

Abstract
While the outcome for pediatric patients with lymphoproliferative disorders (LPD) or lymphoid malignancies, such as acute lymphoblastic leukemia (ALL), has improved dramatically, patients often suffer from therapeutic sequelae. Additionally, despite intensified treatment, the prognosis remains dismal for patients with refractory or relapsed disease. Thus, novel biologically targeted treatment approaches are needed. These targets can be identified by understanding how a loss of lymphocyte homeostasis can result in LPD or ALL. Herein, we review potential molecular and cellular therapeutic strategies that (i) target key signaling networks (e.g., PI3K/AKT/mTOR, JAK/STAT, Notch1, and SRC kinase family-containing pathways) which regulate lymphocyte growth, survival, and function; (ii) block the interaction of ALL cells with stromal cells or lymphoid growth factors secreted by the bone marrow microenvironment; or (iii) stimulate innate and adaptive immune responses.
Keywords: Acute lymphoblastic leukemia, Lymphoproliferative disorders, ALPS, PI3K/AKT/mTOR pathway, JAK/STAT pathway, Notch 1 pathway, Gamma secretase inhibitors, Rapamycin, CpG ODN, NOD/SCID mice, Bone marrow microenvironment
Introduction
There are a wide variety of diseases characterized by abnormal proliferation of lymphocytes, with differing clinical manifestations and existing along a spectrum from benign to malignant. Underlying all these disorders, however, are subversions of normal lymphocyte signaling pathways that contribute to the pathologic process, can be characterized, and potentially be targeted. On one end of this spectrum, acute lymphoblastic leukemia (ALL) is the most common cancer of childhood [1]. The majority of pediatric patients with ALL are cured with current treatment regimens. However, for the 20–30% of pediatric patients who relapse or are refractory to conventional therapy and for adults with ALL, the prognosis remains dismal despite intensified therapy [2, 3]. Relapsed ALL, counted as a separate disease, would be the 5th most common pediatric malignancy and the number one cause of pediatric cancer mortality [4]. Patients with nonmalignant lymphoproliferative disorders (LPD) can be difficult to diagnose, suffer from chronic infections that can be life-threatening, have a decreased life expectancy, and often require long-term treatment with immunosuppressive agents, including steroids, which can result in long-term debilitating consequences. In order to improve outcome and potentially reduce side effects and long-term sequelae from current cytotoxic therapy, the development of novel, biologically relevant, molecular and cellular targeted therapies is critical. Our general approach to identifying these potential biologic targets is to study how dysfunctional B and T cell development and growth relates to normal lymphoid signaling and cellular processes. Normal B and T cell development is tightly regulated by a host of molecules and processes (e.g., lymphoid growth factors secreted by bone marrow and thymic stroma, adhesion molecule receptors, signals related to antigen receptors and the processes involved in limiting the proliferative burst of lymphocytes involved in effector responses) in order to maintain B- and T-lymphocyte homeostasis [5]. An imbalance of these signals can lead to LPD or lymphoid malignancies.
Potential targeted therapies can be identified and tested using in vitro assays, in vitro bone marrow stromal cell culture systems, murine models of ALL and LPD and xenografts of primary human ALL. This review will focus on recent work in the lab to characterize important signal transduction pathways and potential therapeutic targets in ALL and LPD. We have employed three approaches to the identification and testing of novel biologically relevant targets by understanding the pathogenesis of ALL and LPD. We will initially discuss targeting key integrated signaling networks that regulate cell growth, survival, proliferation, and function of these abnormal lymphocytes. Then, we will describe the contribution of lymphoid growth factors secreted by the bone marrow and thymic stromal microenvironment to sustaining abnormal lymphoid cell survival. Finally, we will discuss potential strategies to exploit the immune response to these abnormal cells.
Novel molecular-targeted therapeutics for ALL and LPD
Signal transduction pathways, including PI3K/AKT/mTOR, JAK/STAT, ABL tyrosine kinase, SRC family of tyrosine kinases, and Notch1, are critical for the orchestration of normal B and T cell development, proliferation, survival, and activation [6–9]. These pathways are highly integrated networks that are tightly regulated by intra- and extracellular signals. Deregulation of these networks are key events in the pathogenesis of LPD and/or leukemia/lymphoma, making intermediates in these pathways excellent candidates for molecular targeting. However, targeting single pathway intermediates may not prove ultimately successful because of the crosstalk that occurs among these pathways. This phenomenon, which may lead to acquired resistance to single signal transduction inhibitors, needs to be anticipated as these targeted agents move from the laboratory to the clinic.
Molecular-targeted therapy for ALL
Molecules that inhibit intermediates of key deregulated signaling networks and/or aberrant expression of protein kinases are attractive therapeutics for the treatment of leukemia. The prototype drug of this new treatment paradigm is the tyrosine kinase inhibitor imatinib, which was originally developed for the treatment of BCR-ABL+ CML. Table 1 summarizes molecular targeted agents that are being actively investigated in the preclinical and/or clinical setting for acute leukemia and LPD.
Table 1.
Summary of molecular targeted therapeutics for the treatment of ALL and LPD
| Inhibitor | Target intermediate or pathway | Current or potential lymphoid malignancy |
|---|---|---|
| Imatinib | BCR-ABL, ABL, PDGFR, c-KIT | Ph+ ALL |
| Nilotinib | BCR-ABL, ABL, PDGFR, c-KIT | Ph+ ALL |
| Dasatinib | BCR-ABL, ABL,SRC kinase family, c-KIT, PDGFR, EphrinR | Ph+ ALL, Ph− ALL |
| Rapamycin | mTORC1 (+/− mTORC2) | B- and T-ALL, Ph+ ALL, LPD, ALPS |
| RAD-001 | mTORC1 (+/− mTORC2) | B- and T-ALL, Ph+ ALL, LPD, ALPS |
| CCI-779 | mTORC1 (+/− mTORC2) | B- and T-ALL, Ph+ ALL, LPD, ALPS |
| PI-103 | PI3K, mTORC1, mTORC2 | B- and T-ALL, Ph+ ALL |
| LY294002 | PI3K | B- and T-ALL, Ph+ ALL |
| Perifostine | AKT | B- and T-ALL, Ph+ ALL |
| Tricibine | AKT | B- and T-ALL, Ph+ ALL |
| A-443654 | AKT | B- and T-ALL, Ph+ ALL |
| SGI-1776 | PIM-1 | B- and T-ALL, Ph+ ALL |
| mAb A7R34 | Antagonistic monoclonal antibody to IL-7Rα chain | B- and T-ALL, Ph+ ALL |
| RCP168 peptide | CXCR4 antagonist | B- and T-ALL, Ph+ ALL, LPD, ALPS |
| Gamma-secretase inhibitor | Notch1 pathway | T-ALL, LPD, ALPS |
Targeting BCR-ABL in Ph+ ALL
BCR-ABL is a non-membrane bound fusion oncoprotein that is aberrantly expressed from the resultant reciprocal translocation between chromosomes 9 and 22 (Ph+ chromosome) [10, 11]. This translocation places the c-ABL tyrosine kinase gene under the transcriptional control of BCR [12]. Depending upon the breakpoint, the presence of Ph+ chromosome results in the constitutively active p210 BCR-ABL tyrosine kinase (detected in >95% of patients with CML) or p190 BCR-ABL tyrosine kinase (found in 3% of pediatric and 20% of adult patients with ALL) [11, 12]. While the outcome of patients with CML is relatively favorable, the prognosis for patients with Ph+ ALL is dismal, with an overall 2-year event free survival rate of only 10–20% ([13] and reviewed in [14]). BCR-ABL upregulates multiple signaling networks, including Ras/Raf/MEK/ERK, PI3K/AKT/mTOR, and JAK/STAT, and is associated with the activation of members of the SRC family of tyrosine kinases (e.g., LYN and HCK) and of MYC [15–19]. Expression of BCR-ABL in hematopoietic cells results in cytokine-independent proliferation, resistance to apoptosis, and altered responses to cell–cell and cell–matrix interactions [20–22]. BCR-ABL’s essential role in malignant transformation has made it an excellent candidate for molecular-targeted therapy for Ph+ leukemia [23]. Imatinib mesylate and nilotinib are tyrosine kinase inhibitors that bind to the ATP-binding site of BCR-ABL, inhibiting proliferation and decreasing survival of BCR-ABL+ cells [24, 25]. In addition to blocking the kinase activity of BCR-ABL, imatinib also inhibits ABL, c-Kit, and PDGFR activity [26]. Clinical trials of imatinib in patients with relapsed or refractory Ph+ ALL have shown a complete remission rate of 20%, with 60% of patients achieving remission or clearing peripheral blasts. However, these effects were only transient in that the median time to disease progression is only 2.2 months, indicating acquired resistance [27]. When used as frontline therapy for Ph+ ALL in combination with chemotherapy or allogeneic stem cell transplantation, the results are much more encouraging, with sustained complete remissions in some cases [28]. Despite initial responses, many patients with Ph+ ALL are refractory or quickly develop resistance to imatinib. Many cases of imatinib resistance have been associated with the emergence of mutations in the tyrosine kinase domain of BCR-ABL [29]. Thus, novel therapeutics that target wild-type and mutant forms of BCR-ABL are being been developed.
