

Abstract
Streptomyces antibiotic regulatory proteins (SARPs) have been shown to activate transcription by binding to a tandemly arrayed set of heptameric direct repeats located around the −35 region of their cognate promoters. Experimental evidence is presented here showing that vlmI is a regulatory gene in the valanimycin biosynthetic gene cluster of Streptomyces viridifaciens and encodes a protein belonging to the SARP family. The organization of the valanimycin biosynthetic gene cluster suggests that the valanimycin biosynthetic genes are located on three potential transcripts, vlmHORBCD, vlmJKL and vlmA. Disruption of vlmI abolished valanimycin biosynthesis. Western blot analyses showed that VlmR and VlmA are absent from the vlmI mutant and that the production of VlmK is severely diminished. These results demonstrate that the expression of these genes from the three potential transcripts is under the positive control of VlmI. The vlmA–vlmH and vlmI–vlmJ intergenic regions both exhibit a pattern of heptameric direct repeats. Gel shift assays with VlmI overproduced in Escherichia coli as a C-terminal FLAG-tagged protein clearly demonstrated that VlmI binds to DNA fragments from both regions that contain these heptameric repeats. When a high-copy-number vlmI expression plasmid was introduced into Streptomyces coelicolor M512, which contains mutations in the undecylprodigiosin and actinorhodin activators redD and actII-orf4, undecylprodigiosin production was restored, showing that vlmI can complement a redD mutation. Introduction of the same vlmI expression plasmid into an S. viridifaciens vlmI mutant restored valanimycin production to wild-type levels.
INTRODUCTION
Streptomyces are filamentous soil bacteria that undergo differentiation and sporulation, and produce a multitude of bioactive compounds. The production of antibiotics and other secondary metabolites in these bacteria is tightly controlled by environmental stimuli and by a complex network of regulatory proteins that function at several hierarchical levels. The highest level of regulation involves pleiotropic genes that govern differentiation and sporulation as well as secondary metabolite production (Bibb, 2005). The lowest level employs pathway-specific regulatory genes that are usually associated with individual biosynthetic gene clusters. The family of proteins known as Streptomyces antibiotic regulatory proteins (SARPs) were the first pathway-specific regulatory proteins to be identified (Wietzorrek & Bibb, 1997). More recent studies have shown that the SARP family contains both pathway-specific regulators and pleiotropic regulatory proteins such as AfsR (Tanaka et al., 2007).
Members of the SARP family are transcriptional activators that exhibit a winged helix–turn–helix motif near their N termini that is similar to a motif found in the C terminus of the OmpR family of regulatory proteins (Wietzorrek & Bibb, 1997). Representative examples of SARPs include RedD and ActII-orf4, which control the production of undecylprodigiosin and actinorhodin, respectively, in Streptomyces coelicolor (Fernandez-Moreno et al., 1991; Narva & Feitelson, 1990), DnrI, which controls the production of daunorubicin by Streptomyces peucetius (Stutzman-Engwall et al., 1992), CcaR, which regulates clavulanic acid and cephamycin C biosynthesis in Streptomyces clavuligerus (Perez-Llarena et al., 1997), and FdmR1, which controls fredericamycin production in Streptomyces griseus (Chen et al., 2008). SARPs are postulated to activate transcription by binding to a tandemly arrayed set of heptameric repeats located around the −35 region of their cognate promoters. This hypothesis has been confirmed by gel shift mobility and DNA footprinting assays with promoter regions from the actinorhodin and daunorubicin biosynthetic gene clusters (Arias et al., 1999; Tang et al., 1996). After binding to the direct repeat region, the SARP regulators are believed to initiate transcription by recruitment of RNA polymerase to the appropriate sites (Tanaka et al., 2007).
The antibiotic valanimycin is a potent antitumor and antibacterial azoxy compound isolated from the fermentation broth of Streptomyces viridifaciens MG456-hF10 by Yamato and co-workers (Yamato et al., 1986). Enzymic and genetic investigations (Parry & Li, 1997a, b; Parry et al., 1997) have led to the cloning and sequencing of the valanimycin biosynthetic gene cluster, which has been found to contain 14 genes (Fig. 1a) (Garg et al., 2002). The functions of the products of eight of these genes have now been established. VlmF, which is a member of the major facilitator family of transport proteins, confers valanimycin resistance (Ma & Parry, 2000). VlmD, VlmH and VlmR catalyse the conversion of valine into isobutylhydroxylamine (Garg et al., 2002), while VlmL catalyses the formation of l-seryl-tRNA from l-serine (Garg et al., 2006). VlmA has been shown to catalyse the transfer of l-serine from l-seryl-tRNA to isobutylhydroxylamine, to produce O-(l-seryl)-isobutylhydroxylamine (Garg et al., 2008), while VlmJ and VlmK catalyse the phosphorylation and subsequent dehydration of the biosynthetic intermediate valanimycin hydrate (Garg et al., 2009). The biosynthetic pathway for valanimycin is shown in Fig. 1(b).
Fig. 1.

(a) Valanimycin biosynthetic gene cluster of S. viridifaciens MG456-hF10. Black arrows indicate approximate locations of VlmI binding sites. (b) Biosynthetic pathway for valanimycin in S. viridifaciens MG456-hF10, showing structures of the primary precursors l-valine and l-serine and structures of known intermediates.
The valanimycin gene cluster appears to contain two regulatory genes. The first of these is vlmE, which encodes a protein in the tetR family of repressor proteins. Evidence from other systems suggests that the vlmE gene product probably regulates the expression of the resistance gene vlmF (Garg et al., 2002). The second regulatory gene is vlmI. The deduced translation product of vlmI exhibits strong similarities to a number of members of the SARP family, including DnrI, RedD and TylT (Garg et al., 2002). In order to further characterize the vlmI gene product and to examine its role in valanimycin production, we have inactivated the vlmI gene, overexpressed VlmI and examined the DNA binding properties of VlmI. The results of these studies are reported here.
METHODS
General.
Unless otherwise indicated, all reagents used in this study were purchased from Sigma, Roche Applied Sciences, Bio-Rad Laboratories or G.E. Healthcare. Oligonucleotides were obtained from Sigma Genosys. Restriction enzymes were obtained from either New England Biolabs or Promega. [α-32P]dCTP was obtained from MP Biomedicals. Competent cells of Escherichia coli DH10B and BL21 (DE3) were purchased from Invitrogen and EMD, respectively, and were used according to the manufacturers' recommendations. The purity and molecular mass of overproduced proteins were evaluated by SDS-PAGE using broad-range protein molecular mass markers from Bio-Rad Laboratories. Prestained protein molecular mass markers from New England Biolabs were used for Western blotting. Protein concentrations were determined with the Advanced Protein Assay Reagent from Cytoskeleton or by use of the BCA reagent from Pierce. BSA was used as a standard.
Bacterial strains and plasmids.
The bacterial strains and plasmids used in this work are summarized in Table 1.
Table 1.
