

Abstract
RNA function is dependent on its structure, yet three-dimensional folds for most biologically important RNAs are unknown. We develop a generic discrete molecular dynamics (DMD)-based modeling system that uses long-range constraints inferred from diverse biochemical or bioinformatic analyses to create statistically significant (p < 0.01) native-like folds for RNAs of known structure ranging from 45 to 158 nucleotides. We then predict the unknown structure of the hepatitis C virus IRES pseudoknot domain. The resulting RNA model rationalizes independent solvent accessibility and cryo-electron microscopy structure information. The pseudoknot positions the AUG start codon near the mRNA channel and is tRNA-like, suggesting the IRES employs molecular mimicry as a functional strategy.
Critical RNA structures directly regulate gene expression, splicing and translation (1), but the structures of most biologically important RNA folds are currently unknown. Recent studies highlight significant successes in ab initio structure prediction of local helical structure and of small RNA motifs (2). However, the ability of current approaches to predict RNA structure accurately decreases rapidly with increasing RNA size. De novo prediction of large RNA structures with complex, non-trivial, three-dimensional folds from sequence alone remains beyond the realm of current automated algorithms. A compelling alternative is to develop modeling methods for facile incorporation of readily obtained experimental information.
Long-range constraints for RNA modeling can be inferred from a variety of biochemical and bioinformatic techniques, ranging from chemical footprinting and crosslinking to sequence covariation (3). Algorithms devised thus far are making significant progress towards the goal of incorporating specific classes of tertiary structure information into RNA structure refinement (4). However, current refinement approaches still make large assumptions about the nature of the constraint information used and are closely tied to the specific techniques employed to infer long-range interactions.
To address these challenges, we develop a generic and efficient approach for accurately predicting RNA folds using tertiary structure information as inferred from diverse biochemical or bioinformatic techniques. Distance constraints are incorporated into a discrete molecular dynamics (DMD) engine (2b) that uses a single refinement approach for all classes of tertiary structure constraint information. RNA nucleotides are represented as three pseudo-atoms corresponding to the phosphate (P), sugar (S), and base (B) moieties (Figure 1A). Three pseudo-atoms are sufficient to develop nucleotide-resolution RNA models with rigid base paired helices and physically meaningful base-stacking interactions, while still allowing large RNAs to be refined efficiently.
Figure 1.
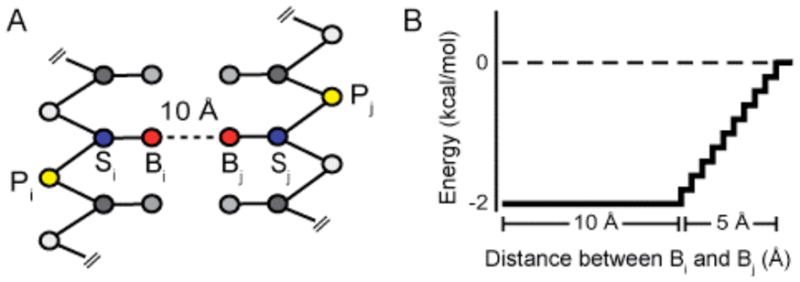
Generic constraint system. (A) Three-bead model for RNA. (B) Interaction potential for distance constraints.
Inferred pair-wise tertiary constraints are incorporated via a generic constraint system that uses a potential well with an effective length of 15.0 Å and a depth of 2.0 kcal/mol between base pseudo-atoms (Figure 1B). This constraint system is compatible with techniques that do not directly provide distance information but instead merely imply pair-wise interactions, as with mutational studies.
Four RNAs were selected to benchmark constrained structure refinement: domain III of the cricket paralysis virus internal ribosome entry site (CrPV) (49 nts), a full-length hammerhead ribozyme from S. mansoni (HHR) (67 nts), S. cerevisiae tRNAAsp (75 nts), and the P546 domain of the T. thermophilia group I intron (P546) (158 nts). Each of these RNAs has a complex three-dimensional fold dependent both on local helical structure and on long-range tertiary interactions. Prior to publication of the high-resolution structures (5), significant biochemical or bioinformatic data describing tertiary interactions were available for each RNA. The secondary structure was also known to good accuracy in each case. Only this prior information (Table S1) was used during refinement.
A single generic and completely automated refinement protocol was applied to each RNA. Simulations begin with the RNA strand in an extended conformation at high temperature. Constraints based on the secondary structure are included, and the molecular system is annealed to allow helices to form. Constraints for inferred tertiary interactions are incorporated, and the RNA is cooled to a final target temperature. RNA structures from this step (100,000) are subjected to automated clustering. The centroid of the most populated cluster is selected as the final predicted structure. Given our refinement model, this structure is representative of the lowest free energy state.
