

Abstract
Tumors usurp established metabolic steps used by normal tissues for glucose utilization and ATP production that rely heavily on mitochondia and employ a route that, although involving mitochondria, includes a much greater dependency on glycolysis. First described by Otto Warburg almost nine decades ago [1], this aberrant phenotype becomes more pronounced with increased tumor malignancy [2]. Thus, while maintaining their capacity for respiration, tumors “turn more parasitic” by enhancing their ability to scavenge glucose from their surroundings. With excess glucose at hand, tumors shunt their metabolic flux more toward glycolysis than do their normal cells of origin, a strategy that allows for their survival when oxygen is limiting while providing them a mechanism to poison their extra-cellular environment with acid, thus paving the way for invasion and metastasis. Significantly, tumors harness a crucial enzyme to regulate and support this destructive path – to entrap and channel glucose toward glycolysis. This enzyme is an isoform of hexokinase, referred to as hexokinase type II, and also in abbreviated form as HK-2 or HK II. Due to many-faceted molecular features at genetic, epigenetic, transcriptional, and enzymatic levels, including sub-cellular localization to mitochondria, HK-2 facilitates and promotes the high glycolytic tumor phenotype [3]. Thus, HK-2 represents a pivotal model gene or enzyme that tumors “select for” during tumorigenesis in order to facilitate their destructive path. In this review, we examine the roles played by mitochondrial bound HK-2 within the context of the highly choreographed metabolic roulette of malignant tumors. Recent studies that outline how the aberrant glycolytic flux can be subverted toward a more “normal” metabolic phenotype, and how the glycolytic flux affects the tumor microenvironment to facilitate tumor dissemination are also described, including how these very features can be harnessed in new metabolic targeting strategies to selectively debilitate tumors.
Keywords: Cancer, Mitochondria, Warburg effect, hexokinase 2, VDAC, 3-bromopyruvate
Introduction
Tumor mitochondria and the associated enzymes and transporters that commit many tumors toward a highly glycolytic phenotype
A universally observed clinical phenotype of highly malignant tumors is their propensity to rapidly proliferate and spread (metastasize) within the tumor-bearing host. Although numerous molecular strategies are harnessed by the tumor to enable them to behave in this insidious manner, a crucial component required is the tumor’s ability to usurp inordinate amounts of glucose from the host’s blood, which in turn is used by the tumor not only to grow, but to also poison its surroundings in order to create a “molecular wasteland” around the tumor, into which it can gradually “metastasize” [4-6].
This propensity to metabolize glucose at excessive rates is the only requirement, and the basis behind the clinical imaging and staging of tumors via positron emission tomography (PET) [7, 8]. Despite the universality of this technique, study of the “bioenergetic” basis behind this phenotype has not been examined that frequently or commonly by the scientific community during the past several decades, except for a few laboratories, one exemplified by a coauthor of this contribution [9]. However, with the advent of new interest in the use of metabolic pathways as useful molecular tools to target tumors, the field has been expanding at a rapid rate in recent years. This review will examine the cellular features in tumors that facilitate the high glycolytic phenotype in malignant tumors, and discuss the possible therapeutic avenues that can be explored in view of findings from research that has been conducted for the past several decades and continues to this day.
Significantly, a study of metabolism in living cells goes back over 100 years to the time of Pasteur, when he described the phenomenon of “fermentation”, i.e., that, in order to survive, cells switch from oxidative phosphorylation (“respiration”) to glycolysis (fermentation) in the absence of adequate oxygen. This phenotype is commonly referred to as “anaerobic glycolysis”, or as the “Pasteur Effect”. Almost seven decades later, Otto Warburg described the same phenomenon in tumor slices demonstrating that it occurred even in the presence of adequate oxygen [1, 10, 11]. This aberrant metabolic feature, also found in rapidly proliferating tumor cells, is now termed the “Warburg Effect” and describes the propensity of malignant tumor cells to consume glucose at abnormally elevated rates and then metabolize and efflux most of it as lactic acid to the surrounding cellular environment [2, 4, 12]. As opposed to the Pasteur Effect, the latter is termed “aerobic glycolysis”. Clearly, fermentation is an inferior and counter-intuitive method to generate energy by a rapidly proliferating tumor cell; yet, it remains a universal phenotype, indicating that other, “non-bioenergetic-based” selective forces are active within the tumor and the tumor-microenvironment to promote this aberrant behavior [13].
It should be emphasized that while malignant tumors have acquired a high glycolytic phenotype, enhancing their glucose uptake by almost 10-fold, their respiration rate remains essentially unchanged from that of surrounding normal tissue, a point eloquently outlined by Koppenol and Bounds in a recent letter to Science [14]. Thus, while a normal cell may synthesize (under normoxia) approximately 32 ATP molecules per glucose molecule, a highly glycolytic tumor may produce an additional 3/4ths of this ATP via glycolysis (2 ATP molecules per glucose molecule). Studies by numerous laboratories to date indicate that this extra glucose intake is most likely diverted by tumors to biosynthetic pathways for fatty acid synthesis and nucleotide synthesis to support their rapid proliferation [4, 13]. Thus, when bioenergetic capacity is considered, tumors are as sufficient in energy generation in comparison to the surrounding normal tissues, if not more.
The key sub-cellular and molecular players that propel the tumor toward this metabolic transformation can be distilled to a few select enzymes; notably hexokinase, several mitochonndrial associated enzymes and plasma membrane transporters, and the crucial organelle involved in energy generation, the mitochondrion.
