

Abstract
Background
In recent years genome sequencing has been used to characterize new bacterial species, a method of analysis available as a result of improved methodology and reduced cost. Included in a constantly expanding list of Vibrio species are several that have been reclassified as novel members of the Vibrionaceae. The description of two putative new Vibrio species, Vibrio sp. RC341 and Vibrio sp. RC586 for which we propose the names V. metecus and V. parilis, respectively, previously characterized as non-toxigenic environmental variants of V. cholerae is presented in this study.
Results
Based on results of whole-genome average nucleotide identity (ANI), average amino acid identity (AAI), rpoB similarity, MLSA, and phylogenetic analysis, the new species are concluded to be phylogenetically closely related to V. cholerae and V. mimicus. Vibrio sp. RC341 and Vibrio sp. RC586 demonstrate features characteristic of V. cholerae and V. mimicus, respectively, on differential and selective media, but their genomes show a 12 to 15% divergence (88 to 85% ANI and 92 to 91% AAI) compared to the sequences of V. cholerae and V. mimicus genomes (ANI <95% and AAI <96% indicative of separate species). Vibrio sp. RC341 and Vibrio sp. RC586 share 2104 ORFs (59%) and 2058 ORFs (56%) with the published core genome of V. cholerae and 2956 (82%) and 3048 ORFs (84%) with V. mimicus MB-451, respectively. The novel species share 2926 ORFs with each other (81% Vibrio sp. RC341 and 81% Vibrio sp. RC586). Virulence-associated factors and genomic islands of V. cholerae and V. mimicus, including VSP-I and II, were found in these environmental Vibrio spp.
Conclusions
Results of this analysis demonstrate these two environmental vibrios, previously characterized as variant V. cholerae strains, are new species which have evolved from ancestral lineages of the V. cholerae and V. mimicus clade. The presence of conserved integration loci for genomic islands as well as evidence of horizontal gene transfer between these two new species, V. cholerae, and V. mimicus suggests genomic islands and virulence factors are transferred between these species.
Background
The genus Vibrio comprises a diverse group of gamma-proteobacteria autochthonous to the marine, estuarine, and freshwater environment. These bacteria play a role in nutrient cycling, degrade hydrocarbons, and can be devastating pathogens for fish, shellfish, and mammals as well as humans [1-5]. From 1981 to 2009, the number of validly described species within the genus increased from 21 to more than 100 [6,7]. The most notorious, V. cholerae, is the etiological agent of the severe diarrheal disease cholera, endemic in southeast Asia for at least 1,000 years and the cause of seven pandemics since 1817. Shown to be autochthonous to the aquatic environment globally, more than 200 serogroups of V. cholerae have been described. Epidemics of cholera are caused by V. cholerae O1 and O139, with V. cholerae non-O1/non-O139 strains associated with sporadic cholera cases and extraintestinal infections [8,9]. Cholera infections have been ascribed to the presence and expression of virulence genes, e.g., ctxA, tcpA, tcpP, and toxT [10,11], which are also harbored by toxigenic strains of V. mimicus, a phylogenetic near-neighbor of V. cholerae. Genomic analyses of V. cholerae and V. mimicus demonstrated significant similarity, suggesting horizontal exchange of virulence factors, such as CTXΦ and VPIs-1 and -2 [12]. Based on results of phylogenetic analyses reported by Thompson et al. [13], V. cholerae and V. mimicus should be assigned to separate genera, a taxonomic assignment not yet resolved.
The aims of this study were to describe the genomes of two Vibrio strains previously characterized as variant V. cholerae by culture-based and molecular methods [14,15], and compare them to closely related Vibrio genomes. Results of this study suggest these two strains represent novel species and demonstrate evidence of horizontal gene transfer with their near-neighbors, V. cholerae and V. mimicus. We present here the genomic characterization of two new Vibrio species, Vibrio sp. RC341 (for which we propose the name Vibrio metecus) and Vibrio sp. RC586 (for which we propose the name Vibrio parilis), that share a close phylogenetic and genomic relationship with V. cholerae and V. mimicus, but are distinct species, based on comparative genomics, average nucleotide identity (ANI), average amino acid identity (AAI), multi-locus sequence analysis (MLSA), and phylogenetic analysis. Also, we present results of a comparative genomic analysis of these two novel species with 22 V. cholerae, two V. mimicus and one each of V. vulnificus and V. parahaemolyticus (see Additional file 1). The new Vibrio species are characterized as Vibrio sp. RC341 and Vibrio sp. RC586, sharing genes and mobile genetic elements with V. cholerae and V. mimicus. These data suggest that Vibrio sp. RC341 and Vibrio sp. RC586 may act as reservoirs of mobile genetic elements, including virulence islands, for V. cholerae and V. mimicus, Horizontal gene transfer among these bacteria enables colonization of new niches in the environment, as well as conferring virulence in the human host. Descriptions of these species and definitions have been provided elsewhere [Haley et al., in preparation].
Results and Discussion
Strains
The two strains analyzed in this study, Vibrio sp. RC341 and Vibrio sp. RC586, were isolated from water samples from the Chesapeake Bay, MD in 1998 and 1999, respectively. Vibrio sp. RC341 and Vibrio sp. RC586 were presumptively classified as variant V. cholerae [14,15], based on similarity to the 16S ribosomal RNA of V. cholerae. Vibrio sp. RC341 appears as yellow V. cholerae-like cells and Vibrio sp. RC586 appears as green V. mimicus-like cells on TCBS agar. Both strains were typeable with V. cholerae antisera, Vibrio sp. RC586 as serogroup O133 and Vibrio sp. RC341 as serogroup O153 [14,15].
General Genome Overview
The genomes of Vibrio sp. RC341 and Vibrio sp. RC586 span 28 and 16 contigs, respectively, and putatively encode 3574 and 3592 ORFs totaling 4,008,705 bp and 4,082,591 bp, respectively. Vibrio sp. RC341 encodes 91 RNAs, 71 of which are tRNAs. Vibrio sp. RC586 encodes 115 RNAs, 91 of which are tRNAs. The %GC content of each genome is ca. 46%, while the %GC content of V. cholerae strains is 47%. Vibrio sp. RC341 encodes 681 hypothetical proteins (19% of total ORFs) and Vibrio sp. RC586 encodes 719 hypothetical proteins (19.6% of total ORFs) determined by subsystem annotation. Twenty-four of these hypothetical proteins of Vibrio sp. RC586 and 48 of Vibrio sp. RC341 showed no homology to any of the sequences in the NCBI database.
Both genomes putatively encode two chromosomes, determined by comparing both chromosomes of V. cholerae N16961 to draft genome sequences of Vibrio sp. RC341 and Vibrio sp. RC586 using the MUMmer program [16] (see Additional files 2 and 3). The smaller chromosome of Vibrio sp. RC586 putatively encodes 1035 predicted ORFs, totaling approximately 1,155,676 bp. By this method, 951 ORFs were detected in Vibrio sp. RC341 totaling 987,354 bp. The smaller size of the second chromosome of Vibrio sp. RC341 can be attributed to low-quality coverage of this genome or uncaptured gaps. Both putative small chromosomes of the two species encode a superintegron region homologous to that of V. cholerae. The superintegron region of Vibrio sp. RC586 is ca. 93.6 kb, putatively encodes 96 ORFs, 66 (69%) of which are hypothetical proteins and the superintegron region of Vibrio sp. RC341 is ca. 68.6 kb, putatively encodes 66 ORFs, only 17 (26%) of which are hypothetical proteins. Interestingly, the superintegron of Vibrio sp. RC341 encodes several membrane bound proteins suggesting their role in the interaction with the extracellular environment.
