

Abstract
This paper describes a detailed structure−activity relationship (SAR) analysis of the metabotropic glutamate receptor 4 (mGluR4) positive allosteric modulator, (−)-N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide (PHCCC). We have now developed compounds with improved potency and efficacy; in addition, compounds are presented that show selectivity for mGluR4 versus the other mGluR subtypes.
Keywords: Metabotropic glutamate receptor 4, allosteric modulation, positive allosteric modulator, PHCCC, Parkinson’s disease, structure−activity relationship, SAR
Parkinson’s disease (PD), with a lifetime risk of 2%, is the second most common neurodegenerative disorder after Alzheimer’s disease (1). PD was first described in 1817 by James Parkinson (2) and results from the degeneration of dopaminergic neurons in the substantia nigra pars compacta. Replacement of lost dopamine using the precursor L-DOPA is still the gold standard treatment of PD. However, there are several known issues with L-DOPA treatment, such as fluctuations in its therapeutic efficacy, appearance of dyskinesias (3), and the potential for further degeneration of nigral neurons (4,5). Due to these limitations, there has been extensive research to develop more effective treatments for PD.
One such area of research that has generated interest is that of the metabotropic glutamate receptors (mGluRs) due their wide distribution in the basal ganglia and their role in neurodegeneration and neuroprotection (6−8). The mGluRs are members of the class C G-protein-coupled receptors (GPCRs) and are characterized by an extracellular domain, which contains the agonist binding site, in addition to seven transmembrane domains (9). There have been eight mGluR subtypes identified, and these are classified into three groups based on sequence homology, G protein coupling profile, and ligand specificity (10). These groups are group I (mGluR1 and mGluR5), group II (mGluR2 and mGluR3), and group III (mGluR4, mGluR6, mGluR7, and mGluR8). Within these three subfamilies, the group III mGluRs still remain the least explored due to the lack of selective ligands (11).
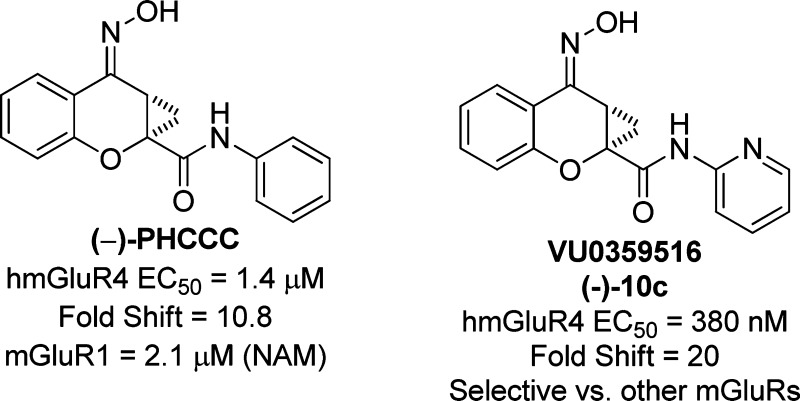
mGluR4 has recently received much attention by the PD field6,12 due to the discovery of the positive allosteric modulator (PAM), (−)-N-phenyl-7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxamide (PHCCC), 1 (Figure 1) (13,14). Since the initial disclosure of PHCCC, 1, there have been numerous publications by a number of groups detailing its efficacy in a wide range of PD rodent models (14−17). In addition to PD paradigms, PHCCC, 1, has been explored for a wide-range of therapeutic potential in a variety of diseases, such as anxiety (18), neuroprotection (19,20), and oncology (20). Despite the numerous studies that have examined biological effects of PHCCC, there still remain several major issues with this scaffold: (1) PHCCC is an mGluR1 antagonist, (2) PHCCC possesses poor physicochemical properties, and (3) PHCCC possesses limited brain penetration. In this paper, we address the structure−activity relationship (SAR) and the selectivity issues associated with the PHCCC scaffold; these studies have now lead to a compound, (−)-10c, VU0359516, exhibiting a 4-fold increase in potency, improved efficacy as an mGluR4 positive allosteric modulator, and selectivity for mGluR4 among the mGluR subtypes.
Figure 1.
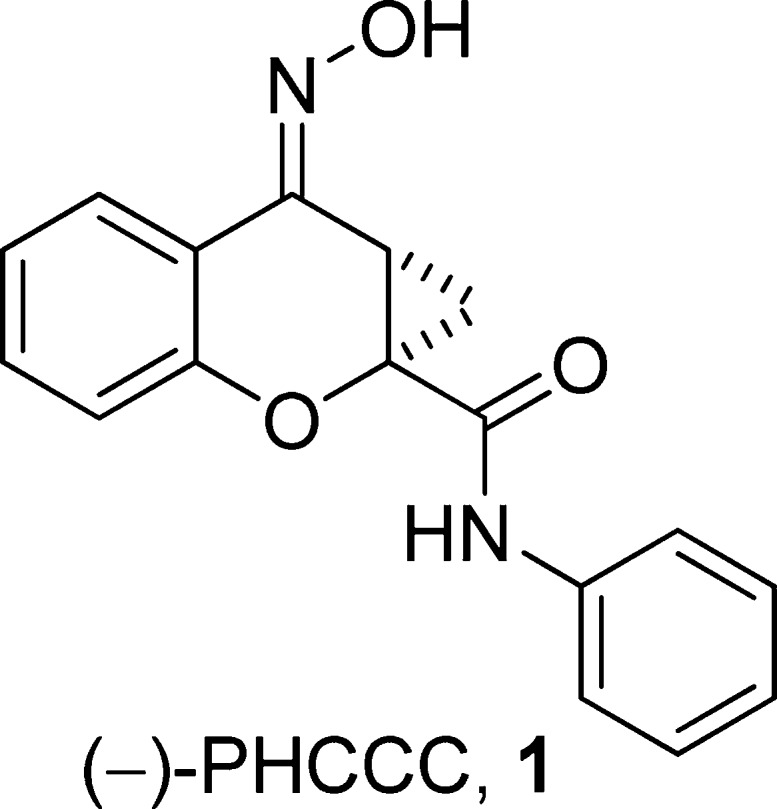
Chemical structure of the prototypical mGluR4 positive allosteric modulator, (−)-PHCCC, 1.