We will first examine the membrane transporters that facilitate influx of glucose and efflux of lactic acid by such tumors, and then describe the mitochondrial-bound hexokinase which acts as a molecular sentinel to coordinate mitochondrial function (ATP generation) and the cytoplasmic glycolytic flux in order for the tumor to rapidly proliferate in the tumor-bearing host.
A. Facilitators of glucose entry and lactate/proton efflux in malignant tumors – glucose, lactate and proton transporters
1] Facilitated influx of glucose
Of the various isoforms of glucose transporters (GLUT) which passively transport glucose across the plasma membrane, several have been implicated to be highly expressed in tumors. Although varying in their kinetic properties for glucose transport, it should be noted that due to the passive nature of glucose entry by these transporters, it is the enzyme hexokinase (discussed later; found predominantly bound to mitochondria in tumors) that “traps” incoming glucose by phosphorylation to impart an ionic charge, and then initiates the entire glycolytic cascade. Thus, in order to enhance glucose entry, a tumor can “evolve” two possible phenotypes; an obvious first step would be to increase the expression of plasma membrane glucose transporters; a second possibility is to switch the glucose transporter isoforms to those that have a higher affinity for glucose. In fact, there have been numerous reports of enhanced expression of glucose transporters in tumors [15-17], with over 10-fold enhancement in their expression levels. With regard to altered expression of transporters that have a higher affinity, of the currently recognized 12 isoforms of GLUT, it is the isoforms 3 and 4 that have the highest affinity for glucose [16]. Not surprisingly, both have been shown to be over-expressed in most human cancers [15]. However, GLUT1, ubiquitously expressed in all normal tissues, is one of the commonly observed over-expressed isoforms in tumors, along with GLUT3 and GLUT4. Thus, it may be that a partial GLUT isoform shift occurs during tumorigenesis, with the “normal” form being maintained albeit at an elevated level in most tumors. This is in marked contrast to hexokinase (discussed later) where a significant isoform switch occurs during tumorigenesis, especially in case of liver derived tumors.
2] Facilitated efflux of lactic acid
The high flux through the glycolytic pathway makes it necessary for the tumor cell to rapidly remove lactic acid, the end product of aerobic glycolysis. Previously thought to be removed via passive diffusion across the plasma membrane in its protonated (charge neutral - undissociated) form, it is now known that a family of transmembrane transporters denoted as monocarboxylate transporters (MCTs) [18] are involved in facilitated efflux of lactic acid to the tumor microenvironment [12]. Among the at least fourteen MCTs that have been identified via the human genome project, only the first four (MCTs 1-4) have been experimentally verified to transport lactate across the plasma membrane. Of the four isoforms, the MCT2 and MCT4 have the highest affinity for lactate, while MCT1, ubiquitously expressed among normal tissues, has the lowest affinity. In a manner similar to that observed for glucose transporters, MCT1 is found over-expressed among tumors, along with MCT2 and 4. Thus, the tumors mirror their selective expression of MCTs vis-a- vis GLUT, with the “normal” isoform being maintained at a higher level, along with co-expression of high affinity lactate transporters to maximize the lactate removal to the tumor microenvironment.
3] Active transport of protons to the tumor microenvironment – pumps that facilitate tumor proliferation?
It is commonly observed that the microenvironment surrounding highly malignant tumors has a lower pH in relation to that within the tumor. This is in stark contrast to the physiological conditions observed between normal tissues and their surroundings, even during hypoxia, where the tissue will have a lower pH, while the surroundings harbor a higher pH. Thus, such tumors play an active role in reversing the pH profile between the tumor and its microenvironment to maintain a hostile pH towards the surrounding normal tissue, which in turn may facilitate tumor dissemination as well as invasion [5, 6, 19]. Which molecules are involved in facilitating this “reversed” pH gradient in tumors? While the lactate transporter is the sole transmembrane channel (symporter) responsible for elimination of lactate produced via glycolysis with release of equivalent amounts of protons, several other transmembrane transporters may play an even greater role, both in facilitating and maintaining this hostile phenotype. Biochemically, an energy expenditure will be required by the tumors to maintain an adverse pH gradient towards the microenvironment. Accordingly, several transmembrane molecules, notably the sodium/proton exchanger (NHE), the chloride/bicarbonate exchanger, the proton/chloride co-transporter, and the transmembrane carbonic anhydrases (CA) in concert play a key role in maintaining this adverse transmembrane pH gradient in tumors [20, 21] (Fig. 1).
Figure 1. The multifaceted events of a tumor cell that require a high glycolytic rate for both tumor survival and proliferation.