Genome Comparisons
The genomes of Vibrio sp. RC341 and Vibrio sp. RC586 were compared with each other and to 22 V. cholerae, two V. mimicus, one V. vulnificus and one V. parahaemolyticus genome sequences by pairwise reciprocal BLAST analysis. Vibrio sp. RC341 and Vibrio sp. RC586 share 2104 non-duplicated ORFs (58% of the Vibrio sp. RC341 protein-coding genome) and 2058 non-duplicated ORFs (57% of the Vibrio sp. RC586 protein-coding genome) with 22 V. cholerae strains. Chun et al. [17] determined that the current V. cholerae core contains 2432 ORFs, indicating a dramatic difference in number of core genes between Vibrio sp. RC341/RC586 and V. cholerae core genomes. Vibrio sp. RC341 shares 2613 ORFs with V. cholerae N16961 (73% of V. sp. RC341), and Vibrio sp. RC586 shares 2581 ORFs with V. cholerae N16961 (71% of Vibrio sp. RC586) (Figure 1). Vibrio sp. RC341 shares 2956 ORFs with V. mimicus MB-451 (82% of Vibrio sp. RC341), and Vibrio sp. RC586 shares 3048 ORFs with V. mimicus MB-451 (84% of Vibrio sp. RC586) (Figure 1). Vibrio sp. RC341 and Vibrio sp. RC586 share 2926 ORFs with each other (81% of ORFs in both genomes) (Figure 1).
Figure 1.
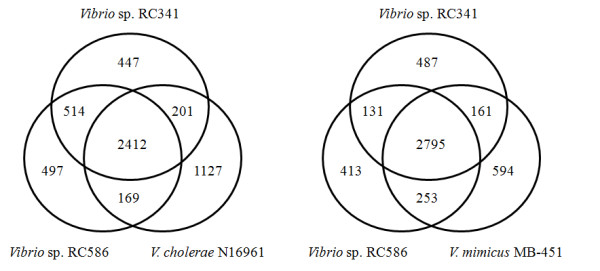
Venn diagrams showing ORFs shared by Vibrio sp. RC341, Vibrio sp. RC586, V. cholerae N16961, and V. mimicus MB-451. The number in the middle shows the conserved number of ORFs shared by the three strains. The numbers show that there are ORFs unique to that strain or that there are ORFs shared.
To determine average nucleotide identity (ANI) and average amino acid identity (AAI) between each genome, the average pairwise similarity between ORFs conserved between the compared genomes was calculated, following methods of Konstantinidis and Tiedje [18] and Konstantinidis et al. [19]. In this approach, two genomes with an ANI >95% and AAI >96% belong to the same species, while those with ANI and AAI below these thresholds, comprise separate species [19,20]. The ANI and AAI between Vibrio sp. RC586 and Vibrio sp. RC341 was 85 and 92%, respectively (see Additional files 4, 5, and 6). The ANIs between Vibrio sp. RC586 and individual V. cholerae ranged between 84 and 86%, while the ANI between Vibrio sp. RC341 and V. cholerae ranged between 85 and 86% (see Additional files 4, 5, and 6). The AAIs between Vibrio sp. RC341 and individual V. cholerae genomes and Vibrio sp. RC341 and V. cholerae were 92% in all comparisons (data not shown). The ANIs between Vibrio sp. RC586 and V. mimicus MB-451 and VM223 were 88% and 87%, respectively, and 86% for Vibrio sp. RC341 and both V. mimicus genomes (see Additional files 4, 5, and 6). The AAI between Vibrio sp. RC341 and V. mimicus strains MB-451 and VM223 was 92% in both comparisons, while the AAI between Vibrio sp. RC586 and both V. mimicus strains was 93% (data not shown).
The V. cholerae genomes had ANI >95% and AAI >96% and both V. mimicus strains a 98% ANI and AAI. The ANI for all V. cholerae and both V. mimicus strains was 86%. Based on these data, it is concluded that Vibrio sp. RC341 and Vibrio sp. RC586 are, indeed, separate species, genetically distinct from V. mimicus and V. cholerae and from each other. Strains of interspecies comparisons shared <95% ANI and <96% AAI with members of other species included in this study, the threshold for species demarcation [19,20], as applied to Vibrio, Burkholderia, Escherichia, Salmonella, and Shewanella spp. [21,19,22]. When Vibrio sp. RC341 and Vibrio sp. RC586 were compared with the more distantly related V. vulnificus and V. parahaemolyticus, Vibrio sp. RC586 showed 72 and 72% ANI and 73 and 73% AAI, respectively and Vibrio sp. RC341 73 and 72% ANI and 73 and 73% AAI with V. vulnificus and V. parahaemolyticus, respectively (see Additional files 4, 5, and 6). Furthermore, comparative analysis of the rpoB sequence demonstrates that Vibrio sp. RC341 and Vibrio sp. RC586 have <97.7% sequence identity with the rpoB sequences of all V. cholerae and V. mimicus strains included in this study. In a comparative DNA-DNA hybridization and ANI analysis, Adékambi et al. [23] demonstrated that rpoB <97.7% correlated with DNA-DNA hybridization <70% and ANI <95%, both being interpreted as demarcation thresholds for bacteria. All V. cholerae strains included in this study showed >99.5% rpoB sequence similarity with V. cholerae N16961 (data not shown). Based on a standard MLSA for the Vibrionaceae [21], Vibrio sp. RC341 and Vibrio sp. RC586 both have <95% pair-wise similarity with V. cholerae, V. mimicus, V. vulnificus, and V. parahaemolyticus strains. All V. cholerae strains and both V. mimicus strains used in this analysis demonstrated >95% similarity between concatenated genes of like-species (data not shown). Karlin's dissimilarity signatures were also calculated between these two genomes and the Vibrio genomes used in this study. Vibrio sp. RC586 shared >10 dissimilarity with all V. cholerae (11.5 to 16.2), V. vulnificus (19.6), and V. parahaemolyticus (41.6) genomes, and > 7 with both V. mimicus strains. Vibrio sp. RC341 shared >10 dissimilarity for all V. cholerae (10.2 to 14) except V. cholerae B33 (9.4) and TMA21 (9.8). Vibrio sp. RC341 shared >10 genome signature dissimilarity with V. parahaemolyticus (40.2), V. vulnificus (16.3), and both V. mimicus (>14) genomes. Vibrio sp RC341 and RC586 shared a genomic dissimilarity of 8.7 with each other. Taken together these data indicate that Vibrio sp. RC341 and Vibrio sp. RC586 are new species with a high genomic relatedness to V. cholerae and V. mimicus.
Evolution of Vibrio sp. RC341 and Vibrio sp. RC586 Lineages
The phylogenies of Vibrio sp. RC341 and Vibrio sp. RC586 were inferred by constructing a supertree, using a 362,424 bp homologous alignment of V. cholerae, V. mimicus, and the new species (Figure 2). Based on the supertree analysis Vibrio sp. RC341 and Vibrio sp. RC586 are deeply rooted in ancestral nodes, suggesting ancient evolution of the two species. Results of this phylogenetic analysis suggest the Vibrio sp. RC341 lineage evolved from a progenitor of the V. cholerae and V. mimicus lineages (Figure 2), a finding supported by strong bootstrap support and further evidenced by the evolutionary distance of V. cholerae and V. mimicus from Vibrio sp. RC341 (see Additional file 7). The two V. mimicus strains are interspersed among V. cholerae, with respect to evolutionary distance, suggesting that evolutionary distances of V. cholerae and V. mimicus are equidistant from Vibrio sp. RC341 (see Additional file 7).