Targeting the src family of kinases
Acquired resistance to imatinib in Ph+ ALL patients may also result from subsequent transforming events involving other key survival kinase pathways, including the SRC family of kinases (SFKs) [14]. SFK members, including LYN, LCK, FYN, HCK, BLK, and SYK, play critical roles in the regulation of lymphoid development, regulation of immune function and cellular homeostasis [6]. Generally, activation of these kinases results in cell growth and survival most likely through interactions with the RAS/RAF/MEK/ERK pathway. Perturbations of SFK-mediated signals have been associated with malignant transformation [30, 31]. For example, the pre-B ALL cell line Nalm 6 expresses high levels of LYN, SYK, and BLK, whereas the T-ALL cell line Jurkat expresses aberrant levels of FYN and LCK [32]. Dasatinib is a dual inhibitor of BCR-ABL and SFKs, with a 325-fold higher potency than imatinib [14, 28]. Dasatinib was subsequently found to inhibit other kinases such as ABL, PDGFRs, and ephrin A receptor kinase. Although the mechanism(s) of action of dasatinib are still being elucidated, dasatinib-mediated inhibition of SFKs leads to cell cycle arrest, apoptosis/cell death, and decreases invasion of solid tumors. Johnson et al. report that while dasatinib rapidly inhibits SFKs and STAT3, the inhibition of SFKs is durable, whereas STAT3 is only transiently inactivated as a result of JAK/STAT pathway activation to compensate for SFK inhibition, resulting in sustained cancer cell survival [33]. Furthermore, the addition of a JAK inhibitor can overcome this apparent resistance to dasatinib. These results suggest that resistance to small molecule inhibitors such as imatinib and dasatinib may be the result of upregulation of compensatory survival signaling networks. The nature of these integrated signals and their response to kinase inhibitors needs to be further elucidated.
Cytokines, such as IL-7 and TSLP, play important roles in promoting normal T- and B cell development as well as sustaining malignant T and B cells [7, 34–37]. Because IL-7-mediated signaling can activate the SFKs to promote cell growth and survival, we have begun to investigate the effect of dasatinib on the proliferation and survival of Ph− ALL [31]. Treatment of ALL cell lines with dasatinib alone can induce profound growth inhibition at concentrations as low as 10 nM in murine ALL cell lines and a fold higher in human lines. In addition, dasatinib can work in concert with the mTOR inhibitor rapamycin to further inhibit the growth of the human pre-B ALL cell line, Nalm 6 (V. Brown, unpublished data). In addition to its in vitro activity, dasatinib shows in vivo activity as a single agent, using a NOD/SCID xenograft model. Immunodeficient mouse models, including NOD/SCID [38], provide powerful tools for preclinical testing of novel therapeutic agents using primary human ALL samples [39, 40]. NOD/SCID xenografted ALL has been shown to maintain its phenotypic and genotypic characteristics even after serial passage into secondary and tertiary hosts [41]. The response of leukemic blasts to chemotherapeutic agents in the NOD/SCID xenograft model has been shown to correlate directly with human response to therapy [42]. In NOD/SCID mice xenografted with cells that were obtained from a patient with relapsed Ph−, pre-B ALL, dasatinib alone profoundly reduces absolute peripheral blast counts and significantly prolongs survival. After 65 days of treatment, all the mice receiving dasatinib are still alive with minimally detectable disease; in contrast, the control mice have a median survival after engraftment of only 13 days (V. Brown, unpublished data). Through the Pediatric Preclinical Testing Program, dasatinib has been reported to have activity in 3 of the 7 ALL xenografts tested, one of which (a Ph+ ALL sample) demonstrated a complete response [43]. Based upon our preclinical pilot data, dasatinib may also have a potential role for the treatment of relapsed ALL patients who are Ph-negative. However, the mechanism of action of dasatinib in Ph-ALL has not been elucidated, and these preclinical investigations are ongoing.
Targeting the PI3K/AKT/mTOR pathway
Because PI3K/AKT/mTOR signaling is aberrantly activated in a variety of cancer types including hematologic malignancies, intermediates of this pathway have become attractive candidates for treatment of these cancers, including leukemia [44]. Most attention has focused on blocking the mammalian target of rapamycin (mTOR). mTOR is a 210 kD intracellular serine-threonine kinase that acts as a central regulator of cell growth, survival, and proliferation [45]. It functions as a sensor to ensure that a cell has the adequate nutrients and energy in order to support cell growth, proliferation, and metabolism before committing to cell growth and division. Activation of mTOR results in ribosomal biogenesis, cell growth, increased transit time through the cell cycle, protein synthesis, angiogenesis, cap-dependent and TOP-protein translation, as well as nutrient and amino acid uptake. This nutrient uptake is critical for cell metabolism and generation of ATP. Conversely, mTOR activation inhibits apoptosis and autophagy. Upstream pathway activators of mTOR include signaling from ras, TCL1, BCR-ABL, and growth factor receptors, such as IL-7R, IGF-1R, c-kit, and flt-3 [46, 47]. Upon engaging its ligand, the growth factor receptor becomes activated, which in turn activates IRS-1, which then leads to PI3K upregulation. PI3K directly associates with many growth factor and cytokine receptors. Upon activation, PI3K generates PIP3 which recruits AKT to the cell membrane for activation by PDK1 and TORC2. The generation of PIP3 is negatively regulated by the tumor suppressor, PTEN. Activated AKT then goes on to phosphorylate TSC2, which inactivates TSC1:TSC2 complex. As a result of this inactivation, RHEB becomes available to activate mTORC1. AKT also promotes the stability of c-myc and CyclinD1 by inactivating GSK3β, a negative regulator of these two pro-survival and pro-proliferation molecules. Also, activated AKT blocks pro-apoptotic molecules through the phosphorylation of FKHRL1 which inactivates it. This integrated signaling network is critical for sustaining cell growth and proliferation [48].
mTOR can form two distinct complexes termed mTORC1 and mTORC2 [49]. mTORC1, composed of mTOR, GβL, Mlst8, PRAS40, and RAPTOR, is sensitive to the mTOR inhibitors (MTIs), such as rapamycin, RAD-001, and CCI-779, and is thought to regulate cell growth, proliferation, autophagy, and translation in response to nutrients and energy availability. Activated mTORC1 goes on to phosphorylate p70S6K (S6K1). Phos-phorylated- (P−) S6K1 induces TOP-translation and ribosomal biosynthesis as well as blocks apoptosis by phosphorylating the pro-apoptotic molecule BAD. S6K1 phosphorylation also acts as a negative feedback mechanism for the PI3K/AKT/mTOR pathway by down-regulating IRS-1 [50]. mTORC2 is composed of mTOR, GβL, mSIN1, RICTOR, and PROTOR/PRR5. mTORC2 works in concert with PDK1 to phosphorylate/activate AKT. Rapamycin does not interfere with mTOR if it is already bound to RICTOR as part of the mTORC2 complex. It is hypothesized that increased AKT activity after MTI exposure is a result of continued mTORC2-mediated AKT phosphorylation and may be a potential mechanism of MTI resistance [51–53]. However, under conditions of chronic MTI exposure, MTIs may inhibit mTORC2 since they are able to bind nascent mTOR prior to its assembly into the mTORC2 complex. mTORC2’s function, regulation, and response to rapamycin remains unclear, and may vary by cell type [54–56].