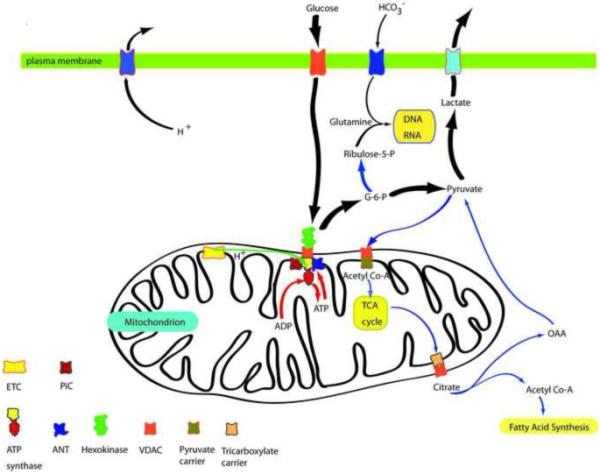
Many tumor cells require a high rate of glycolysis to meet both bioenergetic (ATP) requirements and biosynthetic requirements as follows: A] Both glycolysis (glucose to lactate pathway), initiated by mitochondrial bound hexokinase (green), and oxidative phosphorylation [supported by NADH and FADH not shown] derived from the TCA cycle and terminating with the ATP synthase F1 headpiece (red)] result in the necessary ATP to meet the tumor’s energy (ATP) requirements. B] The high glycolytic flux initiated and facilitated by G-6-P formation via mitochondrial bound hexokinase (green) also leads to additional metabolites for both RNA/DNA biosynthesis and fatty acid synthesis. These are essential for tumor cell proliferation resulting in the subjugation of the surrounding normal tissue. C] By efflux of both lactic acid and protons, the tumor creates a “poisonous” environment for the surrounding normal tissue, most likely to promote a “death zone” around the tumor into which it can slowly encroach. [Abbreviations: ETC, electron transport chain; PIC, phosphate carrier; ANT, adenine nucleotide transporter (carrier); VDAC, voltage dependent anion channel; G-6-P, glucose-6-phosphate; OAA, oxaloacetic acid.] (Note: The ATP synthase is now known to be in complex formation with the phosphate carrier (PIC) and the adenine nucleotide carrier (ANC) to give an ATP synthase/PIC/ANC complex called the “ATP synthasome” (Ref.: Ko, Y.H., Delannoy, M., Hullihen, J., Chiu, W., and Pedersen, P.L. (2003) J. Biol. Chem. 278, 12305-12309).
B. The enemy within – tumorsmitochondria, their bound hexokinase and the role of both in maintaining the high glycolytic phenotype of tumors
Although the plasma membrane proteins discussed above facilitate glucose influx into the tumor or proton and lactate efflux, the key cellular components responsible for coordinating the high glycolytic flux across the tumor as well as bioenergetic homeostasis belongs to the mitochondrion and the enzyme hexokinase II found bound primarily to its outer membrane (OMM) [22-25].
1] The biochemistry of tumor hexokinase II
In fact, the critical step of trapping and committing the incoming glucose to the glycolytic cascade in a tumor cell is facilitated via this crucial phenotype well observed in malignant tumors; that of direct/close interaction of hexokinase II with mitochondrial outer membrane porins (VDACs, voltage gated anion channels) to facilitate access of mitochondrial synthesized ATP for the hexokinase catalyzed reaction below.
This essentially irreversible metabolic first step of the glycolytic cascade entraps and commits incoming glucose to initiate the glycolytic flux across the tumor cell. In addition to mitochondrial binding, tumors have optimized for maximum efficiency of glucose turnover via utilization of additional molecules and selective “switching” of gene isoforms;
2] The enzymology of tumor hexokinase II
Of the four known isoforms of mammalian hexokinases [26, 27], it is isoforms I to III that harbor a significantly higher affinity for glucose (~250-fold higher). Thus, not surprisingly, malignant tumors switch their hexokinase expression pattern predominantly to one of these high-affinity isoforms. This is seen clearly in the case of rapidly growing hepatomas; whereas normal liver expresses glucokinase (or hexokinase IV), which has a high Km (~5 mM; low affinity) for glucose [3], the hepatoma predominantly express the type II isoform. The hexokinase types I-III appear to be duplication products of an ancestral gene similar to the current glucokinase [27, 28]. Thus, while glucokinase is comprised of a single protein domain, the hexokinases harbor twin domains. Among the three high affinity hexokinase isoforms, type II hexokinase harbors the potential for an enhanced glucose turnover rate due to another crucial biochemical phenotype related to the twin domains; although the three hexokinases are composed of two nearly identical protein domains, each with the potential to catalyze the phosphorylation of glucose, in hexokinases type I and III, the N-terminal domain has lost its catalytic role and acquired a regulatory role, thus subjecting these enzyme isoforms to feedback regulation by ADP, inorganic phosphate and by glucose -6-phosphate. In contrast, with type II hexokinase retaining the catalytic capacity in both domains [29], the isoform effectively retains the capacity to double the rate of formation of glucose-6-phosphate, relative to isoforms I and III. Therefore, it is not surprising that type II hexokinase is the isoform predominantly expressed in highly malignant tumors [30, 31].
Another crucial feature that is common at least to hexokinases I and II are their N- termini which are hydrophobic. This allows these enzymes to anchor themselves to the outer mitochondrial membrane via porins (VDACs; voltage-gated anion channels) to gain immediate access to mitochondrial generated ATP [32]. In addition, binding of hexokinase II in this capacity almost completely prevents its product inhibition by glucose-6-phosphate [33], thus allowing the mitochondrial bound enzyme to initiate and then maintain a high glycolytic flux rate in tumors.