Bacterial strains and plasmids used in this study
Abbreviations: Amp, ampicillin; Apr, apramycin; Km, kanamycin; Spec, spectinomycin; Tet, tetracycline; Tsr, thiostrepton.
| Strain or plasmid | Relevant characteristics | Reference or source |
|---|---|---|
| Strains | ||
| S. viridifaciens MG456-hF10 | Valanimycin producer: wild-type | Yamato et al. (1986) |
| S. viridifaciens MG456-hF10 vlmI− mutant | vlmI : : pKC1139ΔvlmI, S. viridifaciens MG456-hF10 with single-crossover disruption of vlmI | This work |
| S. coelicolor strain M512 | ΔredD, ΔactII-orf4 | Fernandez-Moreno (1991) |
| S. coelicolor strain M145 | SCP1−, SCP2− | Kieser et al. (2000) |
| E. coli DH10B | Cloning host | BRL |
| E. coli BL21 (DE3) | Protein overexpression strain, TetR | Novagen |
| E. coli 12567 | RP4-Tc : : Mu-Km : : Tn7, SpecRdam−, dcm−, hsdM− | MacNeil et al. (1992) |
| Plasmids | ||
| pUZ8002 | Non-transmissible, oriT mobilizing plasmid, KmR | Kieser et al. (2000) |
| pKC1139 | E. coli–Streptomyces shuttle plasmid for conjugal transfer, with temperature-sensitive Streptomyces replicon, AprR | Bierman et al. (1992) |
| pWHM3 | E. coli–Streptomyces shuttle vector, TsrR, AmpR | Vara et al. (1989) |
| pWHM3-OriT | E. coli–Streptomyces shuttle vector, oriT, TsrR, AmpR | This work |
| pPM801 | E. coli–Streptomyces shuttle vector, oriT, TsrR | Mazodier et al. (1989) |
| pSET152 | Integrating Streptomyces plasmid, lacZ, intΦC31, repUC, AprR | Bierman et al. (1992) |
| pSET152-Tsr | pSET152 with TsrR and AmpR genes from pXY300 inserted | This work |
| pSET152-ErmE* | pSET152-Tsr with ermE* promoter inserted | This work |
| pBS-VII-41F | ermE* promoter cloned into multiple cloning site of pGEM-3zf, AmpR | B. Shen, University of Wisconsin-Madison |
| pVal35 | pVal38 with ∼3 kb SpeI fragment removed from the 3′ end of insert by digestion and religation | Garg et al. (2002) |
| pXY300 | Derivative of pKC1139, TsrR | Yin et al. (2003) |
| pGEM-T | TA cloning vector, AmpR | Promega |
| pCR2.1 –TOPO | TOPO cloning vector, KmR | Invitrogen |
| pFLAG-CTC | FLAG-tag expression vector, trc promoter, AmpR | Sigma Chemical Company |
| pMECA | Derivative of cloning vector pUC19 with large multiple cloning site, AmpR | Thomson & Parrott (1998) |
| pFLAG-CTC-VlmI-(L) | Longer version of vlmI spanning nucleotides 14 748–15 735 cloned into NdeI and EcoRI sites of pFLAG-CTC | This work |
| pFLAG-CTC-VlmI-(S) | Shorter version of vlmI spanning nucleotides 14 748–15 570 cloned into HindIII and EcoRI sites of pFLAG-CTC | This work |
| pKC1139 ΔvlmI | Internal 478 bp fragment of vlmI spanning nucleotides 14 963–15 442 cloned into XbaI site of pKC1139 | This work |
| pVlmI-CP2-WHM3 | Complementation plasmid produced by subcloning of ermE* promoter-VlmI-L cassette from pVlmI-CP2 into pWHM3 | This work |
| pVlmI-CP2-WHM3-OriT | Complementation plasmid produced by subcloning of ermE* promoter-VlmI-L cassette from pVlmI-CP2 into pWHM3-OriT | This work |
| pVlmK-pET28b | vlmK cloned into expression vector pET28b, KmR | X. Zhang and R. J. Parry, unpublished results |
| pET VlmRN1RC1 | vlmR cloned into pET28b, KmR | Parry et al. (1997) |
| pGEMTΔvlmR | Internal 414 bp fragment of vlmR spanning nucleotides 7080–7493 cloned into pGEM-T | This work |
| pVlmR-BS | Internal 414 bp vlmR fragment spanning nucleotides 7080–7493 cloned into XbaI/HindIII-digested plasmid BS-VII-41F | This work |
| pKC1139ΔvlmR | ermE* promoter and internal 414 bp fragment of vlmR spanning nucleotides 7080–7493 cloned into EcoRI/HindIII-digested pKC1139 | This work |
| pVlmA-CTC | vlmA cloned into pFLAG-CTC, AmpR | Garg et al. (2008) |
| pVlmAH | 443 bp intergenic region between vlmH and vlmA spanning nucleotides 4947–5389 cloned into pGEM-T | This work |
| pVlmIJ | 2.5 kb intergenic region between vlmI and vlmJ spanning nucleotides 15 723–17 754 cloned into pGEM-T | This work |
| pIJMECA-1 | 1.2 kb KpnI fragment of pVlmIJ cloned into KpnI site of pMECA | This work |
| pIJMECA-2 | Plasmid derived from pIJMECA-1 by digestion with SmaI and PmlI DNA followed by religation. Contains 231 bp vlmI–vlmJ intergenic region | This work |
DNA manipulations.
Genomic DNA was prepared from S. viridifaciens MG456-hF10 using DNAzol reagent (MRC) after macerating mycelium frozen with liquid nitrogen. Plasmid DNA was purified with a QIAprep Spin Plasmid kit (Qiagen). DNA fragments were isolated from agarose gels with a QIAquick Gel Extraction kit (Qiagen). PCR products were separated on agarose gels and purified from the gels. Digestion with restriction endonucleases and ligation experiments were carried out by standard procedures under conditions recommended by the manufacturers. Automated DNA sequencing was performed at Lone Star Sequencing Laboratories by using universal and synthetic oligonucleotide primers. End labelling to produce radioactive DNA fragments was carried out using [α-32P]dCTP and Klenow DNA polymerase (New England Biolabs) as per the manufacturer's recommendations. The purified, end-labelled DNA was digested with various restriction enzymes in the manufacturer's recommended buffers to produce the labelled DNA fragments needed to map the VlmI binding sites.
Isolation of intergenic DNA.
A 443 bp intergenic region between vlmA and vlmH in the valanimycin biosynthetic gene cluster was amplified from plasmid pVal35 (Garg et al., 2002) by PCR using the following primers: 5′-CTCGTATCGCGCTCAGTAC-3′ and 5′-ATCGAGAGAACGCATTCTGG-3′. The PCR product was gel-purified and cloned into pGEM-T to produce plasmid pVlmAH. The DNA insert in pVlmAH was released as a NotI fragment for end labelling. Similarly, a 2.5 kb DNA fragment between vlmI and vlmJ was amplified using the primers 5′-TTCTCCTTGCCATCGCTCATAGTT-3′ and 5′-AAGATCATTGATGGTCATGAAGA-3′, and the resulting fragment was cloned into pGEM-T. The resulting plasmid pVlmIJ was digested with NotI, and two DNA fragments corresponding to 1.0 and 1.5 kb in size were gel-purified and used for end labelling. A 1.2 kb KpnI fragment of intergenic DNA from pVlmIJ was gel-purified and cloned into the KpnI site of pMECA (Thomson & Parrott, 1998) to produce plasmid pIJMECA-1. pIJMECA-1 was then digested with SmaI and PmlI (for deletion of NotI–PmlI intergenic DNA) and religated to produce plasmid pIJMECA-2, which contains a 231 bp PmlI–KpnI fragment of DNA from the intergenic region between vlmI and vlmJ. This fragment was end-labelled after isolation as a HindIII–SpeI fragment.
Cloning of VlmI.