Figure 2.
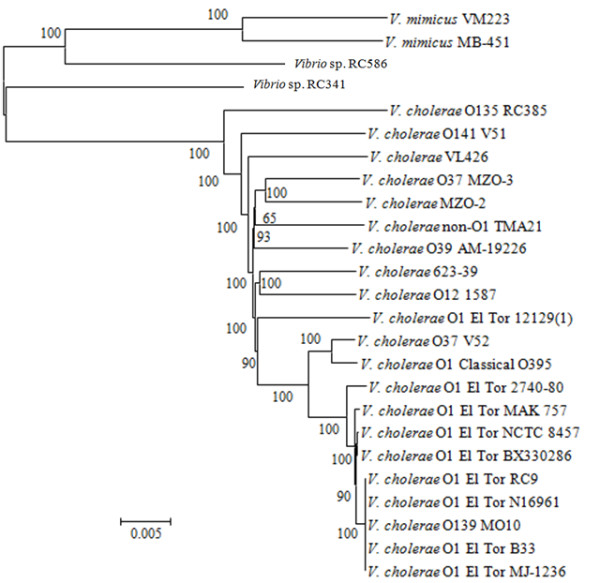
Neighbor-joining tree based on 362,424 bp alignment of homologous sequences using the Kimura-2 parameter for nucleotide substitution. The bootstrap supports, as percentage, are indicated at the branching points. Bar represents 0.005 substitutions per site.
The phylogeny of Vibrio sp. RC586 suggests it evolved from an ancestral member of the V. mimicus lineage after the lineage evolved from a progenitor of V. mimicus/V. cholerae (Figure 2). These iterations are supported by strong bootstrap support calculations. A close evolutionary relationship for Vibrio sp. RC586 and V. mimicus is also supported by shorter evolutionary distances between the Vibrio sp. RC586 and V. mimicus strains (see Additional files 8 and 9). The evolutionary distance of all genomes used in this study from V. cholerae BX 330286, a putative progeny of the progenitor of the 7th pandemic clade [17,24], is shown in Additional file 10.
Virulence Factors
Both Vibrio sp. RC586 and Vibrio sp. RC341 genomes encode several virulence factors found in toxigenic and non-toxigenic V. cholerae and V. mimicus. These include the toxR/toxS virulence regulators, multiple hemolysins and lipases, VSP-I and II, and a type 6 secretion system. Both VSP islands are also present in pathogenic strains of the seventh pandemic clade [25]. Although neither genome encodes CTXΦ phage, the major virulence factor encoding the cholera toxin (CT) that is responsible for the profuse secretory diarrhea caused by toxigenic V. cholerae and V. mimicus, both genomes do have homologous sequences of the chromosomal attachment site for this phage. Although these genomes do not encode TcpA, the outer membrane protein that CTXΦ attaches to during its infection cycle and ToxT, involved in CTXΦ replication and activation, they do encode several other mechanisms necessary for the complete CTXΦ life cycle and both CT production and translocation, including TolQRA, inner membrane proteins involved in CTXΦ attachment to the cell, XerCD tyrosine recombinases, which catalyze recombination between CTXΦ and the host genome, LexA, involved in CTXΦ expression, and EspD, involved in the secretion of the CTXΦ virion and CT translocation into the extracellular environment.
Neither Vibrio sp. RC341 nor Vibrio sp. RC586 encode VPI-1 or VPI-2, but Vibrio sp. RC341 encodes one copy of both VSP-I (VCJ_003466-VCJ_003480) and VSP-II (VCJ_000310 to VCJ_000324) and Vibrio sp. RC586 encodes one copy of VSP-I (VOA_002906-VOA_002918). However, neither of these strains encodes complete VSP islands, but rather variants of canonical VSP islands. Incomplete VSP islands have been frequently found in environmental V. cholerae and V. mimicus isolates [26] [Taviani et al, unpublished].
The toxR/toxS virulence regulators, hemolysins, lipases, and type 6 secretion system are present in all pathogenic and non-pathogenic strains of V. cholerae and both VSP islands are present in pathogenic strains of the seventh pandemic. Presence of these virulence factors in V. cholerae genomes sequenced to date, as well as their divergence consistent with the conserved core of Vibrio sp. RC341 and Vibrio sp. RC586, suggests that they comprise a portion of the backbone of many Vibrio species. Their widespread occurrence suggests the ability of all vibrios to be potential pathogens, but more likely, these factors have an important role in their ecology.
Natural Competence
Analysis of the 22 V. cholerae genomes that have been sequenced revealed the presence of type IV pili genes, involved in natural transformation of Haemophilus spp. and Neisseria spp. and other competent Bacteria [27,28]. Vibrio sp. RC341 and Vibrio sp. RC586 also encode this system. Moreover, both species encode all 33 ORFs described by Meibom et al. [29,30] that comprise the chitin utilization program for induction of natural competence. The presence of these systems in the two new species and in V. cholerae indicates natural competence is widely employed by vibrios to incorporate novel DNA into their genomes and, thereby, enhance both adaption to new environments and in evolution. Furthermore, the well-established association of these bacteria with chitinous organisms and with high densities in biofilms [31] supports the notion that natural competence and horizontal gene transfer are both highly expressed and common in vibrios.
Genomic Islands and Integration Loci for Exogenous DNA
Analysis of 23 complete and draft V. cholerae genomes by Chun et al. [17] showed 73 putative genomic islands to be present. By pairwise reciprocal comparison, the genomes of Vibrio sp. RC341 and Vibrio sp. RC586 are concluded to encode several of these genomic islands, as well as many of the insertion loci of V. cholerae genomic islands [17], indicating extensive horizontal transfer of genomic islands. V. cholerae insertion loci are not specific to individual genomic islands, but can act as integration sites for a variety of islands [17]. Vibrio sp. RC586 contains 33 putative GI insertion loci and Vibrio sp. RC341 contains 40 that are homologous to those found in V. cholerae. In addition to having highly similar attachment sequences and insertion loci, as found in V. cholerae, most of the homologous tRNA sequences between Vibrio sp. RC341, Vibrio sp. RC586, and V. cholerae are identical. However, three glutamine-tRNA and one aspartate-tRNA sequence of Vibrio sp. RC586 and four glutamine-tRNA and four aspartate-tRNA sequences of Vibrio sp. RC341 show between 99 and 97% similarity with homologous V. cholerae tRNA sequences. These sites serve as integration loci for many pathogenicity islands. Interestingly, all tRNA-Ser, the loci most commonly targeted by island encoded integrases of mobile elements in V. cholerae [32], were 100% similar between all strains. This high similarity of platforms serving to insert exogenous DNA suggests that the same or highly similar genomic islands are readily shared. Sequences that are characteristic of GIs and islets with homologous V. cholerae insertion loci and putative function and annotations are described in Additional files 11, 12, and 13.
Vibrio sp. RC586 encodes eighteen sequences that are characteristic of genomic islands and islets that are also found in V. cholerae (see Additional file 12). Of these, VSP-I, islet-2 and GIs-2, -4, -33, -34, -35, -41, -62, -64, -73, and Vibrio sp. RC586-GI-1 are located on the large chromosome and islets-3 and 4, and GIs-9, -10, -20, and -61 are located on the small chromosome (see Additional file 12). The VSP-I island is located at the homologous insertion locus for VSP-I (VOA_002906-VOA_002918) in V. cholerae strains, but is a variant of the canonical island having a deletion in VC0175 (deoxycytidylate deaminase-related protein) and 90% sequence similarity to the canonical island.