We recently reported a brief SAR analysis of the PHCCC scaffold; however, those modifications led to compounds that either were inactive or exhibited reduced activity compared with PHCCC (21). Moreover, folklore surrounding the PHCCC scaffold within the mGluR community suggests that the SAR is flat and that the scaffold is intolerant of chemical optimization. In this report, we further detail our efforts investigating the PHCCC chemotype in an attempt to improve the potency of PHCCC, as well as the selectivity against mGluR1. The areas of the PHCCC scaffold that we have now explored are shown in Figure 2.
Figure 2.

Areas of planned SAR exploration.
Previously, our efforts led to the synthesis of a limited number of amide analogs of the PHCCC chemotype; however, the initial compounds were not improved in activity relative to PHCCC (21). Because the amide portion of the molecule lends itself to library synthesis, we sought to expand our previous efforts in order to more fully explore the amide SAR. To this end, chromone-2-carboxylic acid, 2, was converted to the cyclopropane analog 3 ((CH3)3SOI (22,23), NaOH, dimethylformamide (DMF), 82%) (Scheme 1). The cyclopropane carboxylic acid analog 3 was subjected to a parallel library synthesis approach to afford the amide analogs 4a−n utilizing 2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate methanaminium (HATU) coupling conditions (20−65%). Final conversion of the ketones to oximes (NH2OH, MeOH, 65 °C, 80−92%) produced the desired compounds 5a−n.
Scheme 1.

Reaction conditions: (a) (CH3)3SOI, NaOH, DMF (82%); (b) RNH2, HATU, DMF (20−65%); (c) NH2OH, MeOH, 65 °C (80−92%). All final compounds were purified by mass-directed prep LC (27).
The initial SAR was centered on substitution of the phenyl ring (5a−5e) (Table 1). In these studies, we measured the potency of each compound in a 10 point concentration−response using Chinese hamster ovary cells expressing human mGluR4 and a chimeric G protein, Gqi5, to couple mGluR4 to calcium mobilization (21). In addition to potency, we determined the percent maximal response of each compound in comparison to the maximal response induced by glutamate alone (% Glu Max). This % Glu Max determination serves as an initial indication of compound efficacy, which can then be further explored for selected compounds (discussed below) to determine the ability of a compound to leftward shift the concentration−response relationship of glutamate (fold-shift determinations). Each experiment included a concentration−response curve determination of the activity of (±)-PHCCC as a positive control. In mGluR4/Gqi5/CHO cells, the maximal response in the presence of positive allosteric modulators often greatly exceeds the maximal response induced by glutamate (Figure 5A) (14,21), suggesting that mGluR4 PAMs have the ability to increase glutamate efficacy beyond that induced by a maximal concentration of glutamate alone in this assay system. Because the % Glu Max in the presence of PHCCC varies slightly from day to day, we also expressed the % Glu Max data relative to the PHCCC control run in each experiment to better compare relative efficacy.
Table 1. Potency Values of Analogs 5a−na.
| compound | R | EC50 (μM) hmGluR421 | % Glu Max | % Glu Max relative to (±)-PHCCC |
|---|---|---|---|---|
| (±)-PHCCC, 1 | Ph | 3.2 | ||
| 5a | 2-F-Ph | >5 | 121.5 ± 4.3 | 72.0 ± 2.5 |
| 5b | 3-F-Ph | >5 | 90.4 ± 7.8 | 73.2 ± 6.3 |
| 5c | 4-F-Ph | 2.0 | 120.9 ± 8.6 | 71.6 ± 5.1 |
| 5d | 3,4-diF-Ph | >5 | 110 ± 27.4 | 48.6 ± 12.0 |
| 5e | 3-Cl-4-F-Ph | b | 16.5 ± 1.1 | 13.3 ± 0/9 |
| 5f | benzyl | b | 25.6 ± 01.3 | 15.1 ± 0.8 |
| 5g | phenethyl | >5 | 92.9 ± 5.9 | 55.0 ± 3.5 |
| 5h | cyclohexyl | >5 | 124.4 ± 1.6 | 73.7 ± 1.0 |
| 5i | 2-pyr | 0.87 | 160.2 ± 10.6 | 94.9 ± 6.3 |
| 5j | 3-pyr | >5 | 61.8 ± 2.3 | 36.6 ± 1.4 |
| 5k | 2-pyr-4-F | 1.8 | 163.3 ± 16.3 | 132.1 ± 13.2 |
| 5l | 2-pyr-4-CH3 | b | 28.1 ± 2.6 | 22.7 ± 2.1 |
| 5m | 2-pyr-4-Cl | >5 | 86.9 ± 10.4 | 70.3 ± 8.4 |
| 5n | 2-thiazole | >5 | 127.1 ± 10.4 | 102.8 ± 8.4 |
Potency data represent a triplicate determination. The % Glu Max is calculated as the percent response versus the response to a maximal (1 mM) concentration of glutamate. (±)-PHCCC is run as a control compound each day, and the maximal response generated in mGluR4 CHO cells in the presence of mGluR4 PAMs varies slightly in each experiment. Therefore, data were further normalized to the relative (±)-PHCCC response obtained in each day’s run. % Glu Max and % Glu Max normalized to PHCCC values represent the mean ± SEM of triplicate determinations.
Inactive.
Figure 5.

Potency and efficacy of the novel PHCCC analog, (−)-10c (VU0359516): (a) Compound 10c (VU0359516) was added in progressively higher concentrations to cells coexpressing human mGluR4 and the chimeric G protein Gqi5 (black boxes) in the presence of an EC20 concentration of glutamate; (b) compound (−)-10c (VU0359516), (−)-PHCCC, or DMSO was added, and after a 2.5 min incubation period, an EC20 concentration of glutamate was added; the trace shows that both (−)-10c and (−)-PHCCC only act in the presence of glutamate in this assay; (c) DMSO vehicle, 30 μM PHCCC (both racemic and enantiopure) or 30 μM (−)-10c (VU0359516) was incubated with human mGluR4/Gqi5 cells, and 2.5 min later increasing concentrations of glutamate were added. (±)-PHCCC shifted the glutamate concentration−response curve 7.8-fold to the left, (−)-PHCCC shifted the curve 10.8-fold to the left, and 10c shifted the curve 20.0-fold to the left.