3] The genetics and epi-genetics behind overexpression of hexokinase II in tumors
Malignant tumors also influence their hexokinase II expression levels via both genetic and epi-genetic alterations to facilitate its overexpression; at the gene level, both normal and tumor hexokinase II harbor nearly identical transcriptional regulatory sequences. However, fluorescence in situ hybridization (FISH) studies have indicated that tumors amplify the hexokinase II gene by at least 5-fold [34]. Furthermore, epigenetic changes, i.e., methylation of the hexokinase II promoter is observed in case of normal liver, indicating that the gene is silenced, at least in part, in this manner in normal tissues [35]. When the proximal promoter sequence of hexokinase II is analyzed, several other features that support its expression even under the most adverse physiological conditions within a tumor also become evident. For example, the most proximal cis-elements in the promoter are those for cyclic AMP (thus, the protein kinase A pathway, upregulated by glucagon during starvation), an unexpected feature, as the hexokinases should be up-regulated via increased glucose levels (and protein kinase C pathway, and insulin) [36]. The promoter also harbors the latter elements as well as those for hypoxia (HIF-1) and tumor suppressor p53 [37, 38]. Physiological analyses have indicated that all these elements are active in the hexokinase II promoter. Thus, these crucial features present a genetic survival mechanism in these malignant tumors which enable them to overexpress hexokinase II under “any” adverse physiological or metabolic condition, in order to scavenge glucose from the host’s systemic circulation for the tumor’s benefit [4].
Overall, the above described genetic, epigenetic and biochemical features may be common to most other key enzymes along the glycolytic metabolic pathway, as most tumors “switch” to the fetal type isoforms of these enzymes. These in turn enable the tumor to maximize its ability to harness and channel key metabolites at the expense of surrounding tissues, which commonly express the “adult” or “differentiated” types of isoforms that are usually subjected to feedback regulation at the enzyme level (or their gene expression patterns are tightly regulated by the physiological condition of the tumor bearing host). Thus, by shifting to the fetal isoforms, the tumors bypass many of the biochemical constraints that regulate metabolism, in order to maximize their survival at great expense to the host.
C. Tumor mitochondria – the central coordinating organelle of tumor metabolism, survival and proliferation
The textbook explanation of the mitochondrion is that it is primarily a site for ATP generation. Although accurate, the anaplerotic and cataplerotic metabolic fluxes across mitochondria and the mitochondrial tricarboxylic acid cycle (TCA) all serve to coordinate several key metabolic pathways crucial for tumor proliferation. For example, synthesis of glutamine via the TCA cycle and NH4+, the pentose phosphate shunt (that produces ribose-5-phosphate used for downstream synthesis of RNA and DNA via incorporation of glutamine and bicarbonate), and synthesis of fatty acids via acetyl coenzyme A generated from citrate, all function to provide biosynthetic precursors necessary for the tumor’s rapid proliferation [39]. The mitochondrion also functions as a key regulator of numerous cellular functions critical for cell survival [40]. Although the metabolic emphasis in tumors has shifted to glycolysis, the mitochondrion also plays a key role in controlling the metabolic fluxes within the tumor cell. Thus, while as much as 40 to 75% of ATP generation in a malignant tumor may occur via glycolysis, the mitochondria still function to generate the remainder of the tumor’s energy requirements.
With direct coupling of mitochondrial generated ATP to incoming glucose via VDAC (porin) bound hexokinase, mitochondria regulate the extra-mitochondrial glycolytic flux with that of the TCA cycle and ATP synthase, to balance the energy requirements of the tumor cell with the biochemical requirements for metabolites (anaplerotic and cataplerotic pathways) or metabolite precursors that are required by the tumor. Thus, both the glycolytic pathway and other seminal metabolic pathways i.e., the pentose phosphate shunt, are regulated via the energy coupling resulting from the VDAC bound hexokinase complex.
D. Molecular choreography between glycolysis, respiration, reversed pH and their impact on tumor survival and dissemination
If a “starting-point” for the initiation of the glycolytic cascade is to be considered, it will be the first step where glucose is phosphorylated by hexokinase to commit this key metabolite to the glycolytic pathway. Here, hexokinase, bound to mitochondrial outer membrane via VDAC, is provided preferential access to the mitochondrial ATP via the adenine nucleotide translocator (ANT) in the IMM to rapidly and quantitatively phosphorylate and “trap” the incoming glucose, essentially initiating and regulating the glycolytic flux across the tumor cytosol. The resultant glycolytic metabolic cascade will provide the tumor but with 2 net ATP molecules per phosphorylated glucose molecule, with the remainder of the tumor’s energy requirements being met by the mitochondria, perhaps involving in part the metabolism of glutamine [41].
The second crucial regulatory step of the glycolytic metabolic flux occurs at the outer membrane of mitochondria, when pyruvate enters the mitochondrion to initiate the TCA cycle (via the yet to be characterized pyruvate transporter?). Here, the pyruvate can enter the mitochondrion, or be shuttled to the lactate dehydrogenase (LDH) complex to be reduced to lactate and effluxed to the tumor microenvironment, with concomitant oxidation (and cycling) of NADH to NAD+ to ensure the continuation of the glycolytic cascade (Fig. 1). Significantly, not only are both pyruvate kinase (PK) and lactate dehydrogenase (LDH) up-regulated by hypoxia (HIF-1) (and thus can channel the glycolytic cascade toward lactate) [42], pyruvate dehydrogenase kinase (PDK), a key modulator of pyruvate dehydrogenase complex activity (PDH; which serves as the gatekeeper for entry of pyruvate into mitochondria), is also up-regulated by hypoxia [43]. Thus, HIF-1 mediated effects act in a synergistic manner to veer the glycolytic cascade towards lactate generation. However, it is still not clear as to whether the same effects are in play in tumors that are under normoxic or near normoxic conditions. Overall, the above effects effectively channel the tumor cell’s glycolytic flux towards generation of lactic acid to be effluxed to the tumor microenvironment. In fact, recent reports indicate that extrusion of protons by the tumor to the tumor microenvironment to reduce extracellular pH may be a crucial strategy by tumors to promote their invasiveness and proliferation [20, 21].