Vibrio sp. RC586 also encodes five sequences with homology to the CTXΦ attachment site, with four of them being tandemly arranged on the putative large chromosome (VOA_000105-VOA_000126). At these loci are four elements with high similarity (82 and 81% AAI) to the RS1Φ phage-like elements (rstA1 and rstB1) of V. cholerae SCE264 [33] and 97 to 100% nucleotide identity to the RS1Φ-like elements in V. cholerae TMA21, TM11079-80, VL426, and 623-39, reported by Chun et al. [17] to be GI-33 (Figure 3). RS1Φ is a satellite phage related to CTXΦ and assists in integration and replication of the CTXΦ [34,35]. However, these V. cholerae strains were either CTXΦ-negative or encode a CTXΦ on the other chromosome, while encoding sequences with high similarity to rstA, and rstB of RS1Φ, RS1-type sequences [33]. Immediately upstream of the rstA1-like sequence is an hypothetical protein and immediately downstream of this rstB1-like sequence is an hypothetical protein with 52% identity with that of Colwellia psychrerythraea 34H, and a sequence with 99% similarity to an end-repeat (ER) region and an intergenic region (ig) of CTXΦ (Figure 3). This region may represent a novel phage containing ORFs with similarity to the RS1Φ satellite phage and ER and ig-1 regions with high similarity to CTXΦ. Absence of an integrase in this region suggests it may integrate into the genome via XerCD tyrosine recombinases, as does CTXΦ. All putative genomic islands shared by V. cholerae and Vibrio sp. RC586 are listed in Additional file 12.
Figure 3.
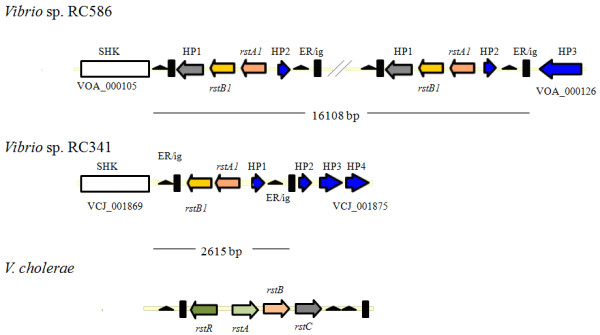
RS1Φ-like elements located at CTXΦ attachment sites on the large chromosomes of Vibrio sp. RC586 and Vibrio sp. RC341 and the canonical RS1Φ of V. cholerae. SHK = sensor histidine kinase, HP = hypothetical protein, ER = end repeat, ig = intergenic region.
Vibrio sp. RC341 putatively encodes 14 sequences that are characteristic of genomic islands and islets that are also found in V. cholerae (see Additional file 11). VSP-I and -II and GIs-1 to 4, 33, and islets-1 to 5 are located on the large chromosome, while GI-9 and 10 are located on the small chromosome (see Additional file 11). These GIs were described by Chun et al. [17] and two are single copies of VSP-I (VCJ_003466 to VCJ_003480) and VSP-II (VCJ_000310 to VCJ_000324). Neither of the VSP islands was present in their entirety, compared to 7th pandemic V. cholerae strains. Similar to the VSP-I variant in Vibrio sp. RC586, the variant in Vibrio sp. RC341 has a deletion of VC0175. Also, ORFs VCJ_003468 to VCJ_003470 are annotated as phage integrase, transposase, and phage integrase, respectively. The homologous ORFs of this VSP-I variant have a 92% sequence similarity to the canonical VSP-I island. Interestingly, VSP-II variant of Vibrio sp. RC341 contains a 10 kb putative phage encoding a type 1 restriction modification system, has a %GC of ca. 38%, and is located at the homologous insertion locus of GI-56 in V. cholerae (tRNA-Met) (Figure 4). This phage shares significant similarity with V. vulnificus YJ016 phage (94% query coverage and 98% sequence similarity). Several variants of VSP-II are encoded in multiple strains of V. cholerae [E. Taviani, unpublished]. However, the variant encoded in Vibrio sp. RC341 is, to date, unique.
Figure 4.
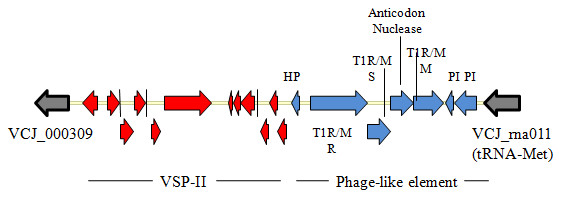
Novel VSP-II variant found in Vibrio sp. RC341. Red arrows represent VSP-II ORFs and blues arrows represent the novel phage-like region in the 3' region of the sequence. Grey arrows represent the adjacent flanking sequences. T1R/M = type I restriction modification system. PI = phage integrase.
Interestingly, Vibrio sp. RC341 encodes V. cholerae GI-33, a ca. 2615 bp region, (VCJ_001870 to VCJ_001874) similar to RS1Φ-like phage in Vibrio sp. RC586, V. cholerae strains VL426, SCE264, TMA21, TM11079-80, and 623-39, showing 93 to 96% nucleotide sequence similarity across 67 to 79% of the phage (Figure 3). This region in Vibrio sp. RC341 encodes only the rstA1 and rstB1 and the 3' hypothetical protein flanked by CTXΦ-like end repeats and an intergenic region, inserted at the homologous CTXΦ attachment site on chromosome I (Figure 3). Analysis of this and similar phages inserting at this locus suggests an extremely high diversity of vibriophages in both structure and sequence in the environment. Putative genomic islands shared by V. cholerae and Vibrio sp. RC341 are listed in Additional file 11.
Horizontal Gene Transfer of Genomic Islands
Homologous genomic islands typically showed higher ANI between strains than the conserved backbone regions of these genomes, an indication of recent transfer of these islands among the same and different species. All GIs shared by Vibrio sp. RC586 and V. cholerae strains were 87 to 100% ANI%, with the exception of two GIs with 77% (GI-9) and 82% (GI-62) ANI (see Additional files 12 and 13). All GIs among Vibrio sp. RC341 and V. cholerae had 87 to 99% ANI, excluding three GIs with 81 to 82% (GIs-3, 9, and 2), and two with and 85% (GI-1, Vibrio sp. RC341 islets -1 and -2) (see Additional files 11 and 13).
Phylogenetic analysis using homologous ORFs of the genomic islands yielded evidence of recent lateral transfer of VSP-I, and GIs-2, 41, and 61 among V. cholerae and Vibrio sp. RC586. In all cases, phylogenies inferred by the ORFs were incongruent with species phylogeny, suggesting the elements were transferred after the species diverged (see Additional files 14, 15, 16, 17, and 18). Using the same methods, we found evidence of recent lateral transfer of VSP-I, GI-4, and islet-3, between V. cholerae and Vibrio sp. RC341. In all cases, phylogenies inferred by the ORFs were incongruent with species phylogeny (see Additional files 16, 17, and 19). Our data suggests that V. cholerae VL426 (V. cholerae biotype albensis) received a VSP-I similar to that of Vibrio sp. RC341 and Vibrio sp. RC586 via horizontal gene transfer. We also found evidence of horizontal transfer of V. cholerae GI-2 from V. cholerae to Vibrio sp. RC586, as well as Vibrio sp. RC341 Islet-3 and V. cholerae GI-4 from Vibrio sp. RC341 to V. cholerae strains.