Mechanisms that lead to mTOR deregulation include overexpression of growth factors (such as IGF), overexpression or mutations of growth factor receptors (e.g., IGFR, HER/EGFR), loss of tumor suppressor genes (e.g., PTEN or TSC1:TSC2 complex), and gain-of-function mutations in mTOR or mTOR-linked pathways (e.g., formation of the aberrant protein BCR-ABL in Ph+ leukemia cells or stimulation of PI3K by aberrant ras/raf/MAPK pathway intermediates) [57, 58]. The most common mechanisms are derived from the over-expression or constitutive activation of PI3K or AKT and/or the loss of the tumor suppressor, PTEN [59, 60]. Deregulated signaling or cross-talk through mTOR-linked pathways can increase mTOR activity. mTOR activation supports cancer cells by stimulating the synthesis of proteins necessary for cell growth, proliferation, survival, angiogenesis, nutrient uptake, and metabolism.
MTIs have shown great promise in the treatment of many cancer types including ALL [44]. While its anti-neoplastic properties were noted shortly after its discovery, the MTI rapamycin (sirolimus) has been most widely used as an immunosuppressive agent and is FDA-approved for this use in adults and children [61]. There is accumulating evidence that MTIs are effective agents in the treatment of cancer [62, 63], with CCI-779 (temsirolimus) most recently FDA-approved for use in the treatment of renal cell carcinoma [64]. Preclinical evidence indicates that MTIs are active in lymphoid malignancies [65–68]. Previously, we have reported that, in vitro, MTIs decrease proliferation, induce apoptosis and cell death, and result in hypophosphorylation of downstream targets of mTORC1 [69–71]. In vivo, MTIs show single agent activity in two mouse models of ALL, the Eμ-RET transgenic mouse model and NOD/SCID xenograft model [69, 71]. Eμ -RET mice, which constitutively express activated RET tyrosine kinase only in B-lineage cells, spontaneously develop lymphoblastic lymphoma/leukemia (ALL) manifested as massive adenopathy, splenomegaly and bone marrow replacement between 3 and 8 months of life [72, 73]. The Eμ-RET mouse model provides a developmentally targeted model of progenitor B cell malignancies that allows us to intervene with therapeutics early and late in the progression of ALL. Models such as these are useful in preclinical evaluation of novel therapeutic strategies and in studying the mechanisms by which leukemia cells develop drug treatment resistance [69]. Treatment of Eμ-RET transgenic mice with advanced ALL with rapamycin significantly decreases tumor burden and prolongs survival [69]. Treatment with the rapamycin analogues RAD-001 (everolimus) and CCI-779 (temsirolimus) yield similar results (V. Brown, unpublished data).
NOD/SCID mice xenografted with primary cells obtained from patients with ALL respond similarly to MTI treatment [71]. In addition to providing an in vivo model to test novel, molecular-targeted agents against primary human ALL cells, the NOD/SCID xenograft model allows for significant expansion of human ALL cells in order to generate sufficient quantities of cells from clinical trial specimens for mechanistic studies. Our laboratory has attempted to establish NOD/SCID xenografts from cryopreserved lymphoblasts from 30 patients with precursor B cell ALL, and have been successful with 70% of samples, numbers equal to or better than most published work (D. Teachey, unpublished data) [38, 41, 42]. The majority of xenografts from these samples had >90% replacement of bone marrow and spleen with human ALL as determined by flow cytometry with anti-human CD45 and CD19. In addition, to demonstrate leukemic stem cell activity, blasts were collected from xenografted mice and injected into secondary and tertiary hosts. Successful engraftment was found with all (N = 16) samples attempted. In addition, there was significant expansion of ALL. Xenografted mice developed between 55 and 720 × 106 lymphoblasts in bone marrow and spleen, representing a 5- to 72-fold increase in ALL in the mice.
While MTI treatment reduces tumor burden (Fig. 1) and prolongs survival, the mice eventually relapse and succumb to their disease [69, 71]. Moreover, ALL cells are protected from MTI-induced effects by the cytokines IL-7 and TSLP in vitro [69, 70], suggesting that these stromal cell-derived cytokines contribute to MTI resistance. The addition of IL-7 or TSLP rescues murine ALL cells from MTI-induced inhibitory effects. This reversal is seen even when the addition of IL-7 or TSLP is delayed by 24–36 hours [69, 70]. Furthermore, antibodies antagonistic to IL-7, TSLP, and their binding to the IL-7Rα chain (i.e., mAB A7R34) inhibit IL-7 and TSLP-induced cell proliferation and block the ability of these cytokines to reverse mTOR inhibition [70]. To directly assess changes in candidate signaling target proteins downstream of mTOR and IL-7R, we have performed immuno-blotting of rapamycin- and IL-7/TSLP-treated cell lysates to detect post-translational modifications of S6K1, S6, 4E-BP1, and STAT5 and have found that P-S6K1 is profoundly decreased in MTI-treated cells [69]. Similar patterns are seen for P-S6 and total S6 proteins [71]. In contrast, both IL-7 and TSLP induce phosphorylation of S6 and 4E-BP1 as well as STAT5 in pre-B ALL cells, again reversing the inhibitory effects of rapamycin [69, 70].
Fig. 1.

Response of ALL to MTI therapy in NOD/SCID xenografts. NOD/SCID mice were xenografted with human ALL cells obtained from patients with ALL. After establishment of ALL (>5% peripheral blasts), mice were randomized to treatment with 5 mg/kg/day CCI-779 (grey bars) or drug vehicle only (white bars). Lymphoblasts in spleen are depicted in this figure (spleen cell count × %CD19/CD45+ cells), comparing treatment to control at sacrifice from eight representative patient samples, showing sensitive and resistant samples. Sample number is depicted on X-axis. Error bars depict standard error of mean
To explore the role of PI3K and AKT activation in MTI resistance, we have inhibited PI3K with the PI3K inhibitor LY294002 and have assessed the effects on cell proliferation and apoptosis. At low micromolar concentrations, LY294002 inhibits proliferation and induces apoptosis/cell death of murine and human ALL cell lines. Furthermore, LY294002 potentiates the inhibitory effects of rapamycin synergistically, as determined by the method of Chou and Talay (V. Brown, unpublished data) [74]. In addition, inhibition of PI3K specifically attenuates the IL-7-mediated rescue of ALL cells from MTIs. These differences in proliferation and cell death are also manifested in changes in S6 and BAD phosphorylation: while treatment with rapamycin or LY294002 alone results in hypophosphorylation of S6 and BAD that is restored by the addition of IL-7, concurrent treatment with all 3 agents results in decreased S6 and BAD phosphorylation, blocking the effect of IL-7’s capacity to rescue ALL cells from the inhibitory effects of rapamycin (V. Brown, unpublished data). In contrast, the phosphorylation of AKT at residue Ser473 follows a different pattern. Treatment with rapamycin or LY294002 alone results in minimal change in phosphorylation of AKT, whereas P-AKT is almost undetectable with concurrent treatment of both inhibitors (V. Brown, unpublished data). While the addition of LY294002 or a combination of LY294002 and rapamycin results in hypophosphorylation of IL-7-treated ALL cells, the addition of LY94002 and rapamycin is still not sufficient to completely block IL-7-induced AKT activation. These data support the hypothesis that IL-7-mediated signaling increases PI3K/AKT, bypassing mTOR inhibition.