The 2-, 3-, and 4-substituted monofluorinated phenyl compounds were active in the micromolar range, with the 4-fluorophenyl analog being equipotent, although slightly less efficacious (71.6% ± 5.1%), compared with PHCCC. Other multihalogenated phenyl analogs either showed weak activity (5d) or were inactive (5e). In an attempt to avoid potential aniline-forming metabolites, we next examined the activity of benzyl, alkyl, or cycloalkyl amides. Each of these substitutions led to compounds with no activity or weak activity at the hmGluR4 receptor (5f−5h). Next, heteroaryl amides were explored with satisfying results. The 2-pyridylamide was found to be the most potent compound in the PHCCC class (5i, potency of 870 nM, % Glu Max relative to PHCCC of 94.9%). Moving the nitrogen around the aromatic ring (3-pyridyl, 5j) led to a less active compound. Halogenated substitution on the 2-pyridyl scaffold also led to active compounds (5k and 5m), although each was reduced in potency compared with the unsubstituted 2-pyridyl derivative; the improved relative efficacy of 5k, however, should be noted. Larger substitution on the 2-pyridyl ring led to a complete loss of activity (5l). Lastly, replacement of the 2-pyridyl with the classical pyridine mimetic 2-thiazole led to a reduction in potency (5i, 870 nM vs 5n, >5 μM). The SAR around the amide substitution highlights the noted “flat” SAR that is often associated with allosteric modulation (see, compounds 5a−5c and 5i−5j). However, despite this shallow SAR, we were able to discover a more potent PHCCC analog (5i).
Moving to the western aromatic SAR, the commercially available chromene-2-one carboxylic acids 2a−i, were converted to the phenyl amides using HATU in DMF (25−70%), which then delivered the cyclopropanated compounds (6a−i) in high yield (62−93%) by application of trimethylsulfoxonium iodide22,23 and anhydrous NaOH in DMF (Scheme 2). The ketone was readily transformed into the corresponding oximes using hydroxylamine in MeOH at 60 °C to generate the E-oxime isomers only, 7a−i (82−98%).
Scheme 2.

Reaction conditions: (a) Aniline, HATU, DMF (25−70%); (b) (CH3)3SOI, NaOH, DMF (62−93%); (c) NH2OH, MeOH, 65 °C (82−98%). All final compounds were purified by mass-directed prep LC (27).
It was observed that substitution at either the 5- or 6-position was tolerated when examined for activity at human mGluR4 (hmGluR4) (Table 2). For the exploration of the western portion, both the equipotent phenyl amide and 4-fluorophenyl amides were used for the synthesis. The 6-Cl, 7f, and 6-Me, 7g, analogs were observed to be equipotent to PHCCC at approximately 3 μM although these compounds were greatly reduced in efficacy (Table 2). The 5-F, 7a, 5-Cl, 7b, 5-Br, 7d, and 6-F, 7e, analogs led to either a minor decrease in potency (>5 μM for 7b and 7e) or loss of activity (7a and 7d). We would note that compound 7e (6-F) did show excellent efficacy of 245% (139% relative to PHCCC). Disubstitution at the 6,8- and 5,7-positions led to inactive compounds (7h, 7i). Even though substitution at the 5- and 6-position of the western aromatic ring did not improve the potency over the parent PHCCC scaffold, this is the first report of a moderate degree of functional group tolerance in this area.
Table 2. Potency Values of Analogs 7a−ia.
| compound | R | R1 | EC50 (μM) hmGluR421 | % Glu Max | % Glu Max relative to (±)-PHCCC |
|---|---|---|---|---|---|
| (±)-PHCCC, 1 | H | H | 3.2 | ||
| 7a | 5-F | H | b | 8.8 ± 1.3 | 3.5 ± 0.5 |
| 7b | 5-Cl | H | >5 | 139.2 ± 7.7 | 54.9 ± 3.0 |
| 7c | 5-Me | H | >5 | 194.0 ± 18.7 | 76.5 ± 7.4 |
| 7d | 5-Br | H | b | 5.1 ± 1.8 | 2.0 ± 0.7 |
| 7e | 6-F | H | >5 | 244.9 ± 24.0 | 139.3 ± 13.6 |
| 7f | 6-Cl | 4-F | 3.4 | 48.4 ± 1.8 | 18.2 ± 0.7 |
| 7g | 6-Me | 4-F | 2.3 | 30 ± 2.1 | 11.2 ± 0.8 |
| 7h | 6,8-diCl | 4-F | b | 27.1 ± 9.7 | 11.9 ± 4.2 |
| 7i | 5,7-diMe | H | b | 14.8 ± 0.9 | 10.8 ± 0.5 |
Potency data represent a triplicate determination. The % Glu Max is calculated as the percent response versus the response to a maximal (1 mM) concentration of glutamate. (±)-PHCCC is run as a control compound each day, and the maximal response generated in mGluR4 CHO cells in the presence of mGluR4 PAMs varies slightly in each experiment. Therefore, data were further normalized to the relative (±)-PHCCC response obtained in each day’s run. % Glu Max and % Glu Max normalized to PHCCC values represent the mean ± SEM of triplicate determinations.
Inactive.
Due to the known toxicity issues associated with cyclohexanone oxime (24), we next looked to replace this group (Figure 3). Unfortunately, all of our efforts to either alkylate or replace the oxime altogether did not lead to any compounds that showed activity at hmGluR4. Annoura and co-workers (25) noted in their initial publication that the oxime was critical for PHCCC’s activity; however, very limited examples have ever been shown.
Figure 3.

Exploration of oxime replacements.