The DNA encoding VlmI was amplified by PCR from plasmid pVal35 with a mixture of Taq and Vent DNA polymerases using an N-terminal primer containing an NdeI site (5′-ATTATCATATGGCAAGGAGAACCCGCA-3′) and a C-terminal primer with an EcoRI site (5′-TTTATGAATTCGCGCCGGGTGCCATG-3′) (restriction sites are indicated by bold italic type). The PCR product was gel-purified and cloned into a pCR2.1-TOPO vector (Invitrogen), and the DNA sequence was verified by sequencing using vector primers. The vlmI gene was subsequently cloned into the NdeI and EcoRI sites of pFLAG-CTC to produce pFLAG-CTC-VlmI-(L). The stop codon of the vlmI gene was removed to place the vlmI gene in-frame with the FLAG-tag of the vector. A smaller version of the vlmI gene with a start site 165 bp downstream from the start site of the larger version was also cloned by PCR. In this case, the N-terminal primer, which contained a HindIII site, had the following sequence: 5′-TATAAAGCTTATGCTGAAGTTCCAGGATTTT-3′, while the C-terminal primer was the same as that used to amplify the longer version of the gene. The resulting PCR product was purified and cloned into pGEM-T, and the DNA sequence was verified by sequencing using vector primers. The shorter version of the vlmI gene was subsequently subcloned into the HindIII and EcoRI cloning sites of pFLAG-CTC to produce pFLAG-CTC-VlmI-(S).
Overexpression, purification and characterization of VlmI.
Overproduction and purification of VlmI-L and VlmI-S from pFLAG-CTC-VlmI-(L) and pFLAG-CTC-VlmI-(S) were carried out according to the protocols outlined in the FLAG vector instruction manual (Sigma). E. coli BL21 (DE3) cells harbouring the desired plasmid were grown overnight in Luria–Bertani (LB) broth supplemented with 100 μg ampicillin ml−1, and then diluted 100-fold into fresh 2× LB broth plus 0.4 % glucose and 100 μg ampicillin ml−1. The cultures were grown at 37 °C until OD600 ∼0.7 was reached, whereupon IPTG was added to a final concentration of 0.5 mM. After 2 h, the cells were harvested and stored at −20 °C until utilized for isolation of the desired protein. The overproduced protein was purified using a column of anti-FLAG antibody resin using a previously described protocol (Garg et al., 2006). The purified protein was stored at 4 °C in Tris-buffered saline buffer containing 15 % (v/v) glycerol. Western blotting with anti-GroEL antibodies (Sigma) was carried out with each protein preparation to check for the presence of GroEL contamination (Couch et al., 2002). Both the smaller and larger versions of VlmI co-purified with an additional two proteins. One of these contaminants appeared to be GroEL, since it reacted with anti-GroEL antibodies.
Disruption of the vlmI gene.
A single-crossover mutation was created to define the role of VlmI in valanimycin biosynthesis. An internal 478 bp fragment of the vlmI gene was amplified by PCR from pVal35 using an N-terminal primer with a SpeI restriction site (5′-ATTATACTAGTGGAGACACTCGTCGACGA-3′) and a C-terminal primer with a SpeI restriction site (5′-ATTATACTAGTATCAGCTGCTGATGAAGC-3′). The PCR product was cloned into pGEM-T, sequenced, and then subcloned into the XbaI site of the E. coli–Streptomyces shuttle vector pKC1139, which contains a temperature-sensitive Streptomyces replicon (Bierman et al., 1992). The resulting plasmid, pKC1139ΔvlmI, was then introduced into S. viridifaciens, and a single-crossover vlmI mutant was recovered using previously described methods (Garg et al., 2002). The genotype of the mutation was verified by Southern blot analysis of KpnI-digested genomic DNA with a 478 bp internal vlmI fragment obtained by EcoRI digestion of plasmid pGEMTΔvlmI. The vlmI mutant (S. viridifaciens-vlmI−) was assayed for valanimycin production in the manner previously described (Garg et al., 2002).
Complementation of S. viridifaciens vlmI mutant and of S. coelicolor M512.
Three complementation plasmids were constructed. The first was derived from the integrating plasmid pSET152 (Bierman et al., 1992). A portion of the vector pXY300 (Yin et al., 2003) carrying ampicillin and thiostrepton resistance genes was amplified by PCR using the following forward and reverse primers: 5′-GACGTCGAGCTCTTACCAATGCTTAATCAGTG-3′ and 5′-GACGTCGAGCTCTTATCGGTTGGCCGCGAGATT-3′, where the bases marked in bold and italic type correspond to a SacI site. The resulting PCR fragment was digested with SacI and then ligated into pSET152 that had been digested with SacI to remove most of the apramycin resistance gene. The resulting plasmid, pSET152-Tsr, was subsequently digested with EcoRI and XbaI, and then ligated to the 0.5 kb ermE* promoter cassette isolated from plasmid pBS-VII-41F (B. Shen, unpublished work) by digestion with the same restriction enzymes. The resulting plasmid was named pSET152-ErmE*. The longer version of the vlmI gene was amplified by PCR from pVal35 with a mixture of Taq and Vent DNA polymerases using the following forward and reverse primers: VlmI N BamHI, 5′-GGATCCGGAGGTACGGACATGGCAAGGAGAACCGC-3′, and VlmI C XbaI, 5′-TCTAGATCAGCGCCGGGTGCCATG-3′, where the bases marked in bold and italic type correspond to BamHI and XbaI sites, respectively. The resulting PCR product was cloned into vector pGEM-T and the insert was sequenced to verify the absence of errors. The vlmI gene was released from the pGEM-T construct by digestion with BamHI and XbaI, and then cloned into BamHI–XbaI-digested pSET152-ErmE* to place the longer version of the vlmI gene downstream of the ermE* promoter. The resulting plasmid was named pVlmI-CP2.
The additional complementation plasmids were derived from the high-copy-number plasmid pWHM3 (Vara et al., 1989). To create a version of pWHM3 that can be transferred by conjugation, an oriT fragment was removed from plasmid pPM801(Mazodier et al., 1989) as a PstI fragment and cloned into PstI-digested pWHM3 to give plasmid pWHM3-OriT. The vlmI gene and upstream ermE* promoter were then removed from pVlmI-CP2 as an EcoRI–XbaI fragment, and cloned into the same sites in pWHM3 and pWHM3-OriT to give complementation plasmids pVlmI-CP2-WHM3 and pVlmI-CP2-WHM3-OriT.