Refined models for all four test RNAs are accurate (Figure 2). Root mean square deviation (RMSD) of the phosphate backbone relative to the accepted structures for the CrPV, HHR, tRNAAsp, and P546 RNAs were 3.6, 5.4, 6.4, and 11.3 Å, respectively. Analysis of the RNA structure prediction significance (p-value) (6) shows that the probabilities that these models result from chance are small, at 2 × 10−3, 2 × 10−5, 3 × 10−6 and ≤ 10−6, respectively.
Figure 2.

Comparison of predicted and experimental RNA structures. Spheres indicate phosphate groups. RMSDs for the CrPV, HHR, tRNA and P546 RNAs are 3.6, 5.4, 6.4, and 11.3 Å.
There are two critical results from this analysis of RNAs with known structures. First, native-like RNA folds were obtained in every case despite the diversity of structural information used to constrain refinement (Table S1). Second, prediction quality was maintained as RNA size increased from a 49 nt pseudoknot to a 158 nt RNA domain with a complex tertiary structure (Figure 2).
Our approach compares favorably to other coarse-grained RNA modeling approaches. Folds for tRNAPhe and the P546 domain have been predicted with the program NAST in which each RNA nucleotide is represented by a single pseudo-atom (4d). NAST modeling was constrained using structure information similar to that used in our refinements. Of the resulting models, the most accurate had RMSDs relative to the accepted structure of 8.0 and 16.3 Å for tRNA and P546, respectively, whereas our approach yields smaller RMSDs of 6.4 and 11.3 Å. NAST simulations used 300 hrs per RNA, as compared to 18 to 40 real-time computing hours for the DMD-based refinements (Figure 2). These comparisons highlight both the accuracy and efficiency of our constrained DMD approach.
Having shown that this fully automated approach recapitulates native-like folds for diverse, well-characterized RNAs, we sought to apply this algorithm to an RNA for which extensive biochemical information exists, but whose structure is unknown. We focused on the pseudoknot domain in the hepatitis C virus (HCV) internal ribosome entry site (IRES).
IRES elements bypass canonical cap-dependent eukaryotic translation initiation by directly recruiting ribosomes to internal sequences in a messenger RNA (7). Structural studies have yielded significant insights for understanding functional mechanisms of IRES elements (5d, 8). High-resolution structures are available for many elements of the HCV IRES (9); however, the three-dimensional fold for the pseudoknot domain (HCV-PK) has not been determined. The pseudoknot domain consists of a pseudoknot at the base of domain III (dIII) and its flanking structures (Figure 3A). Mutation of the pseudoknot inhibits translation initiation in HCV replication (10). Compensatory mutations that restore the pseudoknot do not always restore HCV translation activity, suggesting that sequence conservation is required for functions beyond base pairing. The pseudoknot domain contains the AUG start codon for translation of the HCV polypeptide (in yellow, Figure 3A). Solvent accessibility experiments show the pseudoknot domain is the most highly structured element in the IRES (11). Extensive available biochemical information and intense biomedical interest make the HCV-PK RNA an ideal candidate for deriving biological insights based on structural modeling.
Figure 3.

RNA structure prediction for the pseudoknot domain of the HCV IRES. (A) Secondary structure (10). (B) Predicted structure of the uncomplexed pseudoknot domain. Spheres indicate phosphate positions; nucleotides are colored as shown in panel A. (C) HCV-PK model placed into the electron density of the IRES-ribosome complex (12b). (D) Superposition of hydroxyl radical protection data (11) on the HCV-PK RNA model. Spheres correspond to protected sugar pseudo-atoms; red and yellow indicate strong and moderate protection, respectively.
A three-dimensional model for the HCV-PK domain RNA was refined using the same fully automated folding algorithm as for the four test RNAs. Base pairs in the pseudoknot were modeled as generic tertiary constraints.
The predicted HCV-PK structure is dominated by two structural features (Figure 3B). The first is the four-way junction comprised of stems at the base of dIII (red and purple), dIIIe (cyan), and dIIIf (blue). The second consists of base stacking interactions between the pseudoknot (blue) and dIV (green). The nucleotide linkages between these two motifs are short and lock the dIV helix in a conformation perpendicular to the plane described by the helices of the four-way junction.
Two classes of independent experiments support the proposed structure for the HCV-PK RNA. First, the predicted tight RNA folding in the 4-way junction and pseudoknot is supported by protection from hydroxyl radical cleavage, indicative of solvent inaccessible regions of the RNA backbone (11). Solvent inaccessible regions fall precisely in the interior of the four-way junction and at the interface of this element with the pseudoknot (red and yellow spheres, Figure 3D).