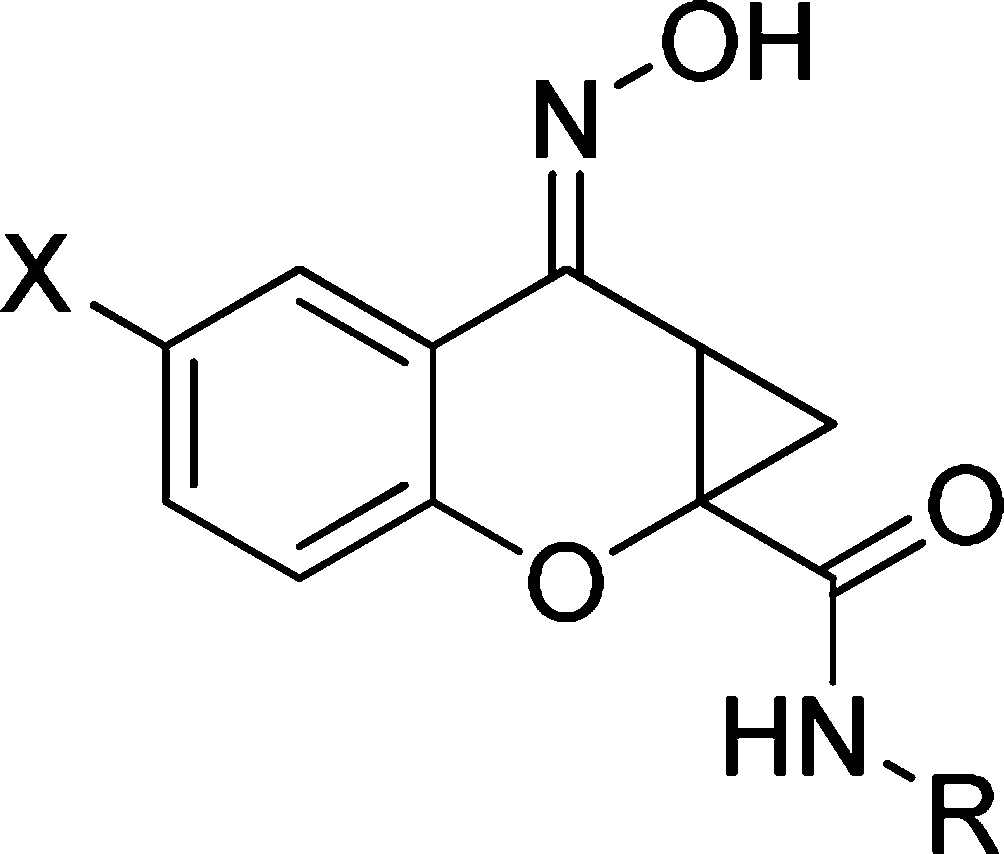
Having evaluated the three portions of the molecule that we outlined in Figure 1, the optimal amide moieties were taken forward and combined with our SAR findings from the western aromatic ring to determine whether there was an additive effect. Additionally, not only were compounds tested in a potency format, but concentration−response curves of glutamate in the presence and absence of a 30 μM concentration of each compound were performed to better assess compound efficacy (“fold shift”, Table 3). Again, the chromone-2-carboxylic acids (2a−d) were converted to the cyclopropane analogs (8a−d) utilizing the conditions outlined previously ((CH3)3SOI (22,23), NaOH, DMF, 75−90%). Coupling the cyclopropane carboxylic acids with the appropriate amines (HATU, DMF, 40−72%), followed by oxime formation (NH2OH, MeOH, 65 °C, 71−98%) afforded the desired compounds 9a−f (Scheme 3).
Table 3. Potency Values of Analogs 9a−fa.
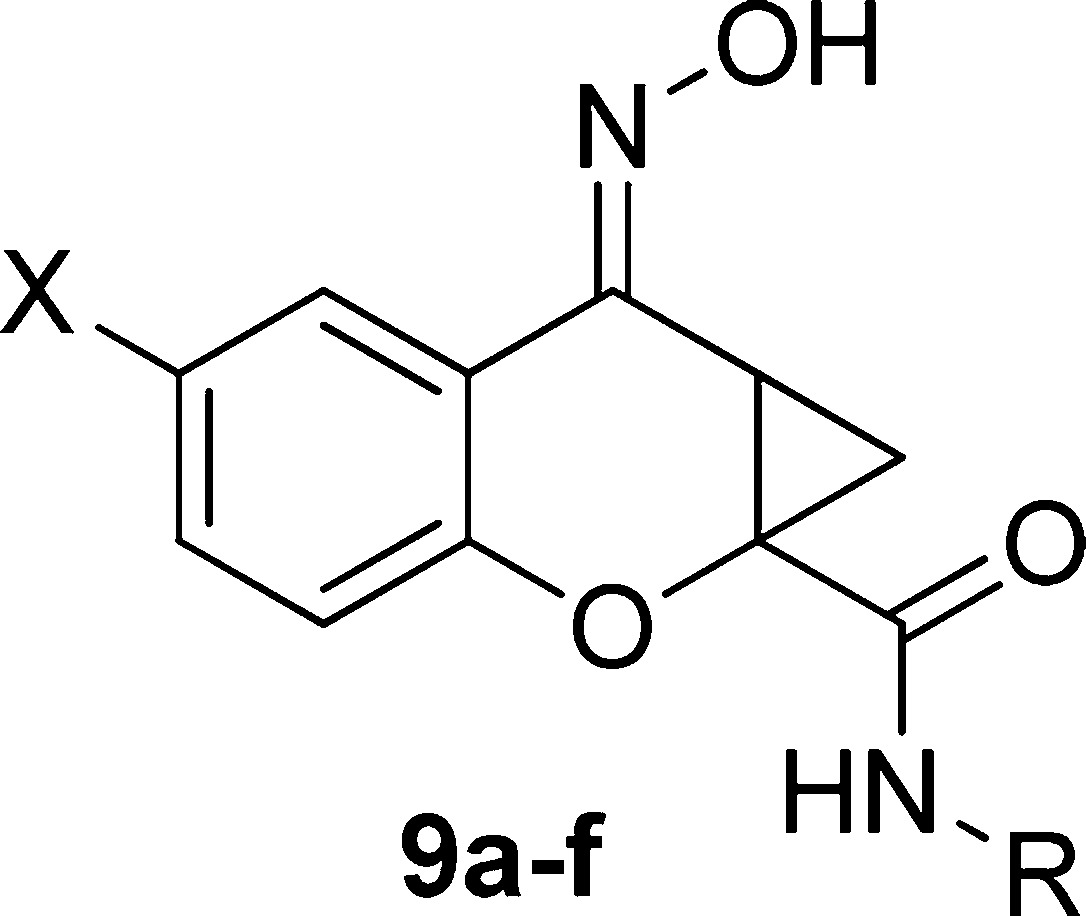
| compound | X | R | EC50 (μM) hmGluR421 | fold shift |
|---|---|---|---|---|
| (±)-PHCCC, 1 | H | Ph | 3.2 | 7.8 ± 0.6 |
| 7e | F | Ph | >5 | b |
| 7f | Cl | 4-F-Ph | 3.4 | 1.8 ± 0.2 |
| 7g | Me | 4-F-Ph | 2.3 | 1.7 ± 0.1 |
| 5c | H | 4-F-Ph | 2.0 | 4.1 ± 0.5 |
| 5i | H | 2-pyr | 0.87 | 9.7 ± 0.4 |
| 5k | H | 2-pyr-4-F | 1.8 | 16.7 ± 0.9 |
| 9a | Cl | 2-pyr-4-F | 0.96 | 7.7 ± 2.5 |
| 9b | Me | 2-pyr | 3.2 | 8.5 ± 0.7 |
| 9c | Me | 2-pyr-4-F | 2.3 | 7.5 ± 1.7 |
| 9d | F | 2-pyr | 2.3 | b |
| 9e | F | 4-F-Ph | >5 | b |
| 9f | F | 2-pyr-4-F | 1.8 | 14.6 ± 1.1 |
Potency data represent triplicate determinations. Fold shift data represent the average of three independent determinations performed in triplicate at a final compound concentration of 30 μM.