Plasmids pVlmI-CP2, pSET152-ErmE* and pVlmI-CP2-WHM3-OriT were each introduced into methylation-deficient E. coli strain ET12567 (pUZ8002) (Kieser et al., 2000; MacNeil et al., 1992) and then transferred to S. coelicolor M512 (Floriano & Bibb, 1996) by conjugation, as previously described (Garg et al., 2002). Plasmids pVlmI-CP2 and pVlmI-CP2-WHM3 were each passed through E. coli strain ET12567 (MacNeil et al., 1992) and then introduced into S. viridifaciens vlmI− by transformation. Exconjugants and transformants were selected for thiostrepton resistance (10 μg thiostrepton ml−1) and purified by two to three rounds of subculturing on R2YE medium (S. coelicolor) or ISP2 medium (S. viridifaciens). S. viridifaciens vlmI− containing pVlmI-CP2 or pVlmI-CP2-WHM3 was assayed for valanimycin production in several ways. TLC analysis of the ethyl acetate extract of the fermentation broth from the complemented strain exhibited a spot with an RF value very closely matching that of the valanimycin standard (Fig. 7). Furthermore, 1H NMR analysis of the compound purified from the complemented strain confirmed its identity to be valanimycin. The 1H spectrum exhibited vinyl resonances for C-3 at 6.32 and 6.25 p.p.m., the isobutyl CH2 resonance for C-4 at 4.12 p.p.m., the isobutyl CH resonance for C-5 at 2.47 p.p.m. and the isobutyl CH3 resonance for C-6 at 1.04 p.p.m. These values are consistent with the proton chemical shifts exhibited by authentic valanimycin (Garg et al., 2009; Ma & Parry, 2000). Finally the compound was shown to be valanimycin by LC-MS analysis of the crude ethyl acetate extract on a Bruker MicroTOF mass spectrometer, as described previously (Garg et al., 2009). The valanimycin exhibited the expected molecular mass: high-resolution ES-MS m/z, 173.09208 (M+H)+, calculated for C7H13N2O3, 173.09207, confirming the production of valanimycin by the complemented strain.
Fig. 7.
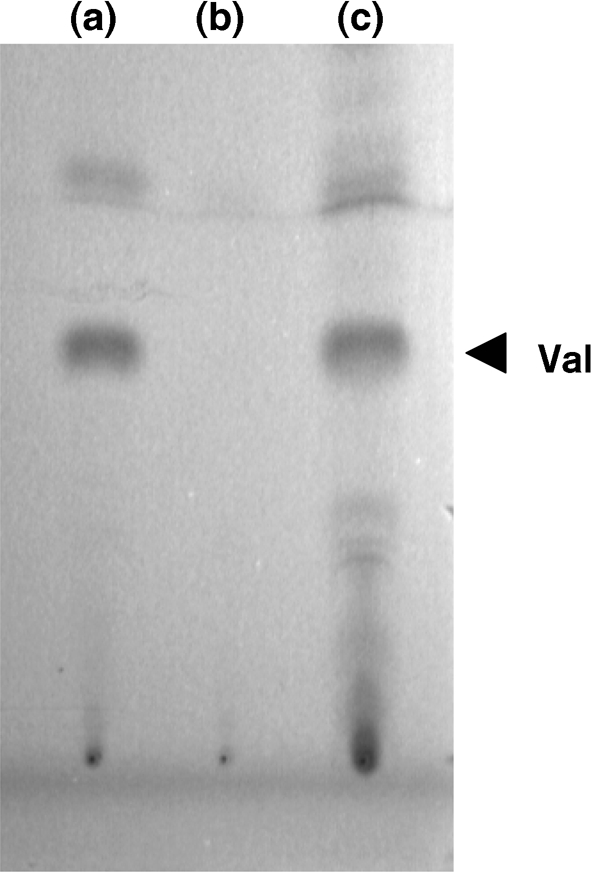
Thin-layer chromatogram (SiO2, CHCl3 : methanol : acetic acid, 5 : 1 : 0.05) of crude ethyl acetate extracts derived from fermentations of (a) wild-type S. viridifaciens, (b) S. viridifaciens vlmI− and (c) S. viridifaciens vlmI− (pVlmI-CP2-WHM3). Valanimycin was visualized with UV light. Val, valanimycin.
S. coelicolor M512 containing pVlmI-CP2, pSET152-ErmE* or pVlmI-CP2-WHM3-OriT was assayed for undecylprodigiosin or actinorhodin production by a modification (A. M. Cerdeno, personal communication) of the procedure of Scheu et al. (1997). Liquid R2YE medium (30 ml) was inoculated with a spore suspension (10 μl) of S. coelicolor M512 containing pVlmI-CP2, pSET152-ErmE* or pVlmI-CP2-WHM3-OriT, and the culture was shaken for 48 h at 30 °C and 250 r.p.m. A 5 ml aliquot of the culture broth was centrifuged and the cell pellet was resuspended in 2.5 ml methanol using a vortex mixer. The mixture was then centrifuged at 3250 g and the methanolic extract was removed. To detect the presence of undecylprodigiosin, concentrated HCl (50 μl) was added to a 1 ml aliquot of the methanol extract and the A530 was determined. To detect the presence of actinorhodin, 10 M NaOH (50 μl) was added to a 1 ml aliquot of the methanol extract and the A630 was measured. The concentration of undecylprodigiosin in a 1 ml aliquot was calculated using the reported molar absorption coefficient for the hydrochloride salt of the antibiotic at 530 nm (Wasserman et al., 1976). The redD+ strain S. coelicolor M145 was assayed for undecylprodigiosin in the same manner. The results are summarized in Table 2.
Table 2.
Complementation of redD mutation in S. coelicolor M512 with vlmI-L
| Strain | Undecylprodigiosin concn (μM)* |
|---|---|
| S. coelicolor M512 (ΔredD) pSETErmE* | 0.1 |
| S. coelicolor M512 (ΔredD) pVlmI-CP2 | 2.2 |
| S. coelicolor M512 (ΔredD) pVlmI-CP2-WHM3-OriT | 700.0 |
| S. coelicolor M145 (redD+) | 431.9 |
*See Methods for details.
Gel mobility shift assay.
DNA gel shift assays were performed as described previously (Garg et al., 2000). Typically, 20 μl assay mixture contained 4000 c.p.m. labelled DNA, 0.2–5 μg protein in 10 mM Tris (pH 7.5), 50 mM KCl, 1 mM EDTA, 2 mM DTT, 2.6 μg BSA and 2 μg sonicated salmon sperm DNA. Unlabelled DNA was added to reactions to check the specificity of DNA–protein interactions. Incubations were carried out at 30oC for 30 min and DNA binding was then analysed by electrophoresis in a Protean II Xi cell (Bio-Rad) using a 5 % native polyacrylamide gel in 0.5× TBE (75 mM Tris, 89 mM boric acid, 2 mM EDTA, pH 8.0) at 100 V for 12–24 h depending upon the size of DNA fragments. Autoradiography of the resulting gels was carried out at −80 °C using Hyperfilm (GE Healthcare).
Disruption of the vlmR gene.
A single-crossover, non-polar mutation with an outgoing ermE* promoter was generated to disrupt the vlmR gene. An internal 414 bp fragment of the vlmR gene was amplified by PCR from pVal35 (Garg et al., 2002) using an N-terminal primer with an XbaI restriction site (5′-TCTAGACGAGCTGCGAGGCCTC-3′) and a C-terminal primer with an HindIII restriction site (5′-AAGCTTCGGCCGTCGAGAACCT-3′). The PCR product was cloned into pGEM-T, sequenced, and then subcloned into XbaI- and HindIII-digested plasmid pBS-VII-41F, resulting in plasmid pVlmR-BS. This plasmid was then digested with EcoRI and HindIII to produce a 0.9 kb DNA fragment that contained the truncated vlmR gene downstream of the ermE* promoter. This DNA fragment was purified on an agarose gel and then subcloned into the E. coli–Streptomyces shuttle vector pKC1139 (Bierman et al., 1992) digested with EcoRI and HindIII. The resulting plasmid, pKC1139ΔvlmR, was introduced into S. viridifaciens by conjugation, and a single-crossover vlmR mutant was recovered using previously described methods (Garg et al., 2002). The genotype of the mutation was verified by Southern blot analysis of KpnI-digested genomic DNA from the mutant using a 414 bp internal vlmR fragment obtained by EcoRI digestion of plasmid pGEMTΔvlmR as the probe (data not shown). The vlmR mutant (S. viridifaciens vlmR−) was assayed for valanimycin production in the manner previously described (Garg et al., 2002) and found to be blocked in valanimycin production.