VSP-II, islets-2, -4, -5, and GIs-1, -2, -3, -9, -10, all present in at least one V. cholerae genome and in Vibrio sp. RC341, showed no evidence of horizontal gene transfer. Most likely there are many undescribed variants of these elements, in both structure and nucleotide sequence, yet to be found in the natural environment, with certain variants more frequently transferred among strains of the same species. Coevolution of the island and host genome over time no doubt occurs. In any case, based on the data reported here V. cholerae is not alone in propagating these elements. They surely cycle among different but closely related species in the environment.
Unique Genomic Islands
Vibrio sp. RC586 putatively encodes five unique genomic islands and islets not yet reported for V. cholerae (see Additional files 12 and 13). Vibrio sp. RC586 GI-2 and islet-5 encode phage-like elements. Interestingly, islet-5 is annotated as probable coat protein A precursor, with similarity to bacteriophage f237 ORF5 of V. campbellii and zona occludens toxin (zot), with high similarity to V. parahaemolyticus and V. harveyi zot (VOA_001598-VOA_001600). This phage-like element is inserted at the homologous locus for V. cholerae O1 Classical CTXΦ insertion (VCA0569-VCA0570). Vibrio sp. RC586 GI-4 encodes sequences homologous to the Tn7 transposition tnsABCDE, a transposon known to integrate into phylogenetically diverse organisms and form genomic islands [36]. Vibrio sp. RC586 GIs-1, -3, -4, and islets-1 through 6 all share homologous insertion loci with previously described V. cholerae GIs (see Additional file 12).
Vibrio sp. RC341 encodes six putative unique genomic islands not reported before (see Additional files 11 and 13). Vibrio sp. RC341 GIs-1, 2, 3, 4, and 7 all encode phage-like/related elements. Vibrio sp. RC341 GI-4 and 7 both encode several transposases and a sequence with homology to an insertion-like sequence in the V. parahaemolyticus insertion sequence element ISV-3L. Vibrio sp. RC341 GI-6 (VCJ_002614 to VCJ002618), ca. 4962 bp region of hypothetical proteins and transposases, is inserted at the homologous locus for V. cholerae O1 Classical CTXΦ, a locus shown to harbor a variety of GIs and phages [17] (see Additional file 11).
Conclusions
The genomes of two new Vibrio species previously characterized as variant V. cholerae, have been sequenced and their sequences used to describe their interesting and important features. The genomes of both species reveal significant nucleotide sequence divergence (12 to 15%) from each other and from V. cholerae and V. mimicus genomes, supporting the conclusion that both represent unique species not described before. Moreover, genes conserved among V. cholerae, V. mimicus, and the two new species varied sufficiently to suggest ancient speciation via genetic drift of the ancestral core genomic backbone. Furthermore, results of our analyses suggest Vibrio sp. RC341 to have evolved from a progenitor of V. cholerae and V. mimicus, whereas Vibrio sp. RC586 is concluded to have evolved from an early V. mimicus clade. Although the ANI of all genomes analyzed in this study demonstrates divergence, putative genomic islands were found to cross species boundaries, often at an higher ANI than the conserved backbone. These data, coupled with phylogenetic analyses, point to lateral transfer of the islands and phages among V. cholerae, V. mimicus, Vibrio sp. RC341, and Vibrio sp. RC586 in the natural environment. Furthermore, homologous GI insertion loci were present in both new species and in the case of V. cholerae, these insertion loci were not GI-specific. The pool of DNA laterally transferred between and among members of the Vibrionaceae strongly suggests that near-neighbors of V. cholerae act as reservoirs of transferable genetic elements and virulence in the environment and that V. cholerae is not alone in propagating these elements therein. Results of this study also demonstrate a widespread allelic variation in these elements and evidence of evolution of mobile genetic elements, including pathogenicity islands, through a multistep mosaic recombination with other elements, including phage. The ability of vibrios to incorporate exogenous DNA at several loci that encode a large combination of GIs, thereby, allows optimization of the genome for success in a specific niche or wider ecology in the natural environment.
Methods
Genome sequencing
Draft sequences were obtained from a blend of Sanger and 454 sequences and involved paired end Sanger sequencing on 8 kb plasmid libraries to 5× coverage, 20× coverage of 454 data, and optional paired end Sanger sequencing on 35 kb fosmid libraries to 1-2× coverage (depending on repeat complexity). To finish the genomes, a collection of custom software and targeted reaction types were used. In addition to targeted sequencing strategies, Solexa data in an untargeted strategy were used to improve low quality regions and to assist gap closure. Repeat resolution was performed using in house custom software [37]. Targeted finishing reactions included transposon bombs [38], primer walks on clones, primer walks on PCR products, and adapter PCR reactions. Gene-finding and annotation were achieved using an automated annotation server [39]. The genomes of these organisms have been deposited in the NCBI Genbank database (accession nos. NZ_ACZT00000000 and NZ_ADBD00000000).
Comparative genomics
Genome to genome comparison was performed using three approaches, since completeness and quality of nucleotide sequences varied from strain to strain in the set examined in this study. Firstly, nucleotide sequences, as whole contigs were directly aligned using the MUMmer program [16]. Secondly, ORFs of a given pair of genomes were reciprocally compared each other, using the BLASTN, BLASTP and TBLASTX programs (ORF-dependent comparison). Thirdly, a bioinformatic pipeline was developed to identify homologous regions of a given query ORF. Initially, a segment on a target contig homologous to a query ORF was identified using the BLASTN program. This potentially homologous region was expanded in both directions by 2,000 bp, after which, nucleotide sequences of the query ORF and selected target homologous region were aligned using a pairwise global alignment algorithm [40]. The resultant matched region in the subject contig was extracted and saved as a homolog (ORF-independent comparison). Orthologs and paralogs were differentiated by reciprocal comparison. In most cases, both ORF-dependent and -independent comparisons yielded the same orthologs, though the ORF-independent method performed better for draft sequences of low quality, in which sequencing errors, albeit rare, hampered identification of correct ORFs.
To determine average nucleotide (ANI) and average amino acid identities (AAI) for the purpose of assigning genetic distances between strains and strains to species groups, a recripocal best match BLASTN analysis was performed for each genome. The average similarity between genomes was measured as the average nucleotide identity (ANI) and average amino acid identity (AAI) of all conserved protein-coding genes, following the methods of Konstantinidis and Tiedje [41]. By this method, AAI>95% and ANI>94% with >85% of protein-coding genes conserved between the pair of genomes, is judged to correspond to strains of the same species, whereas AAI<95% and ANI <94% and <85% conservation of protein-coding genes indicate different species. Dinucleotide relative abundances were determined for each genome used in this analysis. Genomic dissimilarities between genomes were determined following the methods of Karlin et al. [42]. A multi-locus sequence analysis (MLSA) was determined following standard methods for the Vibrionaceae [21]. Data for the MLSA were reported as percent similarity between concatenated homologous ORFs for the genomes which encoded these ORFs. These criteria were applied to results of the analyses employed in this study.
Identification and annotation of genomic islands
Putative genomic islands (GIs) were defined as a continuous array of five or more ORFs discontinuously distributed among genomes of test strains following the methods of Chun et al [17]. Correct transfer or insertion of GIs was differentiated from deletion events by comparing genome-based phylogenetic trees and complete matrices of pairwise orthologous genes between test strains. Identified GIs were designated, and annotated using the BLASTP search of its member ORFs against the Genbank nr database. Arrays of continuous unique ORFs annotated as encoding phage-related elements and/or transposases were also identified as putative genomic islands. Genomic islets were identified as regions less than 5 ORFs and flanked by genomic island insertion loci [17]. Putative genomic islands were also investigated using the web-based application IslandViewer [43].