The dual inhibitor PI-103 is known to inhibit PI3K, mTORC1, and mTORC2 [63, 75, 76]. Single agent PI-103 profoundly inhibits proliferation of murine and human ALL cell lines (V. Brown, unpublished data). Treatment of murine ALL cells with less than 1 μM PI-103 results in profound growth inhibition. In comparison, human ALL lines require higher concentrations (2.5–5 μM) to achieve the same level of inhibition. Concurrent treatment with PI-103 and rapamycin results in almost complete inhibition. While the addition of PI-103 to IL-7-treated cells only partially overcomes IL-7’s proliferative effects, the combination of PI-103 and rapamycin induces profound inhibition in the presence of IL-7, but to a lesser extent than when IL-7 is absent. PI-103 and rapamycin in combination also induces greater cell death, counteracting the pro-survival effects of IL-7. These findings correlate to the changes in S6 phosphorylation after treatment for 24 h with combinations of rapamycin, LY294002, IL-7, and PI-103. Treatment with PI-103 alone or in combination with rapamycin or LY294002 results in hypophosphorylation of S6. In contrast, treatment with IL-7 sustains S6 phosphorylation in the presence of PI-103. The IL-7-induced S6 phosphorylation is abrogated when the cells are co-treated with rapamycin or LY294002 in combination with PI-103. The greatest degree of AKT hypophosphorylation is induced by a combination of PI-103 and rapamycin. In the presence of IL-7, PI-103 alone or in combination with either rapamycin or LY294002 does not decrease P-AKT as compared to cells treated in the absence of IL-7. These data indicate that IL-7 stimulation of ALL cells activates the PI3 K/AKT/mTOR pathway and can continue to sustain cell survival and proliferation, although to a lesser degree, even when this pathway is inhibited at multiple intermediates, suggesting that a mechanism for survival involves an alternative pathway. These data also suggest that targeting multiple nodes along these integrated survival signaling networks is superior to the blocking of one pathway intermediate alone.
Targeting the JAK/STAT pathway
Cytokine stimulation of lymphoid cells leads to the activation of the JAK/STAT pathway [34]. For example, binding of the cytokine IL-7 induces the heterodimerization of the IL-7Rα and γc chains to form the IL-7R at the cell membrane that triggers the activation of the receptor associated tyrosine kinases JAK1 and JAK3 [77]. Activated JAK3 phosphorylates the IL-7Rα chain at residue Tyr449, creating docking sites for both STAT5 and PI3K [78, 79]. STAT5 and PI3K have different binding kinetics to this docking site, thereby yielding a potential mechanism for regulating the extent and timing of these two pathways and thus influencing the downstream integration of these survival signals [80]. TSLP transmits its signal through the TSLP receptor complex (TSLPR) composed of the IL-7Rα and TSLPR chains [81]. TSLP-mediated signaling also results in STAT5 activation, but does so through an alternative pathway [82]. Once STAT5 is activated, it homodimerizes and translocates to the nucleus where it modulates the expression of a multitude of pro-survival and cell proliferation genes, including BCL-XL, c-myc, Cyclin D1, and PIM-1 [9].
The role of the bone marrow microenvironment in promoting ALL cell survival and resistance to therapy
Bone marrow (BM) niches are critical for normal lymphopoiesis and in sustaining ALL cell survival [83]. Normal B cell development requires a complex microenvironment that consists of an extracellular matrix, stroma consisting of a heterogeneous population of accessory cells such as reticular fibroblasts, osteoblasts, and endothelial cells, and diffusible factors, including cytokines, other growth factors, and chemokines [83]. These components make up specific niches in the BM that are critical for B lymphopoiesis. The BM microenvironment also contributes to the progression and maintenance of hematologic malignancies, promoting cell survival, blunting the effects of chemotherapeutics, and providing a sanctuary for minimal residual disease. While primary ALL cells typically undergo spontaneous apoptosis when cultured in media alone, co-culturing with bone marrow stromal cells (BMSCs) increases survival and proliferation of leukemia cells preferentially, with the greatest viability when leukemic blasts are in direct contact with BMSCs [84, 85]. Co-culture of leukemia cells with BMSCs blunts the cytotoxic effects of chemotherapeutic agents in vitro. While the mechanisms by which the BM stroma modulate the response of ALL cells to chemotherapy is not fully understood, there is evidence that the interactions of leukemia cells with extrinsic factors of the stroma provide signals that promote cell survival and ultimately drug resistance [86]. CXCL12 (also known as SDF-1α) is a chemokine that is normally expressed on BMSCs and causes BM retention of hematopoietic stem cells (HSC), B cell progenitors and mature B cells as well as ALL cells. Binding of CXCL12 to CXCR4 on HSC, developing B cells and ALL cells results in the adhesion to BMSCs and subsequent survival of these cells [87]. CXCR4 antagonists that interfere with this cell–cell adhesion can mobilize ALL cells to the peripheral blood, inhibit engraftment of ALL cells, and overcome atroma-mediated chemoresistance [88, 89]. Bertrand and colleagues have found that pre-B ALL cell survival in the presence of BMSCs is mediated by crosstalk among PI3K, mTOR, and the MEK pathways [90, 91]. The binding of VLA-4 (a member of the β1 integrin family that is used by normal B cell progenitors to adhere to BMSCs) expressed on pre-B ALL cells can lead to activation of PI3K/AKT that results in cell survival and apparent drug resistance [84, 92–94].
IL-7 and TSLP are important regulators of normal lymphoid development, and therefore may influence growth of ALL cells [34, 95]. IL-7 and TSLP are closely related, yet distinct, in biologic function. IL-7 is required for normal murine B- and T-cell development. IL-7 promotes the proliferation of pro-B cells, progression from pro-B to pre-B cells and the formation of a functional pre-B cell receptor [96]. Although IL-7 is absolutely required only for T cell development in humans, it still plays an important role in human B cell development, providing a survival signal for B lymphoid precursors. TSLP also supports early B cell proliferation and development but differentially affects B cell progenitors of fetal and adult origin, acting earlier in the development of fetal-derived B cells [97, 98]. IL-7 has been shown to rescue T-ALL lymphoblasts from apoptosis [36, 99, 100]. IL-7 also stimulates growth of human pre-B ALL [101]. Evidence suggests that the addition of IL-7 to co-cultures of BMSCs and ALL cells of B or T origin results in augmented ALL cell survival with increased phosphorylation of AKT, ERK1/2, and p38 [88, 102]. The addition of α-IL7 or α-IL-7Rα antibodies leads to a significant decrease in BMSC-induced survival, but this blockade only partially inhibits survival, suggesting that there are other contributory factors [35]. We have also reported that IL-7 or TSLP can stimulate proliferation and survival of pre-B ALL cell lines and primary human lymphoblasts of some, but not all, specimens tested in BM stromal cell co-cultures [69–71]. These data suggest that blockade of the actions of IL-7 or TSLP by antagonistic antibodies of IL-7, TSLP, or the IL-7Rα chain may be an effective strategy in treating ALL.