Thus, close coupling between the proton-motive forces across the mitochondrial membranes and the plasma membrane will be a necessity for a tumor cell to efflux protons to the microenvironment, in order to maintain a reduced pH in the extracellular milieu. Therefore, a close coordination must exist between key metabolic pathways of the tumor and mitochondrial functions in order for both glycolysis and adverse ΔpH gradients across the plasma membrane to be maintained by the tumor.
E. Therapeutic strategies against tumors targeted at their aberrant metabolic flux
The aberrant metabolic pathways that exist within the tumor as well as reported altered pH profiles within the tumor microenvironment provide new therapeutic strategies to be used against tumors while leaving the surrounding normal tissue relatively unscathed. Some of these have been examined at pre-clinical stages while a few are being tested in Phase I-III clinical trials. These are briefly discussed below. A more thorough review of the various potential small-molecule drugs that can be used to target the glycolytic pathway is given in a recent review by Sheng and co-workers [44].
An obvious first choice to block the initiating steps of glycolysis (Fig. 1) would be the use of analogs of 18F-deoxy glucose (FDG) routinely used in PET imaging of tumors where the non-hydrolyzable analog is prevented from advancing beyond the hexokinase mediated phosphorylation step. Thus, deoxyglucose [45], and two other direct inhibitors of hexokinase, Lonidamine [46] and 3-bromopyruvate [47, 48] have been successfully tested in pre-clinical trials against malignant tumors. Of these, 3-bromopyruvate was shown to be especially effective, eradicating advanced tumors in 19/19 animals, without harm to the animals and without return of the tumors during their life.
Pyruvate kinase is another key enzyme along the glycolytic pathway, which involves an essentially irreversible step of converting phosphoenolpyruvate (PEP) to pyruvate, prior to mitochondrial entry. Thus, it also has presented a suitable drug target to debilitate tumors. Several analogs of PEP have been developed and tested against tumors to test their effects in inhibiting tumor proliferation [49, 50].
Several other attractive targets are presented at the metabolic routes immediately adjacent to mitochondrial pyruvate entry. These involve lactate dehydrogenase (LDH) and an enzyme that regulates the activity of pyruvate dehydrogenase (PDH), pyruvate dehydrogenase kinase (PDK). Both LDH and PDK have been targeted [51, 52], the former utilizing interference RNA techniques, while the latter has been successfully targeted using di-choloroacetate (DCA), an analog of pyruvate, with Phase II/III clinical trials ongoing at one medical center [53, 54].
Targeting lactate efflux is another avenue to inhibit the overall glycolytic pathway to bring about tumor cell-death. This has been successfully tested pre-clinically against malignant tumors both in vitro and in vivo utilizing both interference RNA techniques and the small-molecule drug alpha cyano 4-hydroxy cinnamic acid (ACCA), to bring about tumor apoptosis and necrosis. In addition, the targeted tumors showed increased sensitivity to radiation treatments, indicating increased susceptibility of such tumors upon debilitation of the glycolytic cascade [12, 55, 56].
Recent reports indicate that neutralizing the low pH of the tumor microenvironment via physiological buffers (in this case bicarbonate) can reduce the invasiveness of tumors and thus minimize the incidence of distant metastases [57]. Denoted bicarbonate therapy, it has been experimentally shown that the treatment reduced tumor presence in lymph nodes in select in vivo model systems. Thus, these studies open up new avenues to control proliferation of malignant tumors via targeting the low pH of the tumor microenvironment, perhaps by modulating the effects of plasma membrane transporters including the sodium/proton exchanger, various bicarbonate exchangers, proton/chloride co-transporters and the plasma membrane carbonic anhydrases [21, 58]. Thus, focused targeting of various key steps in the glycolytic pathway of tumor cells to either inhibit the metabolic flux, or to re-route it towards mitochondrial respiration has shown positive results in controlling tumor malignancy in both pre-clinical and Phase II/III clinical stages.
Conclusions
When the war on cancer was first declared over four decades ago, targeting metabolism should have been an obvious first choice, as many research groups had at that time, a better understanding of the biochemical signatures of cancers. However, with the advent of molecular biology and signal transduction research, metabolism-based research was discarded to the “backbenches”, except in a few dedicated laboratories. With the increasing understanding of the difficulty in targeting signal transduction of tumors without significant side-effects, cancer research is being redirected towards targeting the aberrant metabolism of tumors. Already, significant results have been achieved by various research groups where multiple “points” on key metabolic pathways have been targeted, with both small-molecule drugs as well as via interference RNA methodology. A key feature that needs to be used as a molecular “yardstick” to judge the success of these endeavors would be to identify to what extent the hexokinase II catalyzed metabolic step is directly impacted or not. Certainly, in the initial study by Ko et al. [48] showing that 3-bromopyruvate is a potent anti-tumor agent, it was shown to markedly inhibit tumor hexokinase.