Phylogenetic analyses employing genome sequences
A set of orthologues for each ORF of V. cholerae N16961 was obtained for different sets of strains, and individually aligned using the CLUSTALW2 program [44]. The resultant multiple alignments were concatenated to generate genome scale alignments that were subsequently used to reconstruct the neighbor-joining phylogenetic tree [45]. The evolutionary model of Kimura was used to generate the distance matrix [46]. The MEGA program was used for phylogenetic analysis [47].
Authors' contributions
J.C., A.H. and R.R.C. designed research; T.S.B., D.C.B., J.C.D., C.S.H., N.A.H. performed research; J.C., C.J.G., N.A.H., B.J.H., and S.Y.C. analyzed data; B.J.H. and R.R.C. wrote the paper. All authors have read and approved the manuscript.
Supplementary Material
Vibrio strains used in the comparative genomics utilized in this study. Species, strain ID, serogroup/serotype and biotype (where available), geographical location and source of isolation and year of isolation are listed in this table. NCBI Genbank accession numbers are listed in the right column.
MUMmer plot of Vibrio sp. RC586 as query and V. cholerae N16961 as reference. Vibrio sp. RC586 contigs are on Y-axis and V. cholerae N16961 chromosomes are on X-axis. V. cholerae N16961 chromosome I begins at XY-intercept and chromosome II is located on the right section of the X-axis.
MUMmer plot of Vibrio sp. RC341 as query and V. cholerae N16961 as reference. Vibrio sp. RC341 contigs are on Y-axis and V. cholerae N16961 chromosomes are on X-axis. V. cholerae N16961 chromosome I begins at XY-intercept and chromosome II is located on the right section of the X-axis.
Average nucleotide identity analysis of Vibrio sp. RC341. Average nucleotide identity (ANI%) between Vibrio sp. RC341 and Vibrio genomes used in this study.
Average nucleotide identity analysis of Vibrio sp. RC586. Average nucleotide identity (ANI%) between Vibrio sp. RC586 and Vibrio genomes used in this study.
Evolutionary distance analysis of Vibrio sp. RC341. Evolutionary distance of strains used in this study from Vibrio sp. RC341 as determined by ANI between Vibrio sp. RC341 and all strains used in this study.
Evolutionary distance analysis of Vibrio sp. RC586. Evolutionary distance of strains used in this study from Vibrio sp. RC586 as determined by ANI between Vibrio sp. RC586 and all strains used in this study.
Evolutionary distance analysis of V. mimicus MB451. Evolutionary distance of Vibrio sp. RC586 and Vibrio sp. RC341 from V. mimicus MB451 as determined by ANI between V. mimicus MB451 and all strains used in this study.
Evolutionary distance analysis of V. cholerae BX 330286. Evolutionary distance of Vibrio sp. RC586 and Vibrio sp. RC341 from strains V. cholerae BX 330286 as determined by ANI between V. cholerae BX 330286 and all strains used in this study.
Putative genomic islands of Vibrio sp. RC341. Putative genomic islands of Vibrio sp. RC341, showing insertion loci, homologous flanking loci in V. cholerae N16961, %GC, other carrier strains used in this study, ANI with homologous islands, δ*, direction of transfer, islands sharing same insertion loci, and annotation.
Putative genomic islands of Vibrio sp. RC586. Putative genomic islands of Vibrio sp. RC586, showing insertion loci, homologous flanking loci in V. cholerae N16961, %GC, other carrier strains used in this study, ANI with homologous islands, δ*, direction of transfer, islands sharing same insertion loci, and annotation.
Phylogeny of the genomic island GI-2. Phylogeny of the genomic island GI-2 as determined by reconstructing a neighbor-joining tree using the Kimura-2 parameter as a nucleotide substitution model.
Phylogeny of the genomic island GI-41. Phylogeny of the genomic island GI-41 as determined by reconstructing a neighbor-joining tree using the Kimura-2 parameter as a nucleotide substitution model.
Phylogeny of the genomic island GI-4. Phylogeny of the genomic island GI-4 as determined by reconstructing a neighbor-joining tree using the Kimura-2 parameter as a nucleotide substitution model.
Phylogeny of VSP-I. Phylogeny of the genomic island VSP-I as determined by reconstructing a neighbor-joining tree using the Kimura-2 parameter as a nucleotide substitution model.
Phylogeny of the genomic island GI-61. Phylogeny of the genomic island GI-61 as determined by reconstructing a neighbor-joining tree using the Kimura-2 parameter as a nucleotide substitution model.
Phylogeny of Vibrio sp. RC341 Islet-3. Phylogeny of Vibrio sp. RC341 Islet-3 as determined by reconstructing a neighbor-joining tree using the Kimura-2 parameter as a nucleotide substitution model.
Contributor Information
Bradd J Haley, Email: bhaley@umd.edu.
Christopher J Grim, Email: grim@umd.edu.
Nur A Hasan, Email: nur_a_hasan@yahoo.co.uk.
Seon-Young Choi, Email: sychoi@umd.edu.
Jongsik Chun, Email: jchun@snu.ac.kr.
Thomas S Brettin, Email: brettin@lanl.gov.
David C Bruce, Email: dbruce@lanl.gov.
Jean F Challacombe, Email: jchalla@lanl.gov.
J Chris Detter, Email: cdetter@lanl.gov.
Cliff S Han, Email: han_cliff@lanl.gov.
Anwar Huq, Email: huqanwar@gmail.com.
Rita R Colwell, Email: rcolwell@umiacs.umd.edu.
Acknowledgements
This work was supported in part by Korea Science and Engineering Foundation National Research Laboratory Program Grant R0A-2005-000-10110-0, National Institutes of Health Grant 1RO1A139129-01; National Oceanic and Atmospheric Administration, Oceans and Human Health Initiative Grant S0660009; Department of Homeland Security Grant NBCH2070002; Intelligence Community Post-Doctoral Fellowship Program; and funding for genome sequencing was provided by the Office of the Chief Scientist and National Institute of Allergy and Infectious Diseases Microbial Sequencing Centers Grants N01-AI-30001 and N01-AI-40001.