Preparation of VlmA, VlmK and VlmR antibodies.
VlmA, VlmK and VlmR were overproduced from the expression plasmids pVlmA-FLAG-CTC for VlmA (Garg et al., 2008), pVlmK-pET28b for VlmK (X. Zhang and R. J. Parry, unpublished data), and pET-VlmRN1RC1 for VlmR (Parry & Li, 1997a), respectively. Each protein was then purified by affinity chromatography as per the manufacturer's recommendations. Although the purified VlmK and VlmR needed no additional purification, VlmA was contaminated with GroEL and required further purification. This was accomplished by SDS-PAGE. The VlmA band was excised from a developed SDS-PAGE gel using UV light for visualization. The gel slice was then macerated with elution buffer (25 mM Tris, 150 mM NaCl, 0.1 %SDS), and the resulting protein solution was concentrated and the SDS removed by repeated filtration through a 10 kDa molecular-mass-cutoff filter. The purified proteins were each suspended in PBS (50 mM Na2HPO4, 0.85 % NaCl, pH 7.4) at a concentration of 1 mg ml−1. Antibodies to VlmK and VlmA were raised by Alpha Diagnostics, while antibodies to VlmR were prepared by Bethyl Laboratories. Rabbits served as the host animals. Serum samples from the second bleed after booster doses were used for Western analysis.
Western blot analyses.
S. viridifaciens spore suspensions (5 μl) derived from the wild-type and mutant strains were each used to inoculate 50 ml valanimycin production medium contained in a 250 ml conical flask and the cultures were then shaken at 37 °C and 250 r.p.m. for 15 h. The cell pellet from 3 ml of each culture was lysed in the manner previously described (Garg et al., 2008). A 100 μl aliquot of each lysate was mixed with an equal amount of 2× Laemmli buffer (Laemmli, 1970) and heated at 100 °C for 5 min. The mixtures were then centrifuged at 13 200 g, and 10–20 μl of each supernatant was loaded onto a 10 % SDS-PAGE gel along with prestained molecular mass markers and purified VlmK, VlmA and VlmR proteins as a positive control. Western blots were then performed as described previously (Garg et al., 1994) and detected with anti-rabbit IgG conjugated to alkaline phosphatase using the Sigma Fast BCIP-NBT reagent following the manufacturer's instructions.
RESULTS
Disruption of vlmI
To confirm that VlmI regulates the expression of the valanimycin biosynthetic gene cluster, a single-crossover disruption was created in the vlmI gene. The disruption plasmid pKC1139ΔvlmI was introduced into S. viridifaciens by conjugation and a single-crossover vlmI− mutant was recovered using previously described methods (Garg et al., 2002). The genotype expected for the mutation was verified by Southern blot analysis of KpnI-digested genomic DNA with a 478 bp internal vlmI fragment obtained by EcoRI digestion of plasmid pGEMTΔvlmI. Two hybridizing bands were observed at the expected positions (Fig. 2). When the vlmI-disrupted mutant was grown in valanimycin production medium and assayed for valanimycin production, no valanimycin production was observed. These results indicate that vlmI is essential for valanimycin biosynthesis.
Fig. 2.

(a) Diagrammatic illustration of single-crossover disruption in the vlmI gene created by plasmid pKC1139ΔvlmI, showing expected restriction fragments. (b) Southern blot analysis of KpnI-digested genomic DNA from wild-type S. viridifaciens MG456-hF10 (WT) and S. viridifaciens MG456-hF10 (vlmI : : pKC1139ΔvlmI). A 478 bp internal vlmI fragment obtained by EcoRI digestion of plasmid pGEMTΔvlmI was labelled with [α-32P]dCTP and used as a probe. KpnI sites are indicated by K.
Influence of VlmI on expression of valanimycin biosynthetic proteins
The organization of the valanimycin gene cluster suggests that the valanimycin biosynthetic genes are located on three potential transcripts: vlmHORBCD, vlmJKL and vlmA. In order to determine whether VlmI regulates the expression of all three of these transcripts, Western blotting experiments were carried out using polyclonal antibodies to proteins encoded by each potential transcript. Anti-VlmR antibodies were used to detect a translation product of the vlmHORBCD transcript, while anti-VlmK and -VlmA antibodies were used to detect a translation product of the vlmJKL and vlmA transcripts, respectively. The Western blot analyses utilized cell-free extracts from wild-type and mutant strains as well as the purified VlmR, VlmK and VlmA proteins as positive controls (Fig. 3). The results showed that extracts prepared from the vlmI mutant contained no cross-reacting material corresponding to the VlmR and VlmA proteins and a very low level of the VlmK protein. Extracts of wild-type S. viridifaciens used as positive control showed the presence of all these proteins, ruling out the possible instability of these proteins in the cell-free extracts. When single-crossover vlmA and vlmR mutants were used as negative controls, Western analysis showed that these proteins were absent, as expected. However, a single-crossover vlmK mutant used as a negative control appeared to show some cross-reacting material. This may result from the tendency of the vlmK mutant to revert (Garg et al., 2002) or from the presence of an antigenic, truncated version of VlmK produced by translational fusion of vlmK with the vector and termination at a vector stop codon. The results from the Western analyses suggest that VlmI regulates the expression of all of the transcripts that encode valanimycin biosynthetic enzymes.
Fig. 3.

Western blot analyses of cell-free extracts of S. viridifaciens MG456-hF10 strains using anti-VlmA, -VlmR and -VlmK antibodies. The strains used include the wild-type (WT), the single-crossover vlmI mutant vlmI : : pKC1139ΔvlmI (VlmI−), a single-crossover vlmA mutant (VlmA−), a single-crossover vlmR mutant (VlmR−) and a single-crossover vlmK mutant (VlmK−). Overproduced VlmA, VlmR and VlmK were used as positive controls. Proteins were detected with anti-rabbit IgG conjugated to alkaline phosphatase using the Sigma-Fast BCIP NBT reagent.
Overexpression and characterization of VlmI
Analysis of the vlmI gene reveals the presence of two potential start sites. Translation from the downstream start site yields a shorter version of VlmI that differs from the longer version by the absence of 55 amino acids at the N terminus. The longer (L) and shorter (S) versions of VlmI were overproduced in E. coli as C-terminal FLAG-tagged proteins using a pFLAG-CTC vector in which expression is driven from the inducible trc promoter. Both proteins were overproduced mainly in soluble form with yields of approximately 2 mg protein l−1 of culture broth. The maximum amounts of protein were observed after 2 h induction at 37 °C. Longer incubation at 37 °C or the use of lower temperatures did not improve the yield or quality of the overproduced proteins. On SDS-PAGE analysis, both the longer and shorter versions of VlmI exhibited the molecular masses expected for the denatured proteins, but the proteins contained contaminating proteins (data not shown). Western blot analysis of VlmI-L using anti-FLAG antibodies detected almost equal quantities of two proteins with molecular masses of about 38 and 32 kDa. These values correspond to the predicted molecular masses for VlmI-L and VlmI-S. The protein with the smaller molecular mass might be produced from an internal promoter that is present in the longer vlmI sequence or by proteolytic degradation of VlmI-L. As expected, overproduced VlmI-S was not contaminated with VlmI-L. Two additional proteins appeared as contaminants in overproduced, affinity-purified VlmI-L and VlmI-S. One protein, with a mass of 59 kDa, appeared to be GroEL, since it reacted with anti-GroEL antibodies, while the identity of the second protein, with a mass of 75 kDa, was not established.