Acknowledgments
S.P.M. is supported by NIH grant R01CA116257 and an endowment by Marvin E. Klein, M.D. Charitable Trust and P.L.P. by NIH grants R01CA08018 and R01CA010951.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Warburg O, Dickens F, B. Kaiser Wilhelm-Institut f?r Biologie . The Metabolism of tumours : Investigations from the Kaiser-Wilhelm Institute for Biology. Constable; Berlin-Dahlem, London: 1930. [Google Scholar]
- [2].Pedersen PL. Tumor mitochondria and the bioenergetics of cancer cells. Prog.Exp.Tumor Res. 1978;22:190–274. doi: 10.1159/000401202. [DOI] [PubMed] [Google Scholar]
- [3].Pedersen PL, Mathupala S, Rempel A, Geschwind JF, Ko YH. Mitochondrial bound type II hexokinase: a key player in the growth and survival of many cancers and an ideal prospect for therapeutic intervention. Biochim.Biophys.Acta. 2002;1555:14–20. doi: 10.1016/s0005-2728(02)00248-7. [DOI] [PubMed] [Google Scholar]
- [4].Mathupala SP, Ko YH, Pedersen PL. Hexokinase-2 bound to mitochondria: cancer’s stygian link to the “Warburg Effect” and a pivotal target for effective therapy. Semin Cancer Biol. 2009;19:17–24. doi: 10.1016/j.semcancer.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Gatenby RA, Gawlinski ET, Gmitro AF, Kaylor B, Gillies RJ. Acid-mediated tumor invasion: a multidisciplinary study. Cancer Res. 2006;66:5216–5223. doi: 10.1158/0008-5472.CAN-05-4193. [DOI] [PubMed] [Google Scholar]
- [6].Gillies RJ, Robey I, Gatenby RA. Causes and consequences of increased glucose metabolism of cancers. J Nucl Med. 2008;49(Suppl 2):24S–42S. doi: 10.2967/jnumed.107.047258. [DOI] [PubMed] [Google Scholar]
- [7].DeLaPaz RL, Patronas NJ, Brooks RA, Smith BH, Kornblith PL, Milam H, Di Chiro G. Positron emission tomographic study of suppression of gray-matter glucose utilization by brain tumors. AJNR Am J Neuroradiol. 1983;4:826–829. [PMC free article] [PubMed] [Google Scholar]
- [8].Di Chiro G, Brooks RA, Patronas NJ, Bairamian D, Kornblith PL, Smith BH, Mansi L, Barker J. Issues in the in vivo measurement of glucose metabolism of human central nervous system tumors. Ann Neurol. 1984;15(Suppl):S138–146. doi: 10.1002/ana.410150727. [DOI] [PubMed] [Google Scholar]
- [9].Pedersen PL, Warburg me and Hexokinase 2: Multiple discoveries of key molecular events underlying one of cancers’ most common phenotypes, the “Warburg Effect”, i.e., elevated glycolysis in the presence of oxygen. J Bioenerg Biomembr. 2007;39:211–222. doi: 10.1007/s10863-007-9094-x. [DOI] [PubMed] [Google Scholar]
- [10].Warburg O. On respiratory impairment in cancer cells. Science. 1956;124:269–270. [PubMed] [Google Scholar]
- [11].Warburg O. On the origin of cancer cells. Science. 1956;123:309–314. doi: 10.1126/science.123.3191.309. [DOI] [PubMed] [Google Scholar]
- [12].Mathupala SP, Colen CB, Parajuli P, Sloan AE. Lactate and malignant tumors: a therapeutic target at the end stage of glycolysis. J Bioenerg Biomembr. 2007;39:73–77. doi: 10.1007/s10863-006-9062-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Koppenol WH, Bounds PL. The Warburg Effect and metabolic efficiency: Re-crunching the numbers. Science. 2009 E-Letter. [Google Scholar]
- [15].Medina RA, Owen GI. Glucose transporters: expression, regulation and cancer. Biol Res. 2002;35:9–26. doi: 10.4067/s0716-97602002000100004. [DOI] [PubMed] [Google Scholar]
- [16].Zhao FQ, Keating AF. Functional properties and genomics of glucose transporters. Curr Genomics. 2007;8:113–128. doi: 10.2174/138920207780368187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yeluri S, Madhok B, Prasad KR, Quirke P, Jayne DG. Cancer’s craving for sugar: an opportunity for clinical exploitation. J Cancer Res Clin Oncol. 2009;135:867–877. doi: 10.1007/s00432-009-0590-8. [DOI] [PubMed] [Google Scholar]
- [18].Halestrap AP, Meredith D. The SLC16 gene family-from monocarboxylate transporters (MCTs) to aromatic amino acid transporters and beyond. Pflugers Arch. 2004;447:619–628. doi: 10.1007/s00424-003-1067-2. [DOI] [PubMed] [Google Scholar]
- [19].Gatenby RA, Gawlinski ET. The glycolytic phenotype in carcinogenesis and tumor invasion: insights through mathematical models. Cancer Res. 2003;63:3847–3854. [PubMed] [Google Scholar]
- [20].Stock C, Cardone RA, Busco G, Krahling H, Schwab A, Reshkin SJ. Protons extruded by NHE1: digestive or glue? Eur J Cell Biol. 2008;87:591–599. doi: 10.1016/j.ejcb.2008.01.007. [DOI] [PubMed] [Google Scholar]