References
- Pacha RE, Kiehn ED. Characterization and relatedness of marine vibrios pathogenic to fish: physiology, serology, and epidemiology. Journal of Bacteriology. 1969;100(3):1242–1247. doi: 10.1128/jb.100.3.1242-1247.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushmaro A, Banin E, Loya Y, Stackebrandt E, Rosenberg E. Vibrio shiloi sp. nov., the causative agent of bleaching of the coral Oculina patagonica. Int J Syst Evol Microbiol. 2001;51:1383–1388. doi: 10.1099/00207713-51-4-1383. [DOI] [PubMed] [Google Scholar]
- Guerinot ML, West PA, Lee JV, Colwell RR. Vibrio diazotrophicus sp. nov., a marine nitrogen-fixing bacterium. International Journal of Systematic and Evolutionary Microbiology. 1982;32(3):350–357. [Google Scholar]
- Hada HS, West PA, Lee JV, Stemmler J, Colwell RR. Vibrio tubiashii sp. nov., a pathogen of bivalve mollusks. International Journal of Systematic and Evolutionary Microbiology. 1984;34(1):1–4. [Google Scholar]
- Hedlund BP, Staley JT. Vibrio cyclotrophicus sp. nov., a polycyclic aromatic hydrocarbon (PAH)-degrading marine bacterium. Int J Syst Evol Microbiol. 2001;51:61–66. doi: 10.1099/00207713-51-1-61. [DOI] [PubMed] [Google Scholar]
- Thompson CCVA, Souza RC, Vasconcelos ATR, Vesth T, Alves N, Ussery DW, Iida T, Thompson FL. Vibrio2009. Rio de Janeiro, Brasil; 2009. Genomic Taxonomy of the Vibrios. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson FL, Iida T, Swings J. Biodiversity of vibrios. Microbiol Mol Biol Rev. 2004;68(3):403–431. doi: 10.1128/MMBR.68.3.403-431.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huq A, Small E, West P, Huq M, Rahman R, Colwell R. Ecological relationship between Vibrio cholerae and planktonic copepods. Appl Environ Microbiol. 1983;45:275–283. doi: 10.1128/aem.45.1.275-283.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair GB, Oku Y, Takeda Y, Ghosh A, Ghosh RK, Chattopadhyay S, Pal SC, Kaper JB, Takeda T. Toxin profiles of Vibrio cholerae non-O1 from environmental sources in Calcutta, India. Appl Environ Microbiol. 1988;54(12):3180–3182. doi: 10.1128/aem.54.12.3180-3182.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BR, Fanning GR, Madden JM, Steigerwalt AG, Bradford HB Jr, Smith HL Jr, Brenner DJ. Characterization of biochemically atypical Vibrio cholerae strains and designation of a new pathogenic species, Vibrio mimicus. J Clin Microbiol. 1981;14(6):631–639. doi: 10.1128/jcm.14.6.631-639.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda S, Nakagawa T, Shi L, Bi K, Kanoh Y, Tomochika K, Miyoshi S, Shimada T. Distribution of virulence-associated genes in Vibrio mimicus isolates from clinical and environmental origins. Microbiol Immunol. 2004;48(7):547–551. doi: 10.1111/j.1348-0421.2004.tb03551.x. [DOI] [PubMed] [Google Scholar]
- Boyd EF, Moyer KE, Shi L, Waldor MK. Infectious CTXΦ and the Vibrio pathogenicity island prophage in Vibrio mimicus: evidence for recent horizontal transfer between V. mimicus and V. cholerae. Infection and Immunity. 2000;68(3):1507–1513. doi: 10.1128/IAI.68.3.1507-1513.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson FL, Swings J. In: Biology of the Vibrios. Thompson FL, Austin B, Swings J, editor. Washington, D.C: ASM Press; 2006. Taxonomy of the Vibrios; pp. 29–43. [Google Scholar]
- Choopun N. PhD Thesis. University of Maryland, College Park, Marine Estuarine and Environmental Science; 2004. The population structure of Vibrio cholerae in Chesapeake Bay. [Google Scholar]
- Zo YG. PhD Thesis. University of Maryland, College Park, Marine Estuarine and Environmental Science; 2005. Phylogenomic and structural analyses of Vibrio cholerae populations and endemic cholera. [Google Scholar]
- Kurtz S, Phillippy A, Delcher A, Smoot M, Shumway M, Antonescu C, Salzberg S. Versatile and open software for comparing large genomes. Genome biology. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun J, Grim CJ, Hasan NA, Lee JH, Choi SY, Haley BJ, Taviani E, Jeon YS, Kim DW. Comparative genomics reveals mechanism for short-term and long-term clonal transitions in pandemic Vibrio cholerae. Proceedings of the National Academy of Sciences. 2009;106(36):15442–15447. doi: 10.1073/pnas.0907787106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT, Tiedje JM. Genomic insights that advance the species definition for prokaryotes. Proceedings of the National Academy of Sciences. 2005;102(7):2567–2572. doi: 10.1073/pnas.0409727102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT, Ramette A, Tiedje JM. The bacterial species definition in the genomic era. Philosophical Transactions B. 2006;361(1475):1929–1940. doi: 10.1098/rstb.2006.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinidis KT, Tiedje JM. Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Current opinion in microbiology. 2007;10(5):504–509. doi: 10.1016/j.mib.2007.08.006. [DOI] [PubMed] [Google Scholar]
- Thompson CC, Vicente ACP, Souza RC, Vasconcelos ATR, Vesth T, Alves N, Ussery DW, Iida T, Thompson FL. Genomic taxonomy of vibrios. BMC Evolutionary Biology. 2009;9(1):258–273. doi: 10.1186/1471-2148-9-258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlaere E, Baldwin A, Gevers D, Henry D, De Brandt E, LiPuma JJ, Mahenthiralingam E, Speert DP, Dowson C, Vandamme P. Taxon K, a complex within the Burkholderia cepacia complex, comprises at least two novel species, Burkholderia contaminans sp. nov. and Burkholderia lata sp. nov. International Journal of Systematic and Evolutionary Microbiology. 2009;59(1):102–111. doi: 10.1099/ijs.0.001123-0. [DOI] [PubMed] [Google Scholar]
- Adekambi T, Shinnick TM, Raoult D, Drancourt M. Complete rpoB gene sequencing as a suitable supplement to DNA-DNA hybridization for bacterial species and genus delineation. International Journal of Systematic and Evolutionary Microbiology. 2008;58(8):1807–1814. doi: 10.1099/ijs.0.65440-0. [DOI] [PubMed] [Google Scholar]
- Haley BJ, Grim CJ, Hasan NA, Taviani E, Chun J, Brettin TS, Bruce DC, Challacombe JF, Detter JC, Han CS. The pre-seventh pandemic Vibrio cholerae BX 330286 El Tor genome: evidence for the environment as a genome reservoir. Environmental Microbiology Reports. 2010;2(1):208–216. doi: 10.1111/j.1758-2229.2010.00141.x. [DOI] [PubMed] [Google Scholar]
- Dziejman M, Balon E, Boyd D, Fraser CM, Heidelberg JF, Mekalanos JJ. Comparative genomic analysis of Vibrio cholerae: genes that correlate with cholera endemic and pandemic disease. Proc Natl Acad Sci USA. 2002;99(3):1556–1561. doi: 10.1073/pnas.042667999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grim CJ, Choi J, Chun J, Jeon YS, Taviani E, Hasan NA, Haley B, Huq A, Colwell RR. Occurrence of the Vibrio cholerae Seventh Pandemic VSP-I Island and a New Variant. OMICS: A Journal of Integrative Biology. 2010;14(1):1–7. doi: 10.1089/omi.2009.0087. [DOI] [PubMed] [Google Scholar]
- Barnhart BJ, Herriott RM. Penetration of deoxyribonucleic acid into Haemophilus influenzae. Biochimica et Biophysica Acta. 1963;76:25–39. doi: 10.1016/0006-3002(63)90004-0. [DOI] [PubMed] [Google Scholar]
- Wolfgang M, Lauer P, Park HS, Brossay L, Hebert J, Koomey M. PilT mutations lead to simultaneous defects in competence for natural transformation and twitching motility in piliated Neisseria gonorrhoeae. Molecular Microbiology. 1998;29(1):321–330. doi: 10.1046/j.1365-2958.1998.00935.x. [DOI] [PubMed] [Google Scholar]
- Meibom KL, Li XB, Nielsen AT, Wu CY, Roseman S, Schoolnik GK. The Vibrio cholerae chitin utilization program. Proc Natl Acad Sci USA. 2004;101(8):2524–2529. doi: 10.1073/pnas.0308707101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meibom KL, Blokesch M, Dolganov NA, Wu CY, Schoolnik GK. Chitin induces natural competence in Vibrio cholerae. Science. 2005;310(5755):1824–1827. doi: 10.1126/science.1120096. [DOI] [PubMed] [Google Scholar]
- Pruzzo C, Vezzulli L, Colwell RR. Global impact of Vibrio cholerae interactions with chitin. Environ Microbiol. 2008;10(6):1400–1410. doi: 10.1111/j.1462-2920.2007.01559.x. [DOI] [PubMed] [Google Scholar]
- Boyd EF, Almagro-Moreno S, Parent MA. Genomic islands are dynamic, ancient integrative elements in bacterial evolution. Trends in Microbiology. 2009;17(2):47–53. doi: 10.1016/j.tim.2008.11.003. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay AK, Chakraborty S, Takeda Y, Nair GB, Berg DE. Characterization of VPI pathogenicity island and CTXΦ prophage in environmental strains of Vibrio cholerae. J Bacteriol. 2001;183(16):4737–4746. doi: 10.1128/JB.183.16.4737-4746.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis BM, Waldor MK. Filamentous phages linked to virulence of Vibrio cholerae. Curr Opin Microbiol. 2003;6(1):35–42. doi: 10.1016/S1369-5274(02)00005-X. [DOI] [PubMed] [Google Scholar]
- Faruque SM, Asadulghani, Kamruzzaman M, Nandi RK, Ghosh AN, Nair GB, Mekalanos JJ, Sack DA. RS1 element of Vibrio cholerae can propagate horizontally as a filamentous phage exploiting the morphogenesis genes of CTXΦ. Infect Immun. 2002;70(1):163–170. doi: 10.1128/IAI.70.1.163-170.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks AR, Peters JE. Tn7 elements: Engendering diversity from chromosomes to episomes. Plasmid. 2009;61(1):1–14. doi: 10.1016/j.plasmid.2008.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han C, Chain P. Finishing repetitive regions automatically with Dupfinisher. 2006: Citeseer. 2006. pp. 142–147.
