalate transport in parallel with SO42− transport. We also report anion substrate-selective asymmetries of anion exchange.
Lack of species differences.
The 78% amino acid sequence identity between mSlc26a6 and hSLC26A6 is associated with substantial differences in anion transport selectivity and affinity (6, 8). Thus the 80% amino acid sequence identity between mSlc26a2 and hSLC26A2/DTDST similarly prompted us to compare functional properties of these two species orthologs. In contrast to anion transport by SLC26A6, we found that anion transport by mouse and human SLC26A2 did not differ in their tested transport properties. This functional similarity strengthens the possibility that study of mSlc26a2 and the hypomorph knock-in mouse (14) will continue to provide data applicable to hSLC26A2 and its deficiency diseases.
Oxalate transport by wild-type and mutant SLC26A6.
The expression of mSlc26a2 polypeptide in the apical pole of colonocytes (20) prompted us to test it as an oxalate transporter possibly contributing to oxalate absorption or to the 25–50% of transepithelial oxalate secretion that is mSlc26a6 independent (15, 27). We demonstrated that both mouse and human SLC26A2 orthologs mediate oxalate uptake cis-inhibited by SO42−, as well as bidirectional, saturable oxalate/SO42−o exchange. [14C]Oxalatei/Clo− exchange by SLC26A2 was more rapid than [14C]oxalatei/SO42−o exchange. However, Cli−/oxalateo exchange efflux rate constants were ∼10-fold lower than for oppositely directed anion exchange. We confirmed the earlier observation (37) that 5 mM oxalate cis-inhibits SO42− uptake but less potently (Fig. 1B) than previously noted.
SLC26A2 is abundant in chondrocytes, suggesting the possibility of chondrocyte involvement in primary hyperoxaluria. Oxalosis can indeed involve bones, joints (32), and skin (40), but oxalate transport by chondrocytes and fibroblasts has not yet been reported. Urinary oxalate excretion has not been measured in the Slc26a2 hypomorph mutant mice, in which nephrocalcinosis has not been radiologically evident (A. Rossi, University of Pavia, personal communication).
SLC26A2 chondrodysplasia mutant polypeptides have been shown previously to exhibit varying degrees of decreased SO42− transport. We studied two human disease mutations, one of which (A386V) corresponds to the mouse knock-in hypomorph mutation but had not been studied as an anion transporter. We found that transport of SO42− and oxalate was reduced in parallel for both mutants, but that only A386V exhibited parallel reduction in abundance at or near the oocyte surface. The reduced function of the R279W mutant in the setting of apparently wild-type surface abundance suggests that the mutation reduces the rate of the conformational cycle that mediates anion transport. The R279W mutant should thus prove a useful point of entry for future structure-function studies of SLC26A2.
SLC26A2-mediated transport of SO42− and Cl−.
Our results extend previous measurements of SO42− transport by SLC26A2 with demonstrations of exchange with oxalate and with Cl−. Whereas the K1/2 of SO42−o for cis-inhibition of [14C]oxalate uptake was 3 mM, the K1/2 of SO42−o for exchange with intracellular [14C]oxalate exhibited a 15- to 20-fold higher affinity. This difference is due in part to the presence or absence of cis-competitor, but with these additional contributions. The oxalate injection required to measure [14C]oxalate efflux is expected to saturate the internal binding site and promote oxalate translocation and exposure of the outward-facing substrate binding site. A large increase in the population of outward-facing sites available to extracellular substrate should increase apparent affinity. In addition, the required oxalate injection likely reduces intracellular [Ca2+]i and may triggers stress responses or other regulatory pathways potentially altering apparent extracellular substrate affinity.
In contrast to the saturable, bidirectional transport of SO42− and oxalate, Clo− exibited nonsaturable uptake. In addition, [Cl−]o was nonsaturable with respect to exchange of 35SO42−i, [14C]oxalatei, and 36Cli−, suggesting that the extracellular anion binding site of SLC26A2 has very low affinity for Cl−. SLC26A2-mediated 36Cl− efflux exhibited trans-anion selectivity, in that exchange with Clo− was approximately 10-fold more rapid than exchange with SO42−o or with [14C]oxalateo. Thus the free energy required for the conformational transition associated with Cli− translocation may vary with the extracellular anion, suggesting possible simultaneous anion binding on both sides of the membrane, an external regulatory anion binding site, or anion-specific influence on substrate off-rate after translocation.