The integration of molecular targeted agents with conventional treatment strategies
Prior to moving any novel agent into clinical trials in patients with ALL, it is necessary to study the new agent in combination with existing agents used to treat patients. This is important for two reasons: because signal transduction inhibitors (despite the experience with imatinib in CML) are unlikely to be used solely as single agents, and to ensure new agents do not antagonize the effects of existing cytotoxic therapies. To this end, we have begun to evaluate MTIs in combination with chemotherapeutics currently in use to treat ALL. We have reported that CCI-779 and methotrexate are synergistic in combination in vivo, using the NOD/SCID xenograft model [103]. MTIs may increase turnover of cyclin D1, a protein involved in dihydrofolate reductase (DHFR) synthesis [104]. High DHFR expression has been shown to correlate with methotrexate resistance [105]. Accordingly, we have shown that MTIs increase the sensitivity of ALL to methotrexate through decreasing DHFR by increasing turnover of cyclin D1 [103].
The glucocorticoid dexamethasone is commonly used in the treatment of ALL. Generally, dexamethasone treatment of ALL cells induces apoptosis. IL-7 has been shown to protect B- and T-cell lineage cells, particularly pro-B cells, from steroid-induced apoptosis [106]. Dexamethasone’s mode of action may also involve the modulation of TOP-translation. Recent data indicate that engagement of the glucocorticoid receptor by dexamethasone downregulates S6K1, inactivating S6K1’s ability to regulate TOP-translation [107]. These data suggest that dexamethasone can be used to modulate ribosomal biogenesis. Because both dexamethasone and rapamycin can interfere with ribosomal biogenesis, we have investigated the effect these agents in combination on ALL cell proliferation. While dexamethasone alone can inhibit ALL cell proliferation, the co-culturing of ALL cells with rapamaycin and dexamethasone results in increased growth inhibition, an effect that is partially reversed by IL-7 (V. Brown, unpublished data).
Molecular-targeted therapy for LPD
ALPS
Autoimmune lymphoproliferative syndrome (ALPS) is a disorder of disrupted lymphocyte homeostasis caused by defective fas-mediated apoptosis. In an important part of the process by which proliferation of cells of the immune system is controlled, activated B lymphocytes upregulate fas expression and activated B and T lymphocytes upregulate expression of fas-ligand [108]. Fas and fas-ligand interact, triggering the extrinsic apoptotic system and activating the caspase cascade, leading to cellular apoptosis. Patients with ALPS have a defect in this apoptotic pathway, leading to chronic lymphoproliferation, autoimmune manifestations, and a propensity to develop secondary malignancies [109].
ALPS is a rare disorder first characterized in the 1990s in a cohort of patients with chronic lymphoproliferation and an increased number of a T cell population termed double negative T cells (DNTs; cell phenotype CD4−/CD8−, CD3+, TCRαβ+) [110]. DNTs typically represent a small subset of T cells (<1%) in peripheral blood in unaffected individuals [111]. These patients have clinical features resembling two mouse models of autoimmunity, lpr and gld. Lpr and gld mice were later shown to have defective Fas-mediated apoptosis with homozygous mutations in the Fas gene and Fas ligand gene, respectively [112, 113]. Patients with lymphoproliferation and autoimmunity were proposed and subsequently confirmed to have similar genetic defects to the lpr and gld mice and were classified as having ALPS [114, 115].
Currently, in order for a patient to be diagnosed with ALPS, he or she must meet three diagnostic criteria: (i) clinically identifiable chronic lymphoproliferation, (ii) elevated DNTs, and (iii) in vitro evidence of defective fas-mediated apoptosis [109]. Supporting evidence for the diagnosis includes genetic mutations in the fas pathway (Fas, Fas ligand, Caspase 8, or Caspase 10), systemic autoimmunity, elevated serum levels of interleukin-10 (IL-10), elevated serum vitamin B12, and hypergammaglobulinemia; however, these are not diagnostic [116].
Clinical manifestations vary, however, by current definition, all patients have chronic non-malignant lymphoproliferation, defined as having hepatomegaly, splenomegaly, and/or lymphadenopathy of at least 2 nodal groups for greater than 6 months duration [117]. In our studies of this disorder, the lymphoproliferative component of ALPS drew our attention, as we hypothesized that pathways we had identified as being important in malignant lymphoproliferation might also be therapeutically important in this abnormally proliferating population of cells. In addition, many patients (>70%) with ALPS develop autoimmune disease, most commonly with autoimmune destruction of blood cells [118]. Most patients have destruction of multiple cell types. In many patients, autoimmunity waxes and wanes and may flare with systemic insult. Other patients have chronic, severe, and debilitating disease [109]. Other autoimmune manifestations can be found less frequently, including autoimmune kidney (nephritis), liver (hepatitis), joint (arthritis), eye disease (uveitis), and skin (urticaria) disease [119]. Finally, patients with ALPS have an increased risk of developing secondary malignancies [118]. The exact risk is unknown but hypothesized to be 10–20% [120]. Most commonly, patients develop lymphoma (non-Hodgkins and Hodgkins), but leukemia(s) and a number of solid tumors (thyroid, breast, and liver carcinoma) have been described [117]. An increased risk of cancer has been described in unaffected family members of patients with ALPS [120].
Some patients with ALPS require no treatment. Many patients, however, require medications directed toward autoimmune manifestations, particularly autoimmune cytopenias. Patients usually respond to short-term treatment with immunosuppressive medications, including corticosteroids [121]. Occasionally, patients with severe cytopenias need more aggressive and prolonged immunosuppressive treatment. Anecdotal reports and small series have described responses to a number of medications, including cyclosporine, mycophenolate mofetil, methotrexate, and rituximab [119, 122–124]. Nevertheless, these agents are often ineffective, have significant side-effects, and are non-specific for ALPS.
Currently a number of more targeted therapies are undergoing preclinical testing and clinical trials. Pyrimethamine and sulphadoxine were shown to significantly reduce lymphoproliferation and autoimmune cytopenias in a small series of patients with ALPS; however, this combination failed to show any response in a different larger clinical trial [125, 126]. In work looking at signal transduction in proliferating lymphocytes in ALPS, we hypothesized that targeting the mTOR pathway might be effective in controlling the LPD, because mTOR inhibitors induce apoptosis in abnormal lymphocytes and may have activity in diseases with dysregulation in Tregs, including ALPS. We found rapamycin was extremely effective in mouse models of ALPS and was more effective than conventional therapies, including mycophenolate mofetil [127]. Using two murine models of ALPS, CBA-lprcg and MRL-lpr, we found rapamycin reduced the LPD, including decreased numbers of DNTs, lymphadenopathy, and splenomegaly. Interestingly, we have also demonstrated that the autoimmune manifestations of ALPS are controlled with rapamycin, including autoantibodies. Based on these results, we are currently conducting a clinical trial using rapamycin in ALPS patients. Thus far six patients have been treated with rapamycin with excellent results. All patients achieved a rapid hematological complete response, had marked decrease in lymphoproliferation, and significant reduction in DNTs (Teachey et al., in preparation; see www.clinicaltrials.gov). Three of these patients had failed numerous therapies including mycophenolate mofetil.