- Goryshin IY, Reznikoff WS. Tn5 in vitro transposition. J Biol Chem. 1998;273(13):7367–7374. doi: 10.1074/jbc.273.13.7367. [DOI] [PubMed] [Google Scholar]
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9(1):75–90. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers EW, Miller W. Optimal alignments in linear space. Comput Appl Biosci. 1988;4(1):11–17. doi: 10.1093/bioinformatics/4.1.11. [DOI] [PubMed] [Google Scholar]
- Konstantinidis KT, Tiedje JM. Towards a Genome-Based Taxonomy for Prokaryotes. J Bacteriol. 2005;187(18):6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlin S, Mrazek J, Campbell AM. Compositional biases of bacterial genomes and evolutionary implications. Journal of Bacteriology. 1997;179(12):3899–3913. doi: 10.1128/jb.179.12.3899-3913.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langille MGI, Brinkman FSL. IslandViewer: an integrated interface for computational identification and visualization of genomic islands. Bioinformatics. 2009;25(5):664–665. doi: 10.1093/bioinformatics/btp030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23(21):2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Molecular Biology and Evolution. 1987;4(4):406–425. doi: 10.1093/oxfordjournals.molbev.a040454. [DOI] [PubMed] [Google Scholar]
- Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. Journal of molecular evolution. 1980;16(2):111–120. doi: 10.1007/BF01731581. [DOI] [PubMed] [Google Scholar]
- Kumar S, Nei M, Dudley J, Tamura K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief Bioinform. 2008;9(4):299–306. doi: 10.1093/bib/bbn017. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Vibrio strains used in the comparative genomics utilized in this study. Species, strain ID, serogroup/serotype and biotype (where available), geographical location and source of isolation and year of isolation are listed in this table. NCBI Genbank accession numbers are listed in the right column.
MUMmer plot of Vibrio sp. RC586 as query and V. cholerae N16961 as reference. Vibrio sp. RC586 contigs are on Y-axis and V. cholerae N16961 chromosomes are on X-axis. V. cholerae N16961 chromosome I begins at XY-intercept and chromosome II is located on the right section of the X-axis.
MUMmer plot of Vibrio sp. RC341 as query and V. cholerae N16961 as reference. Vibrio sp. RC341 contigs are on Y-axis and V. cholerae N16961 chromosomes are on X-axis. V. cholerae N16961 chromosome I begins at XY-intercept and chromosome II is located on the right section of the X-axis.
Average nucleotide identity analysis of Vibrio sp. RC341. Average nucleotide identity (ANI%) between Vibrio sp. RC341 and Vibrio genomes used in this study.
Average nucleotide identity analysis of Vibrio sp. RC586. Average nucleotide identity (ANI%) between Vibrio sp. RC586 and Vibrio genomes used in this study.
Evolutionary distance analysis of Vibrio sp. RC341. Evolutionary distance of strains used in this study from Vibrio sp. RC341 as determined by ANI between Vibrio sp. RC341 and all strains used in this study.
Evolutionary distance analysis of Vibrio sp. RC586. Evolutionary distance of strains used in this study from Vibrio sp. RC586 as determined by ANI between Vibrio sp. RC586 and all strains used in this study.
Evolutionary distance analysis of V. mimicus MB451. Evolutionary distance of Vibrio sp. RC586 and Vibrio sp. RC341 from V. mimicus MB451 as determined by ANI between V. mimicus MB451 and all strains used in this study.
Evolutionary distance analysis of V. cholerae BX 330286. Evolutionary distance of Vibrio sp. RC586 and Vibrio sp. RC341 from strains V. cholerae BX 330286 as determined by ANI between V. cholerae BX 330286 and all strains used in this study.
Putative genomic islands of Vibrio sp. RC341. Putative genomic islands of Vibrio sp. RC341, showing insertion loci, homologous flanking loci in V. cholerae N16961, %GC, other carrier strains used in this study, ANI with homologous islands, δ*, direction of transfer, islands sharing same insertion loci, and annotation.
Putative genomic islands of Vibrio sp. RC586. Putative genomic islands of Vibrio sp. RC586, showing insertion loci, homologous flanking loci in V. cholerae N16961, %GC, other carrier strains used in this study, ANI with homologous islands, δ*, direction of transfer, islands sharing same insertion loci, and annotation.
Phylogeny of the genomic island GI-2. Phylogeny of the genomic island GI-2 as determined by reconstructing a neighbor-joining tree using the Kimura-2 parameter as a nucleotide substitution model.
Phylogeny of the genomic island GI-41. Phylogeny of the genomic island GI-41 as determined by reconstructing a neighbor-joining tree using the Kimura-2 parameter as a nucleotide substitution model.
Phylogeny of the genomic island GI-4. Phylogeny of the genomic island GI-4 as determined by reconstructing a neighbor-joining tree using the Kimura-2 parameter as a nucleotide substitution model.
Phylogeny of VSP-I. Phylogeny of the genomic island VSP-I as determined by reconstructing a neighbor-joining tree using the Kimura-2 parameter as a nucleotide substitution model.
Phylogeny of the genomic island GI-61. Phylogeny of the genomic island GI-61 as determined by reconstructing a neighbor-joining tree using the Kimura-2 parameter as a nucleotide substitution model.
Phylogeny of Vibrio sp. RC341 Islet-3. Phylogeny of Vibrio sp. RC341 Islet-3 as determined by reconstructing a neighbor-joining tree using the Kimura-2 parameter as a nucleotide substitution model.