Not determined.
Scheme 3.

Reaction conditions: (a) (CH3)3SOI, NaOH, DMF, 75−90%; (b) RNH2, HATU, DMF, 40−72%; (c) NH2OH, MeOH, 65 °C, 71−98%. All final compounds were purified by mass-directed prep LC (25).
Combining the 6-substitution with the appropriate amide component led to compounds that either maintained mGluR4 PAM activity or had a slight diminution in potency. When features of our best compound, 5i (870 nM), were combined with 6-halogenated or 6-methyl substitution, the resulting compounds were equipotent to PHCCC (9b and 9d), suggesting that the 6-substitution did not aid in potency optimization. All other compounds synthesized were equipotent with the phenyl analogs, indicating that the combination of the 6-substitution with the optimal amide moieties did not lead to increased potency at the hmGluR4 receptor.
The studies of Maj and co-workers13 demonstrated that the two enantiomers of PHCCC (Figure 4) exhibited different potencies, with the (−)-enantiomer being the only active isomer. The racemic compounds 5i and 5k were then subjected to separation via chiral HPLC in order to determine the potency of each enantiomer. The racemates of both 5i and 5k were resolved by chiral chromatography using an AD Chirapak column (with 5% isopropyl alcohol (IPA)/hexanes as an eluant). In each instance, the (−)-isomer was eluted second from the chiral prep column. As with PHCCC, the resolved compounds showed preference for the (−)-enantiomer, with the (+)-isomer being inactive for each compound ((+)-10a, inactive, vs (−)-10c (VU0359516), 380 nM, Table 4, Figure 5a) and ((+)-10b, >5 μM vs (−)-10d, 1.3 μM, Table 4). As can be seen in Figure 5b, addition of a 30 μM concentration of compound alone in each case (Figure 5b, first arrow) did not induce a response in calcium mobilization studies, suggesting that racemic PHCCC, (−)-PHCCC, and (−)-10c do not exhibit agonist activity in these experiments and instead act by potentiating the activity of glutamate at mGluR4 (Figure 5b, second arrow). At higher concentrations (>30 μM), however, we would note that, in contrast to PHCCC, we did observe responses induced by (−)-10c when added alone (data not shown), suggesting that the compound does possess some intrinsic agonist activity in this assay when high concentrations are used; this phenomenon will be further explored in future detailed pharmacology studies. Fold shift experiments for (−)-10c and (−)-10d showed that they have a greater ability to potentiate the glutamate response as compared with PHCCC ((−)-10c (VU0359516), 20-fold shift of the glutamate concentration−response curve; (−)-10d, 22 fold shift; (−)-PHCCC, 10-fold shift; Table 4, Figure 5c). With (−)-PHCCC, very little enhancement in the glutamate fold shift was observed over the racemate, 10.8 versus 7.8, whereas (−)-10c (VU0359516) gave a 2-fold enhancement over the racemic analog, 5i. Compound 10c (VU0359516) represents a major step forward in the SAR of the (−)-PHCCC scaffold because this compound has a >4-fold increase in potency against the hmGluR4 receptor and exhibits a 2-fold enhancement in efficacy compared with (−)-PHCCC.
Figure 4.

The (+)- and (−)-isomers of lead PHCCC analogs as eluted from the chiral prep column.
Table 4. Potency Values and Fold Shifts of Chirally Resolved Analogsa.
| compound | Y | EC50 (μM) hmGluR421 | fold shift |
|---|---|---|---|
| (±)-5i | H | 0.87 | 9.7 ± 0.4 |
| (+)-10a | H | b | |
| (−)-10c, VU0359516 | H | 0.38 | 20.0 ± 1.6 |
| (±)-5k | F | 1.8 | 16.7 ± 0.9 |
| (+)-10b | F | >5 | 5.0 |
| (−)-10d | F | 1.3 | 22.6 |
| (±)-PHCCC | 3.2 | 7.8 ± 0.6 | |
| (−)-PHCCC | 1.4 | 10.8 ± 0.8 |
Potency data represent triplicate determinations. Fold shift data represent the average of three independent determinations performed in triplicate at a final compound concentration of 30 μM.
Inactive.
Finally, having discovered compounds with improved potency and efficacy compared with PHCCC, we next determined the selectivity against mGluR1 since PHCCC is a known partial antagonist of this receptor (IC50, 2.1 μM, Table 5). Evaluation of the compounds in Table 5 reveals several interesting trends. Compound 7e (6-F) still retains mGluR1 antagonist activity (IC50 > 5 μM); however, increasing the size of the 6-substituent (7f,g) results in compounds that no longer possess mGluR1 inhibitory activity. In addition, replacing the phenyl amide of PHCCC with the pyridyl amide also leads to compounds devoid of any mGluR1 activity (5i, 5k, 9a, 9b−d, 9f). Analogs that simply substitute the phenyl amide still antagonize mGluR1 (5c and 9e). However, upon combination of a 6-chloro substitution with a substituted phenyl, the compounds are devoid of mGluR1 activity (7f).