- [21].Stock C, Schwab A. Protons make tumor cells move like clockwork. Pflugers Arch. 2009;458:981–992. doi: 10.1007/s00424-009-0677-8. [DOI] [PubMed] [Google Scholar]
- [22].Bustamante E, Morris HP, Pedersen PL. Energy metabolism of tumor cells. Requirement for a form of hexokinase with a propensity for mitochondrial binding. J.Biol.Chem. 1981;256:8699–8704. [PubMed] [Google Scholar]
- [23].Bustamente E, Morris HP, Pedersen PL. Hexokinase: the direct link between mitochondrial and glycolytic reactions in rapidly growing cancer cells. Adv.Exp.Med.Biol. 1977;92:363–380. doi: 10.1007/978-1-4615-8852-8_15. [DOI] [PubMed] [Google Scholar]
- [24].Nakashima RA, Paggi MG, Scott LJ, Pedersen PL. Purification and characterization of a bindable form of mitochondrial bound hexokinase from the highly glycolytic AS-30D rat hepatoma cell line. Cancer Res. 1988;48:913–919. [PubMed] [Google Scholar]
- [25].Nakashima RA, Scott LJ, Pedersen PL. The role of mitochondrial hexokinase binding in the abnormal energy metabolism of tumor cell lines. Ann.N.Y.Acad.Sci. 1986;488:438–450. doi: 10.1111/j.1749-6632.1986.tb46577.x. [DOI] [PubMed] [Google Scholar]
- [26].Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J.Exp.Biol. 2003;206:2049–2057. doi: 10.1242/jeb.00241. [DOI] [PubMed] [Google Scholar]
- [27].Wilson JE. Hexokinases. Rev.Physiol Biochem.Pharmacol. 1995;126:65–198. doi: 10.1007/BFb0049776. [DOI] [PubMed] [Google Scholar]
- [28].Tsai HJ, Wilson JE. Functional organization of mammalian hexokinases: characterization of chimeric hexokinases constructed from the N- and C-terminal domains of the rat type I and type II isozymes. Arch.Biochem.Biophys. 1995;316:206–214. doi: 10.1006/abbi.1995.1029. [DOI] [PubMed] [Google Scholar]
- [29].Tsai HJ, Wilson JE. Functional organization of mammalian hexokinases: both N- and C-terminal halves of the rat type II isozyme possess catalytic sites. Arch.Biochem.Biophys. 1996;329:17–23. doi: 10.1006/abbi.1996.0186. [DOI] [PubMed] [Google Scholar]
- [30].Rempel A, Bannasch P, Mayer D. Differences in expression and intracellular distribution of hexokinase isoenzymes in rat liver cells of different transformation stages. Biochim.Biophys.Acta. 1994;1219:660–668. doi: 10.1016/0167-4781(94)90225-9. [DOI] [PubMed] [Google Scholar]
- [31].Rempel A, Bannasch P, Mayer D. Microheterogeneity of cytosolic and membrane-bound hexokinase II in Morris hepatoma 3924A. Biochem.J. 1994;303(Pt 1):269–274. doi: 10.1042/bj3030269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Pedersen PL. Voltage dependent anion channels (VDACs): a brief introduction with a focus on the outer mitochondrial compartment’s roles together with hexokinase-2 in the “Warburg effect” in cancer. J Bioenerg Biomembr. 2008;40:123–126. doi: 10.1007/s10863-008-9165-7. [DOI] [PubMed] [Google Scholar]
- [33].Bustamante E, Pedersen PL. High aerobic glycolysis of rat hepatoma cells in culture: role of mitochondrial hexokinase. Proc.Natl.Acad.Sci.U.S.A. 1977;74:3735–3739. doi: 10.1073/pnas.74.9.3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rempel A, Mathupala SP, Griffin CA, Hawkins AL, Pedersen PL. Glucose catabolism in cancer cells: amplification of the gene encoding type II hexokinase. Cancer Res. 1996;56:2468–2471. [PubMed] [Google Scholar]
- [35].Goel A, Mathupala SP, Pedersen PL. Glucose metabolism in cancer. Evidence that demethylation events play a role in activating type II hexokinase gene expression. J.Biol.Chem. 2003;278:15333–15340. doi: 10.1074/jbc.M300608200. [DOI] [PubMed] [Google Scholar]
- [36].Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells. Isolation, sequence, and activity of the promoter for type II hexokinase. J.Biol.Chem. 1995;270:16918–16925. doi: 10.1074/jbc.270.28.16918. [DOI] [PubMed] [Google Scholar]
- [37].Mathupala SP, Rempel A, Pedersen PL. Glucose catabolism in cancer cells: identification and characterization of a marked activation response of the type II hexokinase gene to hypoxic conditions. J.Biol.Chem. 2001;276:43407–43412. doi: 10.1074/jbc.M108181200. [DOI] [PubMed] [Google Scholar]
- [38].Mathupala SP, Heese C, Pedersen PL. Glucose catabolism in cancer cells. The type II hexokinase promoter contains functionally active response elements for the tumor suppressor p53. J.Biol.Chem. 1997;272:22776–22780. doi: 10.1074/jbc.272.36.22776. [DOI] [PubMed] [Google Scholar]
- [39].Hsu PP, Sabatini DM. Cancer cell metabolism: Warburg and beyond. Cell. 2008;134:703–707. doi: 10.1016/j.cell.2008.08.021. [DOI] [PubMed] [Google Scholar]