Of note, the low-affinity [Cl−]o dependence of mSlc26a2 for 35SO42− efflux exhibited a sigmoid pattern suggestive of positive cooperativity (Fig. 4D), as was also observed for hSLC26A2-mediated 36Cl−/Cl− exchange. However, the [Cl−]o dependence of hSLC26A2-mediated [14C]oxalate efflux did not show this sigmoid pattern. Thus, in some conditions, extracellular Cl− may exert a regulatory function beyond that of substrate. Complex, condition-dependent substrate cooperativity for electroneutral cation exchange has been observed previously for NHE Na+/H+ exchangers (17) and NCX Na+/Ca2+ exchangers (3).
SLC26A2-mediated anion exchange is not detectably electrogenic.
SO42−o/Cli− exchange and oxalateo/Cli− exchange were unaccompanied by measurable currents in two-electrode voltage-clamped oocytes (Fig. 6). An electrogenic SLC26A2-mediated SO42− influx of magnitude equivalent to that measured in Fig. 1 should be reflected in a DIDS-sensitive outward current of 30–40 nA at the resting potential of approximately −30 to −40 mV. This level of current is detectable by two-electrode voltage clamp at that potential but was not evident (Fig. 6). We therefore conclude that SLC26A2-mediated SO42−o/Cli− exchange is very likely electroneutral. This electroneutral exchange could be SO42−o/2Cli− exchange or H+/SO42−o/Cli− exchange.
An electrogenic SLC26A2-mediated oxalate influx of magnitude equivalent to that measured in Fig. 2 should be reflected in an outward current of only 4 nA at resting potential. This level of current is below the detection threshold (Fig. 6). Thus our present results do not allow a definitive conclusion that SLC26A2-mediated oxalateo/Cli exchange is electroneutral. Nonetheless, SLC26A2-mediated oxalate/Cl− exchange and SO42−/Cl− exchange likely employ the same mechanism. In contrast, electrogenic oxalate/Cl− exchange has been observed in oocytes expressing mSlc26a6 (8), nonmammalian orthologs of Slc26a5/prestins (38), and rat Slc26a1/Sat-1 (30).
Regulation of SLC26A2 by pH and PKC.
Acidic pHo strongly suppresses SLC26A2-mediated exchange of Clo− for intracellular 35SO42−, 36Cl−, and [14C]oxalate (Fig. 7). In contrast, acidic pHi activated 35SO42−i/Clo− exchange but had no effect on 36Cli−/Clo− exchange and [14C]oxalatei/Clo− exchange (Fig. 8). These data are compatible with mechanisms of SO42−/OH− exchange (or the formally equivalent SO42−/H+ cotransport) but not with oxalate2−/OH− exchange. However, the great reduction in SO42− transport rate by Cl− removal suggests that pHo-sensitive SO42−/Cl− exchange is predominant. Regulation of SLC26A2-mediated anion exchange by NH4+ also exhibits substrate selectivity, but not of the same type as regulation by pHi (supplemental Fig. 6).
Inhibition by acidic pHo is of pathophysiological importance in the hypoxic and elevated lactate conditions of inflamed and septic joints. Septic or inflammatory joint synovial fluid pH can reach pH 6.6 or below (42). The cells of the hypoxic nucleus pulposus of vertebral bodies lack carbonic anhydrase activity and exhibit resting pHi of 6.7, with pHo as low as 6.4 (34). In this acidic environment, reduced SLC26A2-mediated SO42− transport by fibroblasts and chondrocytes in these acidic environments could thus impair local glycosaminoglycan biosynthesis. However, potential activation of SO42− uptake by acidic pHi could counter this impairment, suggesting a balance of two counter-regulatory effects.
PKC activation markedly inhibits SLC26A2, in part by reducing its abundance at the oocyte surface. This mode of regulation resembles that previously shown for the apical anion exchanger SLC26A6 but is unlike the insensitivity to PKC shown by the apical anion exchanger SLC26A4/pendrin (21). Thus activation of chondrocyte SLC26A2 through administration of PKC inhibitors might be envisioned as a useful component of treatment for degenerative joint diseases. Future experiments could determine the specific PKC type involved in regulation of SLC26A2.
Possible roles of SLC26A2 in intestine and kidney.