Table 5. mGluR1 Activity on Select PHCCC Analogsa.
| compound | X | R | mGluR1 EC50 (μM) | % Glu Max | categoryb |
|---|---|---|---|---|---|
| (±)-PHCCC, 1 | H | Ph | 2.1 | 23 | NAM |
| 7e | F | Ph | >5.0 | 49 | NAM |
| 7f | Cl | 4-F-Ph | c | ||
| 7g | Me | 4-F-Ph | c | ||
| 5c | H | 4-F-Ph | 1.2 | 16 | NAM |
| 5i | H | 2-Pyr | c | ||
| 5k | H | 2-Pyr-4-F | c | ||
| 9a | Cl | 2-Pyr-4-F | c | ||
| 9b | Me | 2-Pyr | >10 | ||
| 9c | Me | 2-Pyr-4-F | >10 | ||
| 9d | F | 2-Pyr | c | ||
| 9e | F | 4-F-Ph | >5.0 | 29 | NAM |
| 9f | F | 2-Pyr-4-F | c | ||
| (−)-10c, VU0359516 | H | 2-Pyr | c | ||
| (−)-10d | H | 2-Pyr-4-F | c |
Data represent the average of at least three experiments performed in triplicate.
NAM = negative allosteric modulator.
Inactive.
Lastly, having shown that two of the more potent compounds lack mGluR1 NAM activity, we next evaluated the selectivity of these compounds (10c, VU0359516, and 10d) against all of the mGluR subtypes. In these selectivity experiments, a full concentration−response curve of agonist was performed in the presence or absence of a 10 μM final concentration of each compound. Compound 10c (VU0359516) was devoid of activity against the other mGluR subtypes tested (Table 6), whereas 10d was selective against all the mGluR subtypes with the exception of weak antagonist activity versus mGluR2 (2-fold shift of the glutamate concentration−response curve to the right with no decrease in maximal glutamate response). Compound 10c (VU0359516) represents a major advancement over PHCCC in terms of potency, efficacy, and selectivity among the mGluR family.
Table 6. mGluR Activity of Select PHCCC Analogsa.
 |
For mGluR4, data are shown for potency at human mGluR4, % Glu Max at 30 μM, and fold shift (FS) of the glutamate concentration−response curve. For the other receptors (except mGluR1), data represent one experiment performed in duplicate as described in Methods. NAM = negative allosteric modulator
In summary, we have presented an SAR evaluation of the prototypical mGluR4 PAM, PHCCC. Our studies revealed that the scaffold tolerates substitution on the western aromatic ring, along with aromatic and heteroaromatic amides. However, all attempts to replace the oxime moiety failed to produce any compounds with activity at the hmGluR4 receptor. From this study, we have discovered compound 10c (VU0359516), an enantiomerically pure 2-pyridyl amide analog of PHCCC, which was shown to exhibit superior potency and efficacy at the hmGluR4 receptor as compared with PHCCC. Lastly, compound 10c (VU0359516) also does not possess any mGluR off-target activity in the assays examined, making 10c (VU0359516) a potentially very important tool compound for advancing mGluR4 pharmacology and demonstrating that the PHCCC scaffold, previously thought difficult to modify with retention of activity, can be optimized.
Methods
Assay Details
Cell Culture of Human mGluR4/Gqi5/CHO Line
Human mGluR4 (hmGluR4)/CHO cells stably transfected to express the chimeric G protein Gqi5 in pIRESneo3 (Invitrogen, Carlsbad, CA) were cultured in 90% Dulbecco’s modified Eagle media (DMEM), 10% dialyzed fetal bovine serum (FBS), 100 units/mL penicillin/streptomycin, 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH 7.3), 1 mM sodium pyruvate, 2 mM glutamine, 400 μg/mL G418 sufate (Mediatech, Inc., Herndon, VA), and 5 nM methotrexate (Calbiochem, EMD Chemicals, Gibbstown, NJ); 30 000 cells/(20 μL·well) were plated in black-walled, clear-bottomed, TC-treated, 384-well plates (Greiner Bio-One, Monroe, North Carolina) in DMEM containing 10% dialyzed FBS, 20 mM HEPES, 100 units/mL penicillin/streptomycin, and 1 mM sodium pyruvate (Plating Medium). The cells were grown overnight at 37 °C in the presence of 5% CO2. During the day of assay, the medium was replaced with 20 μL of 1 μM Fluo-4, AM (Invitrogen, Carlsbad, CA), prepared as a 2.3 mM stock in DMSO and mixed in a 1:1 ratio with 10% (w/v) pluronic acid F-127 and diluted in assay buffer (Hank’s balanced salt solution, 20 mM HEPES, and 2.5 mM Probenecid (Sigma-Aldrich, St. Louis, MO)) for 45 min at 37 °C. Dye was removed and replaced with 20 μL of assay buffer. Test compounds were transferred to daughter plates using an Echo acoustic plate reformatter (Labcyte, Sunnyvale, CA) and then diluted into assay buffer. Ca2+ flux was measured using the Functional Drug Screening System 6000 (FDSS6000, Hamamatsu, Japan). Baseline readings were taken (10 images at 1 Hz, excitation, 470 ± 20 nm, emission, 540 ± 30 nm), and then 20 μL/well of test compounds was added using the FDSS’s integrated pipettor. For concentration−response curve experiments, compounds were serially diluted 1:3 in DMSO into 10 point concentration−response curves and were transferred to daughter plates using the Echo (240 nL compound transfer followed by 40 μL assay buffer dilution). Cells were incubated with compounds for approximately 2.5 min, and then an EC20 concentration of glutamate was applied; 2 min later an EC80 concentration of glutamate was added. For fold shift experiments, compounds were added at 2× their final concentration, and then increasing concentrations of glutamate were added in the presence of vehicle or the appropriate concentration of test compound. All data were normalized to the percent maximal glutamate response (% Glu Max). Curves were fitted using a four-point logistical equation using Microsoft XLfit (IDBS, Bridgewater, NJ) or GraphPad Prism (La Jolla, CA).