Carrying our studies of signal transduction pathways which may be important in LPD forward, we have also recently shown that targeting the notch signaling pathway may be beneficial in preclinical models of ALPS [128]. Similarly, other groups have shown that arsenic and HDAC inhibitors may be effective using such murine models [129, 130]. In our notch studies, we hypothesized that inhibiting notch signaling would be effective in ALPS because notch signaling has several key roles in T-cell function, including DNT transition in T-cell development and T-cell activation. Thus, by inhibiting this pathway, the production of abnormal DNTs and activation of abnormal T cells would decrease [128]. Notch signaling is mediated through a pathway of four mammalian transmembrane Notch receptors (Notch 1–4) [131]. After ligand binding, two cleavages of the Notch receptors occur, first by a metalloprotease and subsequently by gamma-secretase, releasing the intracellular domain (ICN) that translocates to the nucleus and binds to the transcription factor CSL (RBP-Jk), activating transcription of a number of key intracellular proteins [131]. Gamma-secretase inhibitors (GSIs) are a class of agents in clinical development in a number of diseases including Alzheimer’s and T cell leukemia. GSIs block the second cleavage of the Notch receptors, preventing the release of ICN and transcriptional activation. We tested one of these inhibitors, DAPT, in the two mouse models and found DAPT reduced lymphadenopathy, splenomegaly, autoantibodies, and glomerulonephritis [128]. Figure 2 depicts representative measure of disease burden (lymphadenopathy), comparing treatment with rapamycin (sirolimus), mycophenolate mofetil, DAPT, and vehicle control in murine ALPS.
Fig. 2.
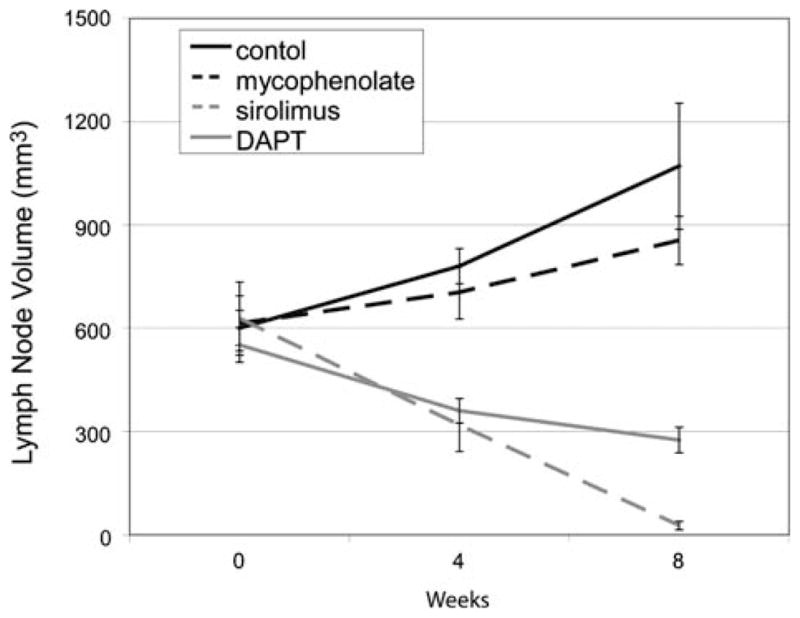
Targeting the mTOR pathway and notch signaling are effective against murine ALPS. CBA-lprcg mice (the most clinically similar mouse model to human ALPS) [116] were treated with sirolimus 5 mg/kg/day, mycophenolate mofetil (MMF) 100 mg/kg/day, DAPT 5 mg/kg/day, or vehicle control. These data depict summary of two different randomizations: sirolimus, MMF, versus control and DAPT versus control. Serial ultrasounds were performed to document lymph node volume in mm3. Mice treated with sirolimus or DAPT showed a statistically significant (P = 0.05) decrease in lymph node volume after only 2 weeks of treatment when compared to control mice. Mice treated with MMF showed no improvement in adenopathy. No statistical difference existed between groups at initiation of treatment. Bars represent mean lymph node volume from mice at each timepoint and error bars represent standard error of the mean. Similar results were found when assessing splenomegaly and autoantibodies [127, 128]
Early reports using GSIs in other disease models delivered drugs at high doses for a short duration and mice developed thymic hypoplasia and intestinal goblet cell hyperplasia within 7 days [132, 133]. These effects were clearly dose and time dependent [134, 135]. In order to reduce toxicity, we used a lower potency GSI and treated mice daily for 5 days a week. Using this approach, we found that inhibiting the notch pathway was both effective and safe. Other groups have been investigating the effects of targeting notch signaling on systemic autoimmunity and have found targeting the notch pathway with GSIs improves autoimmune diseases, including autoimmune encephalomyelitis and uveoretinitis [136, 137]. Nevertheless, different studies have shown that targeting the notch pathway through alternative means, including global knock-down of presenilins (active centers of the gamma-secretase complex) and keratinocyte-specific deletion of notch signaling, can promote autoimmunity [138, 139]. As the notch signaling pathway has multiple complex roles in lymphocyte and immune system homeostasis, it is possible that targeting this pathway could improve some autoimmune diseases and worsen others, depending on the degree and type of suppression. Thus, further preclinical work is needed prior to targeting this pathway in patients.
Novel cellular-targeted therapeutics for ALL
While immune-based, cellular strategies to treat leukemia are often not included in discussions of targeted therapy for ALL, these approaches capitalize on the ability of the immune system to detect the presence of leukemia-related abnormalities in order to target transformed cells specifically. The strength of this cellular targeted approach lies in its use of innate biological mechanisms to identify and attack abnormal cell types. This permits anti-leukemic activity to be generated in the absence of the detailed knowledge of underlying disease mechanisms that is required for molecular targeted therapies. The discovery of techniques to increase the sensitivity of innate and adaptive immune surveillance for leukemia presents the greatest challenge to development of successful clinical applications. However, the notion that an appropriately activated immune system can contribute to the control of acute leukemia is supported by the observation that the graft-versus-leukemia effect that can occur after stem cell transplantation in which donor-derived immune cells can eliminate residual ALL cells in the recipient [140–142].
In the context of immune surveillance, immune effector cells theoretically can be attracted to numerous abnormalities present in leukemia cells, including over-expressed proteins, mutated proteins (such as those produced by chromosomal translocations), and stress response proteins. Indeed, T cells specific for epitopes derived from such proteins have been detected in leukemia patients [143, 144], and have been shown to be capable of exerting significant anti-leukemia activity in xenograft models [145]. However, attempts to generate effective, sustained clinical anti-leukemia immune responses have been undermined by the immune subversion mechanisms present in patients at advanced stages of disease, including Th2 skewing [146], loss of costimulatory molecule expression on leukemia cells [147], dendritic cell dysfunction [148], and an insensitivity of leukemia cells to immune effector mechanisms [149, 150].
In an attempt to develop an approach that is capable of eliciting strong anti-leukemia responses in the immune compromised setting of the leukemia patient, we have focused on toll-like receptor (TLR) ligands, particularly synthetic DNA oligonucleotides containing unmethylated CpG motifs (CpG ODN). TLRs bind structural and genetic components of pathogenic organisms, including bacteria and viruses, and mediate an activation signal to the host immune system [151]. The synthetic CpG ODN mimic the activity of bacterial DNA and serve as a “danger” signal for the immune system, triggering rapid responses from innate immune cells, including plasmacytoid dendritic cells and NK cells, that lead to the release of IFN-α, IFN-γ, and IL-12 [152]. As a result of this early response, CpG ODN enhance the development of Th1 adaptive immune responses. Thus, the use of CpG ODN may provide a non-specific approach to enhancing the innate immune anti-leukemia activity in patients and promote the generation effective Th1 adaptive responses without the need for prior identification of specific candidate antigens.