Second, the HCV-PK model is consistent with cryo-EM electron density maps of the IRES-ribosome complex. Our model of the HCV-PK is that of the uncomplexed IRES, and conformational changes occur in both the ribosome and IRES when the IRES interacts with ribosomal subunits and translation initiation factors (12). For example, domain IV likely unfolds to allow positioning of the start codon in the P-site (13) (see Supporting Information). Nevertheless, the core of our model fits well in the density assigned to the pseudoknot domain in the cryo-EM electron density maps of the IRES-ribosome complex (12b). The critical correlations are that dII and dIII (in orange and purple, respectively, Figures 3C & S1) are positioned to connect sensibly with the rest of the IRES, and the perpendicular orientation of the pseudoknot (in blue) allows the AUG start codon in dIV (in yellow, Figures 3C & 4) to be positioned in or near the mRNA channel. Our HCV-PK model also fits well with high resolution IRES structures positioned in the cryo-EM density (Figure S2).
Figure 4.
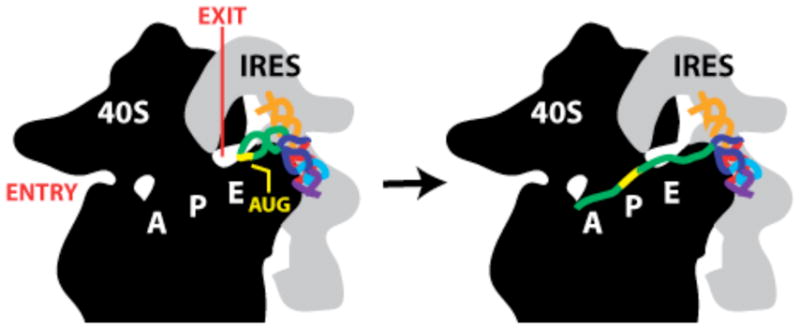
Docking of HCV IRES RNA into the mRNA channel of the 40S ribosome. Cartoons of the 40S subunit (black) and HCV IRES (gray) are based on cryo-EM studies (12). The AUG start site codon, mRNA entry and exit sites, and tRNA binding sites are labeled on the 40S subunit. The HCV-PK model is colored and positioned in the same orientation as in Figure 3C.
Several functional hypotheses are consistent with the predicted model. First, the HCV-PK RNA is L-shaped, similar to tRNA, and can be aligned to yeast tRNAAsp (not shown). Formation of a tRNA-like structure is consistent with biochemical studies showing that the HCV IRES is cleaved by the tRNA-recognizing ribonuclease RNase P (14). tRNA mimicry also rationalizes the presence of a seven-nucleotide loop at the end of domain IV, a structural feature that is generally uncommon in RNA but present in the anti-codon loops of most tRNAs.
A recent structural study also found evidence for tRNA mimicry in domain III of the CrPV IRES (5d). Though the HCV-PK model and CrPV experimental structure have distinct folds, both support tRNA mimicry as a common strategy employed by IRES structures and are consistent with extensive examples of tRNA mimicry in biologically diverse RNAs (15).
Second, the perpendicular orientation of the pseudoknot relative to the 4-way junction may function to position the AUG start codon for translation initiation. In cryo-EM maps of both the 40S- and 80S-IRES complexes, density corresponding to the pseudoknot domain is adjacent to the channel occupied by the mRNA template during translation (12). Thus dIV and, specifically, the AUG start codon will be positioned near the ribosome mRNA exit site.
These observations support a model in which the IRES pseudoknot domain docks initially near the ribosome exit channel, facilitated by its tRNA-like structure (Figure 4). Our model suggests additional conformational changes are required in the IRES and ribosome for the AUG start codon to fully occupy the mRNA channel. A modest unfolding of the dIV helix would then allow this element to serve as the mRNA template for translation of the HCV polyprotein (Figure 4, right).
RNA structure refinement using inferred constraints consistently yields native-like models for RNAs spanning 49 to 158 nts. This approach does not require a specific optimized form for the long-range constraints but does require knowledge of through-space tertiary interactions. The success of this approach implies that knowledge of only a few long-range constraints is sufficient to refine accurate folds for many RNAs with complex structures.
The HCV-PK domain model rationalizes substantial pre-existing biochemical information for this RNA and provides specific and novel functional insights useful for guiding future hypotheses and experiments. RNA structure refinement using inferred constraints holds significant promise for understanding the functions of many biologically important RNAs whose analysis is recalcitrant to high-resolution approaches.