- [40].Mathupala SP, Ko YH, Pedersen PL. Hexokinase II: cancer’s double-edged sword acting as both facilitator and gatekeeper of malignancy when bound to mitochondria. Oncogene. 2006;25:4777–4786. doi: 10.1038/sj.onc.1209603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, Wehrli S, Thompson CB. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A. 2007;104:19345–19350. doi: 10.1073/pnas.0709747104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Feron O. Pyruvate into lactate and back: from the Warburg effect to symbiotic energy fuel exchange in cancer cells. Radiother Oncol. 2009;92:329–333. doi: 10.1016/j.radonc.2009.06.025. [DOI] [PubMed] [Google Scholar]
- [43].Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- [44].Sheng H, Niu B, Sun H. Metabolic targeting of cancers: from molecular mechanisms to therapeutic strategies. Curr Med Chem. 2009;16:1561–1587. doi: 10.2174/092986709788186255. [DOI] [PubMed] [Google Scholar]
- [45].Zhang XD, Deslandes E, Villedieu M, Poulain L, Duval M, Gauduchon P, Schwartz L, Icard P. Effect of 2-deoxy-D-glucose on various malignant cell lines in vitro. Anticancer Res. 2006;26:3561–3566. [PubMed] [Google Scholar]
- [46].Oudard S, Poirson F, Miccoli L, Bourgeois Y, Vassault A, Poisson M, Magdelenat H, Dutrillaux B, Poupon MF. Mitochondria-bound hexokinase as target for therapy of malignant gliomas. Int.J.Cancer. 1995;62:216–222. doi: 10.1002/ijc.2910620218. [DOI] [PubMed] [Google Scholar]
- [47].Ko YH, Smith BL, Wang Y, Pomper MG, Rini DA, Torbenson MS, Hullihen J, Pedersen PL. Advanced cancers: eradication in all cases using 3-bromopyruvate therapy to deplete ATP. Biochem Biophys Res Commun. 2004;324:269–275. doi: 10.1016/j.bbrc.2004.09.047. [DOI] [PubMed] [Google Scholar]
- [48].Ko YH, Pedersen PL, Geschwind JF. Glucose catabolism in the rabbit VX2 tumor model for liver cancer: characterization and targeting hexokinase. Cancer Lett. 2001;173:83–91. doi: 10.1016/s0304-3835(01)00667-x. [DOI] [PubMed] [Google Scholar]
- [49].Garcia-Alles LF, Erni B. Synthesis of phosphoenol pyruvate (PEP) analogues and evaluation as inhibitors of PEP-utilizing enzymes. Eur J Biochem. 2002;269:3226–3236. doi: 10.1046/j.1432-1033.2002.02995.x. [DOI] [PubMed] [Google Scholar]
- [50].Gosalvez M, Lopez-Alarcon L, Garcia-Suarez S, Montalvo A, Weinhouse S. Stimulation of tumor-cell respiration by inhibitors of pyruvate kinase. Eur.J.Biochem. 1975;55:315–321. doi: 10.1111/j.1432-1033.1975.tb02165.x. [DOI] [PubMed] [Google Scholar]
- [51].Fantin VR, Leder P. Mitochondriotoxic compounds for cancer therapy. Oncogene. 2006;25:4787–4797. doi: 10.1038/sj.onc.1209599. [DOI] [PubMed] [Google Scholar]
- [52].Fantin VR, St-Pierre J, Leder P. Attenuation of LDH-A expression uncovers a link between glycolysis, mitochondrial physiology, and tumor maintenance. Cancer Cell. 2006;9:425–434. doi: 10.1016/j.ccr.2006.04.023. [DOI] [PubMed] [Google Scholar]
- [53].Michelakis ED, Webster L, Mackey JR. Dichloroacetate (DCA) as a potential metabolic-targeting therapy for cancer. Br J Cancer. 2008;99:989–994. doi: 10.1038/sj.bjc.6604554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37–51. doi: 10.1016/j.ccr.2006.10.020. [DOI] [PubMed] [Google Scholar]
- [55].Colen CB, Seraji-Bozorgzad N, Marples B, Galloway MP, Sloan AE, Mathupala SP. Metabolic remodeling of malignant gliomas for enhanced sensitization during radiotherapy: an in vitro study. Neurosurgery. 2006;59:1313–1323. doi: 10.1227/01.NEU.0000249218.65332.BF. discussion 1323-1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mathupala SP, Parajuli P, Sloan AE. Silencing of monocarboxylate transporters via small interfering ribonucleic acid inhibits glycolysis and induces cell death in malignant glioma: an in vitro study. Neurosurgery. 2004;55:1410–1419. doi: 10.1227/01.neu.0000143034.62913.59. discussion 1419. [DOI] [PubMed] [Google Scholar]
- [57].Robey IF, Baggett BK, Kirkpatrick ND, Roe DJ, Dosescu J, Sloane BF, Hashim AI, Morse DL, Raghunand N, Gatenby RA, Gillies RJ. Bicarbonate increases tumor pH and inhibits spontaneous metastases. Cancer Res. 2009;69:2260–2268. doi: 10.1158/0008-5472.CAN-07-5575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Pastorekova S, Zatovicova M, Pastorek J. Cancer-associated carbonic anhydrases and their inhibition. Curr Pharm Des. 2008;14:685–698. doi: 10.2174/138161208783877893. [DOI] [PubMed] [Google Scholar]