DNA gel shift mobility assays with overproduced VlmI protein
As previously reported (Garg et al., 2002), the intergenic region between vlmA and vlmH exhibits three imperfect heptameric repeats with a conserved spacing found in other SARP binding sites (TCACGTG-15x-TCAGGTG-4x-TCAGGGA). A similar group of three imperfect heptameric repeats with a conserved spacing occurs upstream of vlmJ (TCACAGG-15x-TCAGAAA-4x-TCAGGAA). A sequence alignment of these heptameric repeats with verified and putative SARP binding sites present in other antibiotic biosynthetic gene clusters is shown in Fig. 4. DNA gel shift assays were conducted to evaluate the putative VlmI binding sites. A 443 bp intergenic region between vlmA and vlmH was amplified by PCR and labelled by end filling with Klenow DNA polymerase and [α-32P]dCTP after restriction enzyme digestion. DNA binding assays were carried out using purified, C-terminal FLAG-tagged forms of VlmI-L and VlmI-S. VlmI-L exhibited tight binding to the 443 bp DNA fragment, with nearly 100 % of the DNA being shifted when 3.2 μg of protein was added. Addition of a 50-fold excess of the unlabelled form of the 443 bp DNA fragment to the binding assay resulted in reversal of VlmI binding to the radiolabelled DNA, thereby showing that the binding is specific (Fig. 5a). In contrast to the behaviour of VlmI-L, VlmI-S did not produce any gel shift with the 443 bp DNA fragment (Fig. 5b). The lack of DNA binding activity displayed by VlmI-S also shows that the DNA binding activity exhibited by overproduced VlmI-L, which is contaminated with VlmI-S, is entirely due to VlmI-L.
Fig. 4.

Sequence alignment of verified and putative SARP binding sites present in the valanimycin (vlm), actinorhodin (act), daunorubicin (dnr) and fredericamycin (fdm) biosynthetic gene clusters (Wietzorrek & Bibb, 1997; Chen et al., 2008). Conserved residues are indicated by asterisks, while spacings between the binding sites are indicated numerically.
Fig. 5.

(a) DNA gel shift assays showing binding of VlmI-L to a 443 bp vlmA–vlmH intergenic DNA fragment. The 443 bp DNA fragment was labelled at both ends with [α-32P]dCTP. The assays in lanes 1–6 show the behaviour when the complete 443 bp fragment was mixed with increasing amounts of VlmI-L. Lane 7 shows the behaviour when the labelled DNA fragment plus 4 μg VlmI-L was mixed with a 50-fold excess of the unlabelled 443 bp DNA fragment. Lanes 8–11 show the results of gel shift assays after the labelled 443 bp fragment was digested with PmlI, and lanes 12–15 show the results of gel shift assays after digestion of the same fragment with SmaI. The quantity of VlmI used in each assay is shown at the top of each lane. S and P show the positions of the SmaI and PmlI sites, respectively. (b) Comparison of DNA gel shifts produced by VlmI-L and VlmI-S with a radiolabelled 443 bp vlmA–vlmH intergenic DNA fragment.
Localization of the VlmI binding site within the vlmA–vlmH intergenic region
The binding of VlmI to the vlmA–vlmH intergenic region having been established, experiments were carried out to localize the binding site within this DNA region. The labelled 443 bp DNA fragment was digested with several restriction enzymes and gel shift assays were carried out with the labelled fragments to determine which fragments bound to VlmI. The clearest results were obtained when the end-labelled 443 bp fragment was digested with PmlI. Since two PmlI sites are present within the 443 bp fragment, digestion resulted in the formation of two end-labelled fragments 258 and 128 bp in size as well as an unlabelled 57 bp fragment. Both of the labelled PmlI DNA fragments failed to show any gel mobility shift (Fig. 5a), thereby suggesting that the 57 bp fragment was most probably essential for VlmI binding. Since the DNA sequence of this 57 bp fragment contains the tandem heptameric repeat sequences TCACGTG-15-TCAGGTG-4-TCAGGGA, these results support the hypothesis that these repeats constitute the VlmI binding site. One SmaI site is present in the 443 bp labelled DNA fragment. Partial digestion of the labelled fragment with SmaI produced two end-labelled fragments of 250 and 193 bp in size.
The 250 bp fragment contains the 57 bp DNA sequence with the potential VlmI binding site. As expected, the undigested 443 bp fragment remaining after partial digestion and the 250 bp fragment both exhibited gel shifts, while the 193 bp fragment exhibited no gel shift (Fig. 5a). This behaviour is consistent with the hypothesis that the tandem heptameric repeat sequences TCACGTG-15x-TCAGGTG-4x-TCAGGGA are responsible for VlmI binding, since this sequence resides on the 250 bp fragment produced by SmaI digestion.
DNA gel shift assay for the intergenic region between vlmI and vlmJ
The 2.5 kb intergenic DNA between vlmI and vlmJ was also evaluated for VlmI binding properties. The 2.5 kb fragment was cleaved into 1.0 and 1.5 kb fragments by digestion with NotI and the fragments were then end-labelled after gel purification. Gel shift assays showed that VlmI-L did not bind to the 1 kb fragment of DNA located immediately upstream of the vlmI start site. However, VlmI-L did exhibit binding to the 1.5 kb segment of DNA that is located immediately upstream from vlmJ. The 1.5 kb fragment was further digested with several restriction enzymes, and gel shift assays were carried out with the labelled fragments to determine which fragments bound to VlmI. The results are summarized in Fig. 6(a). When the 1.5 kb segment was digested with PmlI, two fragments with sizes of 1.2 and 0.33 kb were produced. Of these two, only the 1.2 kb fragment exhibited a gel mobility shift (Fig. 6b). When the 1.5 kb fragment was digested with KpnI, a 0.57 kb fragment was produced that showed a positive gel shift with VlmI, while the remaining KpnI fragments showed no affinity to VlmI (Fig. 6b). A 231 bp PmlI–KpnI fragment common to the 1.2 and 0.57 kb fragments was then subcloned and used for gel shift assays. This fragment exhibited a strong gel shift in the presence of VlmI-L (Fig. 6b). This result is consistent with the fact that the 231 bp fragment contains a set of heptameric repeats predicted to be a VlmI binding site (TCACAGG-15x-TCAGAAA-4x-TCAGGAA). Additional evidence to support the hypothesis that these heptameric repeats constitute a VlmI binding site was obtained by digestion of the 231 bp fragment with BsaHI, which cleaves the DNA between the TCAGAAA and TCAGGAA repeats. Neither of the two DNA fragments produced by BsaHI digestion exhibited a gel mobility shift (Fig. 6b).
Fig. 6.

(a) Summary of DNA gel shifts produced by VlmI-L with the 2.5 kb vlmI–vlmJ intergenic region and its digestion products. B, N, P and K indicate results of digestion with BsaHI, NotI, PmlI and KpnI, respectively. (b) DNA gel shifts produced by Vlm-L with a radiolabelled 1.5 kb NotI fragment from the vlmI–vlmJ intergenic region and fragments derived from it by digestion with PmlI, KpnI and BsaHI. Fragment sizes are given in kb. The main text provides additional details.