Using the NOD/SCID xenograft model, we have begun to investigate the ability of CpG ODN to elicit effective innate immune anti-leukemia responses [153]. Because of its dysfunctional innate immune system and lack of adaptive immune system, the NOD/SCID model represents a stringent test of the effectiveness of CpG ODN. Despite these immune deficiencies, administration of CpG ODN to mice bearing a significant leukemic burden causes a dramatic reduction in the blast population in blood, spleen, and liver [153]. In contrast, there is a lack of activity in the bone marrow, as expected, given that immune therapies are generally inefficient when the ratio of immune effector cells to their targets is low, as in this case in which the density of leukemic blasts in the marrow overwhelmed the relatively small number of effector cells. Moreover, when CpG ODN is administered earlier in disease engraftment, i.e. when the disease burden in the marrow compartment is lower, survival is significantly improved (Fig. 3; [153]). The CpG ODN-mediated anti-leukemic activity is at least partially dependent on the presence of NK cells and correlates with the production of IFN-α and IFN-β by the host immune system. Importantly, while some leukemia cells respond directly to CpG ODN by upregulating cell surface costimulatory molecules [154], we observed anti-leukemia activity independently of the cell lines’ direct responses. Following this in vivo demonstration of CpG ODN induced activity, we confirmed in vitro that such an NK-mediated effect could also be generated using human immune effector cells [153]. These results demonstrate that (i) CpG ODN can stimulate the innate immune system to mount an anti-ALL immune response and (ii) primary human ALL blasts are sensitive to the immune effector mechanisms mediating the CpG ODN effect.
Fig. 3.

CpG ODN treatment delays disease progression in NOD/SCID mice bearing human leukemia. NOD/SCID mice (5 per group) were injected with 107 human ALL cell line 380 cells and then injected with CpG ODN (open squares) or PBS (filled squares) on days 2, 10, and 20 after ALL cell injection (indicated by arrows). Progression of disease, as measured by the presence of human ALL cells in peripheral blood, was significantly delayed in the treated group, as was overall survival as compared to control (not shown)
ALL blasts are often inefficient stimulators of the immune system, inducing either anergy or Th2 responses; therefore, the ability to augment the generation of Th1 responses to ALL may be clinically beneficial [146, 155]. To investigate the influence of CpG ODN on adaptive immune responses, we have directly assessed the ability of CpG ODN to alter ALL blast immunogenicity [154]. Many, but not all, ALL cell lines and primary patient samples tested are capable of responding directly to CpG ODN in vitro; this response correlates with their expression of TLR9, the receptor for CpG ODN [154]. Responsive ALL cells produce cytokines, most notably IL-6 and IL-8, and upregulate expression of CD40. When cultured in the presence of allogeneic T cells, CpG ODN-treated ALL cells stimulate the production of IFN-γ and decrease production of IL-5 by T cells, indicative of a shift toward a Th1 response. The ability to alter ALL immunogenicity is not unique to CpG ODN, indicating that other members of the TLR family may also provide contributions to the generation of anti-leukemia activity [156].
A critical component of the effective use of immune-based cellular therapy is choosing when to implement the therapy. As we observed with our late administration of CpG ODN to NOD/SCID xenografts, immunotherapeutic approaches are generally ineffective when given at a time of high disease burden, especially with a rapidly growing cell population like ALL. However, the ability of induction or reinduction therapy to get >90% of children with ALL into complete remission provides a setting in which immune therapy can be applied at very low disease burden when the effector:target ratio will be at its greatest, enhancing the chances of success for immune intervention. With this in mind, we are currently assessing the effectiveness of CpG ODN when applied in a post-chemotherapy environment, a time when the necessary immune effector populations will be compromised but also a time when the tumor burden is sufficiently low. If CpG ODN generates effective immune activity as an adjuvant to chemotherapy, the ability to stimulate both early innate responses and subsequent adaptive responses may be highly effective in achieving long-term disease control. Leukemia-specific T cells have recently been shown to be present at higher numbers in patients who are in first remission compared to a control population in both AML and ALL [143, 157], and at least in the case of AML that the presence of these T cells correlates with sustained remission [157]. Given the results of our work to date, we hypothesize that the application of CpG ODN at the completion of chemotherapy may be of particular value in patients with minimal residual disease, as an immune approach may be more effective against this ostensibly chemo-resistant population of cells. Furthermore, by stimulating an enhanced Th1 adaptive response against leukemia antigens, we would predict a significant improvement in the duration of remission for this refractory group. Work is currently underway to test this hypothesis.
Summary
Patients with ALL or LPD have benefited greatly from the dramatic advances in therapy over the past three decades. For example, improvement in treatment for pediatric ALL has led to event free survival rates of over 80% overall [1]. However, 20–30% of children with ALL will develop recurrent disease, with the number of children with relapsed acute leukemia exceeding the incidence of most other pediatric malignancies [3, 158]. Despite strategies of risk stratification and modern intensified chemotherapy and stem cell transplantation, the prognosis for these pediatric and adult patients remains poor. At present, children who have bone marrow or combined bone marrow and extramedullary relapses while on therapy have a 5–20% likelihood of long-term survival [2, 4, 13]. As initial therapy is intensified, there is a concern that recurrent disease may be more refractory to traditional chemotherapy. Thus, the identification and then the incorporation of novel biologically based molecular and cellular targeted agents into current treatment strategies is crucial. However, both de novo and acquired resistance to these targeted agents must be carefully studied with lessons learned from the use of imatinib for the treatment of Ph+ CML and ALL. In addition, these targeted agents need to be tested in combination with other targeted therapeutics and/or conventional chemotherapeutics. Despite the prospect of resistance, many molecular targeted therapies, including small molecule inhibitors and antagonistic antibodies that interfere with the signaling networks necessary for cell survival and proliferation or with the interaction of these abnormal lymphocytes with its stromal environment as well as immune-based cellular targeted therapies, such as the use of CpG ODN to stimulate innate and adaptive immune responses, show great promise in the treatment of ALL and LPD.
Acknowledgments
This article was supported by grants NIH 1 K08 CA104882-01A1, grant # IRG-78-002-30 from the American Cancer Society, the Children’s Cancer Fund, and the Florence R.C. Murray Program at the Children’s Hospital of Philadelphia (VIB); the WW Smith Charitable Trust (VIB and SAG); NIH CA102646, CA1116660, ACS RSG0507101, and the Sanford Chair and Weinberg Funds of the Children’s Hospital of Philadelphia (SAG), ASCO Young Investigator and Career Development Awards, and the Larry and Helen Hoag Foundation Clinical Translational Research Career Development Award (DTT); NIH R03 CA123554, Foerderer-Murray Program at the Children’s Hospital of Philadelphia (GSDR).