Although SLC26A2 can exchange oxalate, it is likely a minor contributor to intestinal transepithelial transport of oxalate under normal conditions. The strong DIDS sensitivity of all SLC26A2 transport modes does not support its contribution to DIDS-insensitive intestinal oxalate absorption (23). In oocytes, SLC26A2-mediated oxalate influx and efflux were both at considerably lower rates than SLC26A6-mediated oxalate influx and efflux (6, 8). Moreover, small interfering RNA (siRNA) knockdown of hSLC26A6 by ∼60% in Caco2 cells decreased unidirectional oxalate fluxes in both directions by 50% (16), suggesting that most oxalate is carried by SLC26A6 in this cell model. The direction of colonic oxalate/Cl− exchange will depend on translumenal membrane gradients of oxalate and Cl−. The Cl− gradient varies along the length of the intestine and with the Cl− secretory state of the enterocyte. The oxalate gradient is a function of dietary oxalate intake, net oxalate transport by upstream epithelia, enterocyte oxalate production, and (in normal colon) bacterial consumption. In the presence of a stable oxalate gradient, SLC26A2-mediated electroneutral oxalate/Cl− exchange might switch from oxalate absorption to secretion upon reduction in intracellular [Cl−] triggered by Cl− secretagogue stimulation. SLC26A2-mediated oxalate transport should be unaffected by elevated colonic lumenal [NH4+] (supplemental Fig. 6D) but reduced by mildly acidic intestinal lumenal pH (Fig. 7).
However, hSLC26A2 could mediate a significant portion of that oxalate secretion dependent on exchange with luminal SO42−, and this exchange mode might be sensitive to inhibition by both the mildly acidic lumenal pH and the elevated [NH4+] of the colon. Although median [SO42−] in municipal water supplies was 0.28 mM, [SO42−] in private well water can exceed 20 mM (11, 19), and common dietary sources rich in SO42− include preserved fruits, Brassica vegetables, and beer. SLC26A2 might thus, also serve, in states of positive SO42− balance, to secrete SO42− in exchange for lumenal Cl−, complementing renal SO42− excretion.
hSLC26A2 mRNA levels have been suggested as a biomarker for the differential diagnosis of Crohn's disease and ulcerative colitis. SLC26A2 mRNA levels are increased severalfold in Crohn's disease and unchanged or strongly decreased in ulcerative colitis (9, 43). SLC26A2 and SLC6A14 mRNA levels used as part of a seven gene panel yielded rates of correct prediction, sensitivity, and specificity higher than with previously available diagnostic indices (43). In addition, SLC26A2 is downregulated in colon cancer biopsies compared with surrounding normal tissue (18).
SLC26A2 mRNA is expressed in the kidney, and SLC26A2 polypeptide has been immunolocalized in the brush border of the rat proximal tubule (5). In that location, oxalate/SO42− exchange by SLC26A2 might contribute to tertiary active Cl− reabsorption across the proximal tubular epithelium, coupled to sodium-sulfate cotransport (1).
GRANTS
This work was supported by National Institutes of Health T32 Grants DK-07094 and DK-07477 (to J. F. Heneghan), R01DK-43495 (to S. L. Alper), and by P30DK-34854 (Harvard Digestive Diseases Center to to S. L. Alper).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
ACKNOWLEDGEMENTS
We thank Johanna Hastbacka (University of Helsinki) for hSLC26A2 cDNA, Jean Schwartzbauer (Princeton University) for hSLC26A2-GFP cDNA, and Andrew K. Stewart (Harvard Medical School) for helpful discussion.