Supplementary Material
Acknowledgments
This work was supported by NIH grant GM064803 (to K.M.W.) and an ARRA supplement (to K.M.W. and N.V.D.).
Footnotes
SUPPORTING INFORMATION AVAILABLE
Details of the refinement algorithm, two figures, biochemical data used to constrain refinements, discussion of placing the HCV-PK model in the cryo-EM density, and PDB files of predicted RNA models. Available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Gesteland RF, et al. Cold Spring Harbor Laboratory Press. 3. Cold Spring Harbor, N.Y: 2006. [Google Scholar]
- 2.(a) Das R, Baker D. Proc Natl Acad Sci U S A. 2007;104:14664–14669. doi: 10.1073/pnas.0703836104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ding F, et al. RNA. 2008;14:1164–1173. doi: 10.1261/rna.894608. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Parisien M, Major F. Nature. 2008;452:51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 3.(a) Ziehler WA, Engelke DR. Curr Protoc Nucleic Acid Chem. 2001;Chapter 6(Unit 6):1. doi: 10.1002/0471142700.nc0601s00. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Juzumiene D, et al. Methods. 2001;25:333–343. doi: 10.1006/meth.2001.1245. [DOI] [PubMed] [Google Scholar]; (c) Gutell RR, et al. Nucleic Acids Res. 1992;20:5785–5795. doi: 10.1093/nar/20.21.5785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Badorrek CS, et al. Proc Natl Acad Sci U S A. 2006;103:13640–13645. doi: 10.1073/pnas.0606156103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Das R, et al. Proc Natl Acad Sci U S A. 2008;105:4144–4149. doi: 10.1073/pnas.0709032105. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Gherghe CM, et al. J Am Chem Soc. 2009;131:2541–2546. doi: 10.1021/ja805460e. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Jonikas MA, et al. RNA. 2009;15:189–199. doi: 10.1261/rna.1270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.(a) Westhof E, et al. Acta Crystal. 1988;A44:112–123. [Google Scholar]; (b) Cate JH, et al. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]; (c) Martick M, Scott WG. Cell. 2006;126:309–320. doi: 10.1016/j.cell.2006.06.036. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Costantino DA, et al. Nat Struct Mol Biol. 2008;15:57–64. doi: 10.1038/nsmb1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hajdin C, et al. RNA. 2010;16 doi: 10.1261/rna.1837410. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Pisarev AV, et al. C R Biol. 2005;328:589–605. doi: 10.1016/j.crvi.2005.02.004. [DOI] [PubMed] [Google Scholar]; (b) Kieft JS. Trends Biochem Sci. 2008;33:274–283. doi: 10.1016/j.tibs.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfingsten JS, et al. Science. 2006;314:1450–1454. doi: 10.1126/science.1133281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Lukavsky PJ. Virus Res. 2008;139(2):166–71. doi: 10.1016/j.virusres.2008.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Filbin ME, Kieft JS. Curr Opin Struct Biol. 2009;19:267–276. doi: 10.1016/j.sbi.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.(a) Wang C, et al. RNA. 1995;1:526–537. [PMC free article] [PubMed] [Google Scholar]; (b) Kieft JS, et al. RNA. 2001;7:194–206. doi: 10.1017/s1355838201001790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kieft JS, et al. J Mol Biol. 1999;292:513–529. doi: 10.1006/jmbi.1999.3095. [DOI] [PubMed] [Google Scholar]
- 12.(a) Spahn CM, et al. Science. 2001;291:1959–1962. doi: 10.1126/science.1058409. [DOI] [PubMed] [Google Scholar]; (b) Boehringer D, et al. Structure. 2005;13:1695–1706. doi: 10.1016/j.str.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 13.(a) Pestova TV, et al. Genes Dev. 1998;12:67–83. doi: 10.1101/gad.12.1.67. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Otto GA, Puglisi JD. Cell. 2004;119:369–380. doi: 10.1016/j.cell.2004.09.038. [DOI] [PubMed] [Google Scholar]; (c) Fraser CS, et al. Nat Struct Mol Biol. 2009;16:397–404. doi: 10.1038/nsmb.1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Nadal A, et al. J Biol Chem. 2002;277:30606–30613. doi: 10.1074/jbc.M203595200. [DOI] [PubMed] [Google Scholar]; (b) Lyons AJ, Robertson HD. J Biol Chem. 2003;278:26844–26850. doi: 10.1074/jbc.M304052200. [DOI] [PubMed] [Google Scholar]
- 15.Hammond JA, et al. RNA. 2009;15:294–307. doi: 10.1261/rna.1360709. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.