Selectivity Experiments
Rat mGluR1/BHK and rat mGluR5/HEK cells were cultured as previously described (ref (21)). Rat mGluR2, -3, -7, and -8 and human mGluR6 were subcloned into pIRES puro 3 and stably expressed in HEK cells coexpressing GIRK 1/2 subunits, which were cultured and plated as described (26). For selectivity experiments for mGluRs other than mGluR1, cells were incubated with a 10 μM concentration of each compound followed by a concentration−response curve of agonist (glutamate for all mGluRs except mGluR7, which was tested using the agonist L-AP4). Compounds were deemed inactive if they did not shift the agonist concentration−response curve more than 2-fold to the left or right or did not change the maximal agonist response by more than 25%. For mGluR1, full concentration−response curves of compounds (serially diluted 1:3, 30 μM top final concentration) were performed. Cells were incubated with compounds for approximately 2.5 min, and then an EC20 concentration of glutamate was applied; 2 min later an EC80 concentration of glutamate was added. Antagonist activity at mGluR1 was determined using the blockade of the response of the EC80 agonist addition. All data were normalized to the percent maximal glutamate response (% Glu Max).
Chemical Synthesis and Purification
Synthesis of 5i, (E)-7-(Hydroxyimino)-N-(pyridin-2-yl)-1,1a,7,7a-tetrahydrocyclopropa[b]chromene-1a-carboxamide (All Compounds Were Synthesized in an Analogous Manner)
To a rt solution of trimethylsulfoxonium iodide (3.63 g, 16.5 mmol) in DMF (40 mL) was added NaOH (1.62 g, 40.5 mmol) followed by chromone-2-carboxylic acid (3.0 g, 16 mmol). The reaction was stirred at rt for 3 h. The reaction was then quenched with 1 N HCl and extracted with EtOAc. The combined organic layers were concentrated to afford an orange solid. The resulting acid (1.1 g, 5.4 mmol) was dissolved in DMF (15 mL), and then HATU (2-(1H-7-azabenzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate methanaminium; 2.27 g, 5.90 mmol) was added followed by 2-aminopyridine (598 mg, 6.43 mmol), and the mixture was stirred at rt overnight. The reaction was diluted with H2O and extracted with EtOAc. The combined organic extracts were washed with H2O and 1 N HCl and then dried over MgSO4 and concentrated. The residue was purified by column chromatography (EtOAc/hexanes, 0 → 60%) to afford the desired compound 4i as a tan solid (630 mg, 2.25 mmol, 14%). LCMS: >98% @ 214 nM. RT = 2.41 min; m/z = 281.2 [M + H]+. 1H NMR (CDCl3, 300 MHz) δ 9.44 (s, 1 H), 8.35 (d, 1 H, J = 3.0 Hz), 8.28 (d, 1 H, J = 6.3 Hz), 7.96 (dd, 1 H, J = 5.9, 1.2 Hz), 7.79 (t, 1 H, J = 6.4 Hz), 7.57 (t, 1 H, J = 6.6 Hz), 7.22−7.13 (m, 3 H), 2.89 (dd, 1 H, J = 11.0, 7.0 Hz), 2.31 (dd, 1 H, J = 11.0, 6.5 Hz), 1.62 (t, 1 H, J = 5.0 Hz).
To a solution of 4i (360 mg, 1.28 mmol) and MeOH (20 mL) was added hydroxylamine (50% w/w in H2O; 126 mg, 1.94 mmol), and the reaction was heated at 60 °C for 20 h. The reaction was concentrated, and the desired product (5i) was isolated as a tan solid (374 mg, 1.27 mmol, 99%). LCMS: >98% @ 214 nm. RT = 2.34 min; m/z = 296.1 [M + H]+. 1H NMR (CDCl3, 300 MHz) δ 9.21 (s, 1 H), 8.37−8.35 (m, 1 H), 8.28 (d, 1 H, J = 8.4 Hz), 7.82−7.74 (m, 2 H), 7.37 (t, 1 H, J = 6.9 Hz), 7.14−7.03 (m, 4 H), 3.51 (dd, 1 H, J = 10.7, 7.7 Hz), 2.20 (dd, 1 H, J = 10.8, 5.9 Hz), 1.51 (t, 1 H, J = 7.0 Hz).
Professors Lindsley and Hopkins directed and designed the chemistry. Professors Conn and Niswender directed the molecular pharmacology. Ms. Luo performed the in vitro molecular pharmacology. Drs. Williams and Zhou helped design compounds and performed synthetic chemistry.
The authors thank the National Institute of Mental Health, the Michael J. Fox Foundation, the Vanderbilt Department of Pharmacology, and the Vanderbilt Institute of Chemical Biology for support of this research.