REFERENCES
- 1. Aronson PS, Giebisch G. Mechanisms of chloride transport in the proximal tubule. Am J Physiol Renal Physiol 273: F179–F192, 1997 [DOI] [PubMed] [Google Scholar]
- 2. Barmeyer C, Ye JH, Sidani S, Geibel J, Binder HJ, Rajendran VM. Characteristics of rat downregulated in adenoma (rDRA) expressed in HEK 293 cells. Pflügers Arch 454: 441–450, 2007 [DOI] [PubMed] [Google Scholar]
- 3. Beauge L, DiPolo R. The squid axon Na/Ca2+ exchanger shows ping pong kinetics only when the Ca2+(i)-regulatory site is saturated. Cell Physiol Biochem 23: 37–42, 2009 [DOI] [PubMed] [Google Scholar]
- 4. Burckhardt BC, Fromter E. Pathways of NH3/NH4+ permeation across Xenopus laevis oocyte cell membrane. Pflügers Arch 420: 83–86, 1992 [DOI] [PubMed] [Google Scholar]
- 5. Chapman JM, Karniski LP. Protein localization of SLC26A2 (DTDST) in rat kidneys (Abstract). FASEB J 21: 937. 27, 2007 [DOI] [PubMed] [Google Scholar]
- 6. Chernova MN, Jiang L, Friedman DJ, Darman RB, Lohi H, Kere J, Vandorpe DH, Alper SL. Functional comparison of mouse slc26a6 anion exchanger with human SLC26A6 polypeptide variants: differences in anion selectivity, regulation, and electrogenicity. J Biol Chem 280: 8564–8580, 2005 [DOI] [PubMed] [Google Scholar]
- 7. Chernova MN, Jiang L, Shmukler BE, Schweinfest CW, Blanco P, Freedman SD, Stewart AK, Alper SL. Acute regulation of the SLC26A3 congenital chloride diarrhoea anion exchanger (DRA) expressed in Xenopus oocytes. J Physiol 549: 3–19, 2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Clark JS, Vandorpe DH, Chernova MN, Heneghan JF, Stewart AK, Alper SL. Species differences in Cl- affinity and in electrogenicity of SLC26A6-mediated oxalate/Cl- exchange correlate with the distinct human and mouse susceptibilities to nephrolithiasis. J Physiol 586: 1291–1306, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Comelli EM, Lariani S, Zwahlen MC, Fotopoulos G, Holzwarth JA, Cherbut C, Dorta G, Corthesy-Theulaz I, Grigorov M. Biomarkers of human gastrointestinal tract regions. Mamm Genome 20: 516–527, 2009 [DOI] [PubMed] [Google Scholar]
- 10. Cornaglia AI, Casasco A, Casasco M, Riva F, Necchi V. Dysplastic histogenesis of cartilage growth plate by alteration of sulphation pathway: a transgenic model. Connect Tis Res 50: 232–242, 2009 [DOI] [PubMed] [Google Scholar]
- 10a. Dawson PA, Russell CS, Lee S, McLeay SC, van Dongen JM, Cowley DM, Clarke LA, Markovich D. Urolithiasis and hepatotoxicity are linked to the anion transporter Sat1 in mice. J Clin Invest 120: 706–712, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Deplancke B, Finster K, Graham WV, Collier CT, Thurmond JE, Gaskins HR. Gastrointestinal and microbial responses to sulfate-supplemented drinking water in mice. Exp Biol Med (Maywood) 228: 424–433, 2003 [DOI] [PubMed] [Google Scholar]
- 12. Dorwart MR, Shcheynikov N, Yang D, Muallem S. The solute carrier 26 family of proteins in epithelial ion transport. Physiology 23: 104–114, 2008 [DOI] [PubMed] [Google Scholar]
- 13. Everett LA, Glaser B, Beck JC, Idol JR, Buchs A, Heyman M, Adawi F, Hazani E, Nassir E, Baxevanis AD, Sheffield VC, Green ED. Pendred syndrome is caused by mutations in a putative sulphate transporter gene (PDS). Nature Gene 17: 411–422, 1997 [DOI] [PubMed] [Google Scholar]
- 14. Forlino A, Piazza R, Tiveron C, Della Torre S, Tatangelo L, Bonafe L, Gualeni B, Romano A, Pecora F, Superti-Furga A, Cetta G, Rossi A. A diastrophic dysplasia sulfate transporter (SLC26A2) mutant mouse: morphological and biochemical characterization of the resulting chondrodysplasia phenotype. Hum Mol Genet 14: 859–871, 2005 [DOI] [PubMed] [Google Scholar]
- 15. Freel RW, Hatch M, Green M, Soleimani M. Ileal oxalate absorption and urinary oxalate excretion are enhanced in Slc26a6 null mice. Am J Physiol Gastrointest Liver Physiol 290: G719–G728, 2006 [DOI] [PubMed] [Google Scholar]