Complementation of S. viridifaciens vlmI mutant and of S. coelicolor redD mutation
A copy of the longer version of vlmI was cloned into the integrating vector pSET152-ErmE* and into the high-copy-number, non-integrating vectors pWHM3 and pWHM3-OriT to produce the complementation plasmids pVlmI-CP2, pVlmI-CP2-WHM3 and pVlmI-CP2-WHM3-OriT, respectively. In order to determine whether VlmI can complement mutations in the SARP genes redD and actII-orf4, which respectively control undecylprodigiosin and actinorhodin production in S. coelicolor, the complementation plasmids were introduced into S. coelicolor M512 (ΔredD, ΔactII-orf4) (Floriano & Bibb, 1996). Spectrophotometric analysis was used to measure the concentration of undecylprodigiosin and actinorhodin in methanol extracts of the mycelium, as described in Methods. When pVlmI-CP2 was introduced into S. coelicolor M512, a low level of undecylprodigiosin production was detectable (Table 2). When pVlmI-CP2-WHM3-OriT was used for complementation, the amount of undecylprodigiosin produced was about 300 times greater than that observed with the integrating plasmid pVlmI-CP2, and it was about 1.5 times higher than the level of undecylprodigiosin produced by the redD+ strain S. coelicolor M145 (Table 2). Production of actinorhodin was not detected with either complementation plasmid. These observations indicate that vlmI can complement the redD mutation, but it is unable to complement the actII-orf4 mutation.
When plasmid pVlmI-CP2 was introduced into the S. viridifaciens vlmI− mutant, it failed to restore valanimycin production. However, the introduction of the high-copy-number plasmid pVlmI-CP2-WHM3 successfully complemented the vlmI mutation. TLC analysis of the ethyl acetate extract of the fermentation broth from the complemented strain exhibited a spot with an RF value very closely matching that of the valanimycin standard (Fig. 7). This compound was confirmed to be valanimycin by NMR spectroscopy and high-resolution electrospray ionization-MS analysis.
DISCUSSION
Genes that encode SARPs have been found in a number of antibiotic biosynthetic gene clusters, including those for actinorhodin, undecylprodigiosin, daunorubicin and cephamycin. Sequence analysis of the vlmI gene from the valanimycin biosynthetic gene cluster indicates that VlmI is a member of the SARP family of transcriptional activators. The vlmI gene contains two potential start sites. Translation from the downstream start site yields a version of VlmI that differs from the longer version by the absence of 55 amino acids at the N terminus. In this study, we have demonstrated by gel shift mobility assays that the longer version of VlmI (VlmI-L) binds to a bidirectional promoter in the intergenic region between vlmA and vlmH as well as to a promoter region upstream of vlmJ. Both of these regions contain SARP boxes that are similar to those that have been shown to be the binding sites for ActII-Orf4 and DnrI (Fig. 4). VlmI therefore controls the expression of most, if not all, of the valanimycin biosynthetic genes. The studies also show that the shorter version of VlmI does not bind to these promoter regions. This suggests that the additional 55 amino acids present in the N terminus region of VlmI-L are important for the proper folding of the protein. The failure of the shorter version of VlmI to bind DNA was unexpected, since the N-terminal region of VlmI-L does not contain any conserved motifs and it does not appear to be present in most of the other SARPs whose sequences have been reported. However, the transcriptional activator for the undecylprodigiosin biosynthetic pathway, RedD, is an exception to this pattern, since it contains an extended N terminus that exhibits some sequence similarities to the N terminus of VlmI-L (Garg et al., 2002). The presence of an extended N terminus in both VlmI-L and RedD may explain our finding that VlmI-L is able to complement a redD mutation in S. coelicolor, but not an actII-orf4 mutation in the same organism. Complementation of the redD mutation was much more efficient when vlmI-L was introduced into the M512 strain using a high-copy-number plasmid instead of an insertional plasmid. This behaviour is presumably due to the higher level of VlmI expression achieved with the high-copy-number vector. Attempts to complement a vlmI mutation in S. viridifaciens with the insertional plasmid bearing vlmI-L were unsuccessful, while the use of a high-copy-number plasmid resulted in wild-type levels of valanimycin production (Fig. 7). These results may also be the consequence of the higher level of VlmI expression produced by the multicopy plasmid. Gel shift studies and Western blot analyses indicate that VlmI-L binds to the vlmA–vlmH intergenic region and activates transcription of both the vlmA and the vlmHORBCD transcripts from the divergently arranged promoters in this region. A precedent for this type of behaviour is provided by DnrI and ActII-Orf4, which have also been shown to activate bidirectional transcription (Arias et al., 1999; Tang et al., 1996). Gel shift studies indicate that VlmI-L binds to a region of heptameric repeats upstream from vlmJ. The location of this VlmI binding site is unusual since it lies about 0.9 kb upstream of the predicted vlmJ translational start site. Analysis of the sequence between the VlmI binding site and the vlmJ start site reveals the presence of seven direct 122 bp repeats. These repeats do not represent an N-terminal extension of VlmJ, since they are not in-frame with VlmJ. An analysis of the repeat sequence with the MFOLD program (http://mfold.bioinfo.rpi.edu/) suggests that each repeat forms a complex hairpin structure. The location of the VlmI binding site at a relatively large distance from the translational start of vlmJ and the presence of these repeats may indicate that another regulatory protein is involved in the regulation of the vlmJKL transcript. This might explain the weaker binding affinity of VlmI to this region and the leaky VlmK phenotype observed in the vlmI disruptant.
Acknowledgments
This work was supported by National Institutes of Health grant GM053818 and Robert A. Welch Foundation grant C-0729. We thank Professor Ben Shen, University of Wisconsin-Madison, for plasmid pBS-VII-41F, Dr Xiujun Zhang, Rice University, for plasmid pVlmK-pET28b, and Professor Mervyn Bibb, John Innes Centre, Norwich, UK, for S. coelicolor M512. We also thank Dr Lawrence Alemany for determination of NMR spectra, Dr Tan Guo for measurement of high-resolution mass spectra, and Dr Jane Coughlin for helpful suggestions on ways to improve the manuscript.