Funding Statement
National Institutes of Health, United States
References
- Hefti F. F. (2005) Parkinson's Disease. Drug Discovery Nerv. Syst. Dis. 183–204. [Google Scholar]
- Parkinson J. (1817) An Essay on the Shaking Palsy, Sherwood, Neely and Jones, London. [Google Scholar]
- Bedard P. J.; Blanchet P. J.; Levesque D.; Soghomonian J. J.; Grondin R.; Morissette M.; Goulet M.; Calon F.; Falardeau P.; Gomez-Mancilla B.; Doucet J. P.; Robertson G. S.; Di Paolo T. (1999) Pathophysiology of L-dopa-induced dyskinesias. Mov. Dis. 14, 4–8. [PubMed] [Google Scholar]
- Graham D. G. (1978) Oxidative pathway for catecholamines in the genesis of neuromelanin and cytotoxic quinones. Mol. Pharmacol. 14, 633–643. [PubMed] [Google Scholar]
- Jankovic J. (2001) Parkinson's disease therapy: Treatment of early and late disease. Chin. Med. J. 114, 227–234. [PubMed] [Google Scholar]
- Hopkins C. R.; Lindsley C. W.; Niswender C. M. (2009) mGluR4 positive allosteric modulation as potential treatment of Parkinson's disease. Future Med. Chem. 1, 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn P. J.; Battaglia G.; Marino M. J.; Nicoletti F. (2005) Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat. Rev. Neurosci. 6, 787–798. [DOI] [PubMed] [Google Scholar]
- Bruno V.; Battaglia G.; Copani A.; D'Onofrio M.; Di Iorio P.; De Blasi A.; Melchiorri D.; Flor J. P.; Nicoletti F. (2001) Metabotropic glutamate receptor subtypes as targets for neuroprotective drugs. J. Cereb. Blood Flow Metab. 21, 1013–1033. [DOI] [PubMed] [Google Scholar]
- Conn P. J.; Pin J.-P. (1997) Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 37, 205–237. [DOI] [PubMed] [Google Scholar]
- Schoepp D. D.; Jane D. E.; Monn J. A. (1999) Pharmacological agents acting at subtypes of metabotropic glutamate receptors. Neuropharmacology 38, 1431–1476. [DOI] [PubMed] [Google Scholar]
- Marino M. J.; Hess J. F.; Liverton N. (2005) Targeting the metabotropic glutamate receptor mGluR4 for the treatment of diseases of the central nervous system. Curr. Top. Med. Chem. 5, 885–895. [DOI] [PubMed] [Google Scholar]
- Lindsley C. W.; Niswender C. M.; Engers D. W.; Hopkins C. R. (2009) Recent progress in the development of mGluR4 positive allosteric modulators for the treatment of Parkinson's disease. Curr. Top. Med. Chem. 9, 949–963. [PubMed] [Google Scholar]
- Maj M.; Bruno V.; Dragic Z.; Yamamoto R.; Battaglia G.; Inderbitzin W.; Stoehr N.; Stein T.; Gasparini F.; Vranesic I.; Kuhn R.; Nicoletti F.; Flor P. J. (2003) (−)-PHCCC, a positive allosteric modulator of mGluR4: Characterization, mechanism of action, and neuroprotection. Neuropharmacology 45, 895–906. [DOI] [PubMed] [Google Scholar]
- Marino M. J.; Williams D. L. Jr.; O'Brien J. A.; Valenti O.; McDonald T. P.; Clements M. K.; Wang R.; DiLella A. G.; Kinney G. G.; Conn P. J. (2003) Allosteric modulation of group III metabotropic glutamate receptor 4: A potential approach to Parkinson's disease treatment. Proc. Natl. Acad. Sci. U.S.A. 100, 13668–13673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O.; Marino M. J.; Wittmann M.; Lis E.; DiLella A. G.; Kinney G. G.; Conn P. J. (2003) Group III metabotropic glutamate receptor-mediated modulation of the striatopallidal synapse. J. Neurosci. 23, 7218–7226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia G.; Busceti C. L.; Molinaro G.; Giagioni F.; Traficante A.; Nicoletti F.; Bruno V. (2006) Pharmacological activation of mGluR4 metabotropic glutamate receptors reduces nigrostriatal degeneration in mice treated with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. J. Neurosci. 26, 7222–7229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenti O.; Mannaioni G.; Seabrook G. R.; Conn P. J.; Marino M. J. (2005) Group III metabotropic glutamate-receptor-mediated modulation of excitatory transmission in rodent substantia nigra pars compacta dopamine neurons. J. Pharmacol. Exp. Ther. 313, 1296–1304. [DOI] [PubMed] [Google Scholar]
- Stachowicz K.; Klak K.; Klodzinska A.; Chojnacka-Wojcik E.; Pilc A. (2004) Anxiolytic-like effects of PHCCC, an allosteric modulator of mGluR4 receptors, in rats. Eur. J. Pharmacol. 498, 153–156. [DOI] [PubMed] [Google Scholar]
- Canudas A. M.; Di Giorgi-Gerevini V.; Iacovelli L.; Nano G.; D'Onofrio M.; Arcella A.; Giangaspero F.; Busceti C.; Ricci-Vitani L.; Battaglia G.; Nicoletti F.; Melchiorri D. (2004) PHCCC, a specific enhancer of type 4 metabotropic glutamate receptors, reduces proliferation and promotes differentiation of cerebellular granule cell neuroprecursors. J. Neurosci. 24, 10343–10352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovelli L.; Arcella A.; Battaglia G.; Pazzaglia S.; Aronica E.; Spinsanti P.; Caruso A.; De Smaele E.; Saran A.; Gulino A.; D'Onofrio M.; Giangaspero F.; Nicoletti F. (2006) Pharmacological activation of mGluR4 metabotropic glutamate receptors inhibits the growth of medulloblastomas. J. Neurosci. 26, 8388–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niswender C. M.; Johnson K. A.; Weaver C. D.; Jones C. K.; Xiang Z.; Luo Q.; Rodriguez A. L.; Marlo J. E.; de Paulis T.; Thompson A. D.; Days E. L.; Nalywajko T.; Austin C. A.; Williams M. B.; Ayala J. E.; Williams R.; Lindsley C. W.; Conn P. J. (2008) Discovery, characterization, and antiparkinsonian effect of novel positive allosteric modulators of metabotropic glutamate receptor 4. Mol. Pharmacol. 74, 1345–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor K. G.; Hobbs W. E.; Clark M. S.; Chaney J. (1972) Carbenoids with neighboring heteroatoms. III. Electrophilic reactions of two α-halocyclopropyllithium compounds. J. Org. Chem. 37, 2436–2443. [Google Scholar]
- Johnson C. R.; Mori K.; Nakanishi A. (1979) Preparation and reaction of N-(p-tolylsulfonyl)sulfilimines. J. Org. Chem. 44, 2065–2067. [Google Scholar]
- Parmar D.; Burka L. T. (1991) Metabolism and disposition of cyclohexanone oxime in male F-344 rats. Drug. Metab. Dispos. 19, 1101–1107. [PubMed] [Google Scholar]
- Annoura H.; Fukunaga A.; Uesugi M. (1996) A novel class of antagonists for metabotropic glutamate receptors, 7-(hydroxyimino)cyclopropa[b]chromen-1a-carboxylates. Bioorg. Med. Chem. Lett. 6, 763–766. [Google Scholar]
- Niswender C. M.; Johnson K. A.; Luo Q.; Ayala J. E.; Kim C.; Conn P. J.; Weaver C. D. (2008) A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol. Pharmacol. 73, 1213–1224. [DOI] [PubMed] [Google Scholar]
- Leister W.; Strauss K.; Wisnoski D.; Zhao Z.; Lindsley C. (2003) Development of a custom high-throughput preparative liquid chromatography/mass spectrometer platform for the preparative purification and analytical analysis of compound libraries. J. Comb. Chem. 5, 322–329. [DOI] [PubMed] [Google Scholar]