- 16. Freel RW, Morozumi M, Hatch M. Parsing apical oxalate exchange in Caco-2BBe1 monolayers: siRNA knockdown of SLC26A6 reveals the role and properties of PAT-1. Am J Physiol Gastrointest Liver Physiol 297: G918–G929, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fuster D, Moe OW, Hilgemann DW. Steady-state function of the ubiquitous mammalian Na/H exchanger (NHE1) in relation to dimer coupling models with 2Na/2H stoichiometry. J Gen Physiol 132: 465–480, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Galamb O, Sipos F, Solymosi N, Spisak S, Krenacs T, Toth K, Tulassay Z, Molnar B. Diagnostic mRNA expression patterns of inflamed, benign, and malignant colorectal biopsy specimen and their correlation with peripheral blood results. Cancer Epidemiol Biomarkers Prev 17: 2835–2845, 2008 [DOI] [PubMed] [Google Scholar]
- 19. Gomez GG, Sandler RS, Seal E., Jr High levels of inorganic sulfate cause diarrhea in neonatal piglets. J Nutr 125: 2325–2332, 1995 [DOI] [PubMed] [Google Scholar]
- 20. Haila S, Hastbacka J, Bohling T, Karjalainen-Lindsberg ML, Kere J, Saarialho-Kere U. SLC26A2 (diastrophic dysplasia sulfate transporter) is expressed in developing and mature cartilage but also in other tissues and cell types. J Histochem Cytochem 49: 973–982, 2001 [DOI] [PubMed] [Google Scholar]
- 21. Hassan HA, Mentone S, Karniski LP, Rajendran VM, Aronson PS. Regulation of anion exchanger Slc26a6 by protein kinase C. Am J Physiol Cell Physiol 292: C1485–C1492, 2007 [DOI] [PubMed] [Google Scholar]
- 22. Hastbacka J, de la Chapelle A, Mahtani MM, Clines G, Reeve-Daly MP, Daly M, Hamilton BA, Kusumi K, Trivedi B, Weaver A. The diastrophic dysplasia gene encodes a novel sulfate transporter: positional cloning by fine-structure linkage disequilibrium mapping. Cell 78: 1073–1087, 1994 [DOI] [PubMed] [Google Scholar]
- 23. Hatch M, Freel RW. Intestinal transport of an obdurate anion: oxalate. Urol Res 33: 1–16, 2005 [DOI] [PubMed] [Google Scholar]
- 24. Hoglund P, Haila S, Socha J, Tomaszewski L, Saarialho-Kere U, Karjalainen-Lindsberg ML, Airola K, Holmberg C, de la Chapelle A, Kere J. Mutations of the Down-regulated in adenoma (DRA) gene cause congenital chloride diarrhoea. Nature Gene 14: 316–319, 1996 [DOI] [PubMed] [Google Scholar]
- 25. Holmes RP, Goodman HO, Assimos DG. Contribution of dietary oxalate to urinary oxalate excretion. Kidney Int 59: 270–276, 2001 [DOI] [PubMed] [Google Scholar]
- 26. Humphreys BD, Chernova MN, Jiang L, Zhang Y, Alper SL. NH4Cl activates AE2 anion exchanger in Xenopus oocytes at acidic pHi. Am J Physiol Cell Physiol 272: C1232–C1240, 1997 [DOI] [PubMed] [Google Scholar]
- 27. Jiang Z, Asplin JR, Evan AP, Rajendran VM, Velazquez H, Nottoli TP, Binder HJ, Aronson PS. Calcium oxalate urolithiasis in mice lacking anion transporter Slc26a6. Nature Gene 38: 474–478, 2006 [DOI] [PubMed] [Google Scholar]
- 28. Karniski LP. Functional expression and cellular distribution of diastrophic dysplasia sulfate transporter (DTDST) gene mutations in HEK cells. Hum Mol Genet 13: 2165–2171, 2004 [DOI] [PubMed] [Google Scholar]
- 29. Karniski LP. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene: correlation between sulfate transport activity and chondrodysplasia phenotype. Hum Mol Genet 10: 1485–1490, 2001 [DOI] [PubMed] [Google Scholar]
- 30. Krick W, Schnedler N, Burckhardt G, Burckhardt BC. Ability of sat-1 to transport sulfate, bicarbonate, or oxalate under physiological conditions. Am J Physiol Renal Physiol 297: F145–F154, 2009 [DOI] [PubMed] [Google Scholar]
- 31. Kujala M, Hihnala S, Tienari J, Kaunisto K, Hastbacka J, Holmberg C, Kere J, Hoglund P. Expression of ion transport-associated proteins in human efferent and epididymal ducts. Reproduction 133: 775–784, 2007 [DOI] [PubMed] [Google Scholar]