Abbreviations
SARP, Streptomyces antibiotic regulatory protein
References
- Arias, P., Fernandez-Moreno, M. A. & Malpartida, F. (1999). Characterization of the pathway-specific positive transcriptional regulator for actinorhodin biosynthesis in Streptomyces coelicolor A3(2) as a DNA-binding protein. J Bacteriol 181, 6958–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bibb, M. J. (2005). Regulation of secondary metabolism in streptomycetes. Curr Opin Microbiol 8, 208–215. [DOI] [PubMed] [Google Scholar]
- Bierman, M., Logan, R., Obrien, K., Seno, E. T., Rao, R. N. & Schoner, B. E. (1992). Plasmid cloning vectors for the conjugal transfer of DNA from Escherichia coli to Streptomyces spp. Gene 116, 43–49. [DOI] [PubMed] [Google Scholar]
- Chen, Y., Wendt-Pienkowski, E. & Shen, B. (2008). Identification and utility of FdmR1 as a Streptomyces antibiotic regulatory protein activator for fredericamycin production in Streptomyces griseus ATCC 49344 and heterologous hosts. J Bacteriol 190, 5587–5596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couch, R., Seidle, H. & Parry, R. J. (2002). Construction of expression vectors to produce affinity-tagged proteins in Pseudomonas. Biotechniques 32, 1230–1236. [DOI] [PubMed] [Google Scholar]
- Fernandez-Moreno, M. A., Caballero, J. L., Hopwood, D. A. & Malpartida, F. (1991). The act cluster contains regulatory and antibiotic export genes, direct targets for translational control by the bldA tRNA gene of Streptomyces. Cell 66, 769–780. [DOI] [PubMed] [Google Scholar]
- Floriano, B. & Bibb, M. (1996). afsR is a pleiotropic but conditionally required regulatory gene for antibiotic production in Streptomyces coelicolor A3(2). Mol Microbiol 21, 385–396. [DOI] [PubMed] [Google Scholar]
- Garg, R. P., Menon, A. L., Jacobs, K., Robson, R. M. & Robson, R. L. (1994). The hypE gene completes the gene cluster for H2-oxidation in Azotobacter vinelandii. J Mol Biol 236, 390–396. [DOI] [PubMed] [Google Scholar]
- Garg, R. P., Yindeeyoungyeon, W., Gilis, A., Denny, T. P., Van Der Lelie, D. & Schell, M. A. (2000). Evidence that Ralstonia eutropha (Alcaligenes eutrophus) contains a functional homologue of the Ralstonia solanacearum Phc cell density sensing system. Mol Microbiol 38, 359–367. [DOI] [PubMed] [Google Scholar]
- Garg, R. P., Ma, Y., Hoyt, J. C. & Parry, R. J. (2002). Molecular characterization and analysis of the biosynthetic gene cluster for the azoxy antibiotic valanimycin. Mol Microbiol 46, 505–517. [DOI] [PubMed] [Google Scholar]
- Garg, R. P., Gonzalez, J. M. & Parry, R. J. (2006). Biochemical characterization of VlmL, a seryl-tRNA synthetase encoded by the valanimycin biosynthetic gene cluster. J Biol Chem 281, 26785–26791. [DOI] [PubMed] [Google Scholar]
- Garg, R. P., Qian, X. L., Alemany, L. B., Moran, S. & Parry, R. J. (2008). Investigations of valanimycin biosynthesis: elucidation of the role of seryl-tRNA. Proc Natl Acad Sci U S A 105, 6543–6547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg, R. P., Alemany, L. B., Moran, S. & Parry, R. J. (2009). Isolation, characterization, and bioconversion of a new intermediate in valanimycin biosynthesis. J Am Chem Soc 131, 9608–9609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F. & Hopwood, D. A. (2000). Practical Streptomyces Genetics. Norwich: The John Innes Foundation.
- Laemmli, U. K. (1970). Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227, 680–685. [DOI] [PubMed] [Google Scholar]
- Ma, Y. & Parry, R. J. (2000). A novel valanimycin-resistance determinant (vlmF) from Streptomyces viridifaciens MG456-hF10. Microbiology 146, 345–352. [DOI] [PubMed] [Google Scholar]
- MacNeil, D. J., Gewain, K. M., Ruby, C. L., Dezeny, G., Gibbons, P. H. & Macneil, T. (1992). Analysis of Streptomyces avermitilis genes required for avermectin biosynthesis utilizing a novel integration vector. Gene 111, 61–68. [DOI] [PubMed] [Google Scholar]
- Mazodier, P., Petter, R. & Thompson, C. (1989). Intergeneric conjugation between Escherichia coli and Streptomyces species. J Bacteriol 171, 3583–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narva, K. E. & Feitelson, J. S. (1990). Nucleotide sequence and transcriptional analysis of the redD locus of Streptomyces coelicolor A3(2). J Bacteriol 172, 326–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parry, R. J. & Li, W. (1997a). An NADPH : FAD oxidoreductase from the valanimycin producer, Streptomyces viridifaciens. Cloning, analysis, and overexpression. J Biol Chem 272, 23303–23311. [DOI] [PubMed] [Google Scholar]
- Parry, R. J. & Li, W. (1997b). Purification and characterization of isobutylamine N-hydroxylase from the valanimycin producer Streptomyces viridifaciens MG456-hF10. Arch Biochem Biophys 339, 47–54. [DOI] [PubMed] [Google Scholar]
- Parry, R. J., Li, W. & Cooper, H. N. (1997). Cloning, analysis, and overexpression of the gene encoding isobutylamine N-hydroxylase from the valanimycin producer, Streptomyces viridifaciens. J Bacteriol 179, 409–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Llarena, F. J., Liras, P., Rodriguez-Garcia, A. & Martin, J. F. (1997). A regulatory gene (ccaR) required for cephamycin and clavulanic acid production in Streptomyces clavuligerus: amplification results in overproduction of both β-lactam compounds. J Bacteriol 179, 2053–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheu, A. K., Martinez, E., Soliveri, J. & Malpartida, F. (1997). abaB, a putative regulator for secondary metabolism in Streptomyces. FEMS Microbiol Lett 147, 29–36. [DOI] [PubMed] [Google Scholar]
- Stutzman-Engwall, K. J., Otten, S. L. & Hutchinson, C. R. (1992). Regulation of secondary metabolism in Streptomyces spp. and overproduction of daunorubicin in Streptomyces peucetius. J Bacteriol 174, 144–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, A., Takano, Y., Ohnishi, Y. & Horinouchi, S. (2007). AfsR recruits RNA polymerase to the afsS promoter: a model for transcriptional activation by SARPs. J Mol Biol 369, 322–333. [DOI] [PubMed] [Google Scholar]
- Tang, L., Grimm, A., Zhang, Y. X. & Hutchinson, C. R. (1996). Purification and characterization of the DNA-binding protein DnrI, a transcriptional factor of daunorubicin biosynthesis in Streptomyces peucetius. Mol Microbiol 22, 801–813. [DOI] [PubMed] [Google Scholar]
- Thomson, J. M. & Parrott, W. A. (1998). pMECA: a cloning plasmid with 44 unique restriction sites that allows selection of recombinants based on colony size. Biotechniques 24, 922–928. [DOI] [PubMed] [Google Scholar]
- Vara, J., Lewandowska-Skarbek, M., Wang, Y. G., Donadio, S. & Hutchinson, C. R. (1989). Cloning of genes governing the deoxysugar portion of the erythromycin biosynthesis pathway in Saccharopolyspora erythraea (Streptomyces erythreus). J Bacteriol 171, 5872–5881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserman, H. H., Rodgers, G. C. & Keith, D. D. (1976). Undecylprodigiosin. Tetrahedron 32, 1851–1854. [Google Scholar]
- Wietzorrek, A. & Bibb, M. (1997). A novel family of proteins that regulates antibiotic production in streptomycetes appears to contain an OmpR-like DNA-binding fold. Mol Microbiol 25, 1181–1184. [DOI] [PubMed] [Google Scholar]
- Yamato, M., Iinuma, H., Naganawa, H., Yamagishi, Y., Hamada, M., Masuda, T. & Umezawa, H. (1986). Isolation and properties of valanimycin, a new azoxy antibiotic. J Antibiot 39, 184–191. [DOI] [PubMed] [Google Scholar]
- Yin, X., O'Hare, T., Gould, S. J. & Zabriskie, T. M. (2003). Identification and cloning of genes encoding viomycin biosynthesis from Streptomyces vinaceus and evidence for involvement of a rare oxygenase. Gene 312, 215–224. [DOI] [PubMed] [Google Scholar]