- 32. Kuo LW, Horton K, Fishman EK. CT evaluation of multisystem involvement by oxalosis. Am J Roentgenol Radium Ther 177: 661–663, 2001 [DOI] [PubMed] [Google Scholar]
- 33. Liu XZ, Ouyang XM, Xia XJ, Zheng J, Pandya A, Li F, Du LL, Welch KO, Petit C, Smith RJ, Webb BT, Yan D, Arnos KS, Corey D, Dallos P, Nance WE, Chen ZY. Prestin, a cochlear motor protein, is defective in non-syndromic hearing loss. Hum Mol Genet 12: 1155–1162, 2003 [DOI] [PubMed] [Google Scholar]
- 34. Razaq S, Urban JP, Wilkins RJ. Regulation of intracellular pH by bovine intervertebral disc cells. Cell Physiol Biochem 10: 109–115, 2000 [DOI] [PubMed] [Google Scholar]
- 35. Rossi A, Superti-Furga A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum Mutat 17: 159–171, 2001 [DOI] [PubMed] [Google Scholar]
- 36. Sasaki S, Ishibashi K, Nagai T, Marumo F. Regulation mechanisms of intracellular pH of Xenopus laevis oocyte. Biochim Biophys Acta 1137: 45–51, 1992 [DOI] [PubMed] [Google Scholar]
- 37. Satoh H, Susaki M, Shukunami C, Iyama K, Negoro T, Hiraki Y. Functional analysis of diastrophic dysplasia sulfate transporter. Its involvement in growth regulation of chondrocytes mediated by sulfated proteoglycans. J Biol Chem 273: 12307–12315, 1998 [DOI] [PubMed] [Google Scholar]
- 38. Schaechinger TJ, Oliver D. Nonmammalian orthologs of prestin (SLC26A5) are electrogenic divalent/chloride anion exchangers. Proc Natl Acad Sci USA 104: 7693–7698, 2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stewart AK, Chernova MN, Shmukler BE, Wilhelm S, Alper SL. Regulation of AE2-mediated Cl- transport by intracellular or by extracellular pH requires highly conserved amino acid residues of the AE2 NH2-terminal cytoplasmic domain. J Gen Physiol 120: 707–722, 2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Takenaka H, Yasuno H, Fukuda T, Ono T, Kishimoto S. Widespread cutaneous oxalosis in a patient with primary hyperoxaluria. Br J Dermatol 149: 1075, 2003 [DOI] [PubMed] [Google Scholar]
- 41. Toure A, Lhuillier P, Gossen JA, Kuil CW, Lhote D, Jegou B, Escalier D, Gacon G. The testis anion transporter 1 (Slc26a8) is required for sperm terminal differentiation and male fertility in the mouse. Hum Mol Genet 16: 1783–1793, 2007 [DOI] [PubMed] [Google Scholar]
- 42. Treuhaft PS, DJMC Synovial fluid pH, lactate, oxygen and carbon dioxide partial pressure in various joint diseases. Arthritis Rheum 14: 475–484, 1971 [DOI] [PubMed] [Google Scholar]
- 43. von Stein P, Lofberg R, Kuznetsov NV, Gielen AW, Persson JO, Sundberg R, Hellstrom K, Eriksson A, Befrits R, Ost A, von Stein OD. Multigene analysis can discriminate between ulcerative colitis, Crohn's disease, and irritable bowel syndrome. Gastroenterology 134: 1869–1881, 2008 [DOI] [PubMed] [Google Scholar]
- 44. Wang Z, Wang T, Petrovic S, Tuo B, Riederer B, Barone S, Lorenz JN, Seidler U, Aronson PS, Soleimani M. Renal and intestinal transport defects in Slc26a6-null mice. Am J Physiol Cell Physiol 288: C957–C965, 2005 [DOI] [PubMed] [Google Scholar]
- 45. Xu J, Song P, Miller ML, Borgese F, Barone S, Riederer B, Wang Z, Alper SL, Forte JG, Shull GE, Ehrenfeld J, Seidler U, Soleimani M. Deletion of the chloride transporter Slc26a9 causes loss of tubulovesicles in parietal cells and impairs acid secretion in the stomach. Proc Natl Acad Sci USA 105: 17955–17960, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xu J, Song P, Nakamura S, Miller M, Barone S, Alper SL, Riederer B, Bonhagen J, Arend LJ, Amlal H, Seidler U, Soleimani M. Deletion of the chloride transporter slc26a7 causes distal renal tubular acidosis and impairs gastric acid secretion. J Biol Chem 284: 29470–29479, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]