Contributor Information
Valerie I. Brown, Email: brownv@email.chop.edu, Division of Oncology, Children’s Hospital of Philadelphia, University of Pennsylvania School of Medicine, ARC 902, 3615 Civic Center Boulevard, Philadelphia, PA 19104, USA
Alix E. Seif, Division of Oncology, Children’s Hospital of Philadelphia, University of Pennsylvania School of Medicine, ARC 902, 3615 Civic Center Boulevard, Philadelphia, PA 19104, USA
Gregor S. D. Reid, Division of Oncology, Children’s Hospital of Philadelphia, University of Pennsylvania School of Medicine, ARC 902, 3615 Civic Center Boulevard, Philadelphia, PA 19104, USA
David T. Teachey, Division of Oncology, Children’s Hospital of Philadelphia, University of Pennsylvania School of Medicine, ARC 902, 3615 Civic Center Boulevard, Philadelphia, PA 19104, USA, Division of Hematology, Department of Pediatrics, Children’s Hospital of Philadelphia, University of Pennsylvania School of Medicine, Philadelphia, PA 19104, USA
Stephan A. Grupp, Division of Oncology, Children’s Hospital of Philadelphia, University of Pennsylvania School of Medicine, ARC 902, 3615 Civic Center Boulevard, Philadelphia, PA 19104, USA, Department of Pathology and Laboratory Medicine, Children’s Hospital of Philadelphia, University of Pennsylvania School of Medicine, Philadelphia, PA 19104, USA
References
- 1.Pui CH, Evans WE. Treatment of acute lymphoblastic leukemia. N Engl J Med. 2006;354:166–78. doi: 10.1056/NEJMra052603. [DOI] [PubMed] [Google Scholar]
- 2.Gaynon PS, Qu RP, Chappell RJ, Willoughby ML, Tubergen DG, Steinherz PG, et al. Survival after relapse in childhood acute lymphoblastic leukemia: impact of site and time to first relapse—the Children’s Cancer Group Experience. Cancer. 1998;82:1387–95. doi: 10.1002/(sici)1097-0142(19980401)82:7<1387::aid-cncr24>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 3.Gurney JG, Severson RK, Davis S, Robison LL. Incidence of cancer in children in the United States. Sex-, race-, and 1-year age-specific rates by histologic type. Cancer. 1995;75:2186–95. doi: 10.1002/1097-0142(19950415)75:8<2186::aid-cncr2820750825>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 4.Chessells JM. Relapsed lymphoblastic leukaemia in children: a continuing challenge. Br J Haematol. 1998;102:423–38. doi: 10.1046/j.1365-2141.1998.00776.x. [DOI] [PubMed] [Google Scholar]
- 5.Stoddart A, Fleming HE, Paige CJ. The role of the preBCR, the interleukin-7 receptor, and homotypic interactions during B-cell development. Immunol Rev. 2000;175:47–58. [PubMed] [Google Scholar]
- 6.Hibbs ML, Harder KW. The duplicitous nature of the Lyn tyrosine kinase in growth factor signaling. Growth Factors. 2006;24:137–49. doi: 10.1080/08977190600581327. [DOI] [PubMed] [Google Scholar]
- 7.Kittipatarin C, Khaled AR. Interlinking interleukin-7. Cytokine. 2007;39:75–83. doi: 10.1016/j.cyto.2007.07.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.LeBien TW. Fates of human B-cell precursors. Blood. 2000;96:9–23. [PubMed] [Google Scholar]
- 9.Paukku K, Silvennoinen O. STATs as critical mediators of signal transduction and transcription: lessons learned from STAT5. Cytokine Growth Factor Rev. 2004;15:435–55. doi: 10.1016/j.cytogfr.2004.09.001. [DOI] [PubMed] [Google Scholar]
- 10.Nowell PC. The minute chromosome (Phl) in chronic granulocytic leukemia. Blut. 1962;8:65–6. doi: 10.1007/BF01630378. [DOI] [PubMed] [Google Scholar]
- 11.Rowley JD. Letter: a new consistent chromosomal abnormality in chronic myelogenous leukaemia identified by quinacrine fluorescence and Giemsa staining. Nature. 1973;243:290–3. doi: 10.1038/243290a0. [DOI] [PubMed] [Google Scholar]
- 12.Clark SS, McLaughlin J, Timmons M, Pendergast AM, Ben-Neriah Y, Dow LW, et al. Expression of a distinctive BCR-ABL oncogene in Ph1-positive acute lymphocytic leukemia (ALL) Science. 1988;239:775–7. doi: 10.1126/science.3422516. [DOI] [PubMed] [Google Scholar]
- 13.Uckun FM, Nachman JB, Sather HN, Sensel MG, Kraft P, Steinherz PG, et al. Clinical significance of Philadelphia chromosome positive pediatric acute lymphoblastic leukemia in the context of contemporary intensive therapies: a report from the Children’s Cancer Group. Cancer. 1998;83:2030–9. [PubMed] [Google Scholar]
- 14.Piccaluga PP, Paolini S, Martinelli G. Tyrosine kinase inhibitors for the treatment of Philadelphia chromosome-positive adult acute lymphoblastic leukemia. Cancer. 2007;110:1178–86. doi: 10.1002/cncr.22881. [DOI] [PubMed] [Google Scholar]
- 15.Ilaria RL, Jr, Van Etten RA. P210 and P190(BCR/ABL) induce the tyrosine phosphorylation and DNA binding activity of multiple specific STAT family members. J Biol Chem. 1996;271:31704–10. doi: 10.1074/jbc.271.49.31704. [DOI] [PubMed] [Google Scholar]
- 16.Nieborowska-Skorska M, Wasik MA, Slupianek A, Salomoni P, Kitamura T, Calabretta B, et al. Signal transducer and activator of transcription (STAT)5 activation by BCR/ABL is dependent on intact Src homology (SH)3 and SH2 domains of BCR/ABL and is required for leukemogenesis. J Exp Med. 1999;189:1229–42. doi: 10.1084/jem.189.8.1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Raitano AB, Halpern JR, Hambuch TM, Sawyers CL. The Bcr-Abl leukemia oncogene activates Jun kinase and requires Jun for transformation. Proc Natl Acad Sci USA. 1995;92:11746–50. doi: 10.1073/pnas.92.25.11746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sawyers CL, Callahan W, Witte ON. Dominant negative MYC blocks transformation by ABL oncogenes. Cell. 1992;70:901–10. doi: 10.1016/0092-8674(92)90241-4. [DOI] [PubMed] [Google Scholar]
- 19.Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, et al. Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3 k/Akt-dependent pathway. EMBO J. 1997;16:6151–61. doi: 10.1093/emboj/16.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McGahon A, Bissonnette R, Schmitt M, Cotter KM, Green DR, Cotter TG. BCR-ABL maintains resistance of chronic myelogenous leukemia cells to apoptotic cell death. Blood. 1994;83:1179–87. [PubMed] [Google Scholar]
- 21.Sirard C, Laneuville P, Dick JE. Expression of bcr-abl abrogates factor-dependent growth of human hematopoietic M07E cells by an autocrine mechanism. Blood. 1994;83:1575–85. [PubMed] [Google Scholar]
- 22.Skorski T, Nieborowska-Skorska M, Wlodarski P, Wasik M, Trotta R, Kanakaraj P, et al. The SH3 domain contributes to BCR/ABL-dependent leukemogenesis in vivo: role in adhesion, invasion, and homing. Blood. 1998;91:406–18. [PubMed] [Google Scholar]
- 23.Heisterkamp N, Jenster G, ten Hoeve J, Zovich D, Pattengale PK, Groffen J. Acute leukaemia in bcr/abl transgenic mice. Nature. 1990;344:251–3. doi: 10.1038/344251a0. [DOI] [PubMed] [Google Scholar]
- 24.Buchdunger E, Zimmermann J, Mett H, Meyer T, Muller M, Druker BJ, et al. Inhibition of the Abl protein-tyrosine kinase in vitro and in vivo by a 2-phenylaminopyrimidine derivative. Cancer Res. 1996;56:100–4. [PubMed] [Google Scholar]
- 25.Druker BJ, Tamura S, Buchdunger E, Ohno S, Segal GM, Fanning S, et al. Effects of a selective inhibitor of the Abl tyrosine kinase on the growth of Bcr-Abl positive cells. Nat Med. 1996;2:561–6. doi: 10.1038/nm0596-561. [DOI] [PubMed] [Google Scholar]
- 26.Cross SA, Lyseng-Williamson KA. Imatinib: in relapsed or refractory Philadelphia chromosome-positive acute lymphoblastic leukaemia. Drugs. 2007;67:2645–54. doi: 10.2165/00003495-200767170-00013. [DOI] [PubMed] [Google Scholar]
- 27.Wassmann B, Pfeifer H, Scheuring U, Klein SA, Gokbuget N, Binckebanck A, et al. Therapy with imatinib mesylate (Glivec) preceding allogeneic stem cell transplantation (SCT) in relapsed or refractory Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL) Leukemia. 2002;16:2358–65. doi: 10.1038/sj.leu.2402770. [DOI] [PubMed] [Google Scholar]
- 28.Piccaluga PP, Rondoni M, Paolini S, Rosti G, Martinelli G, Baccarani M. Imatinib mesylate in the treatment of hematologic malignancies. Expert Opin Biol Ther. 2007;7:1597–611. doi: 10.1517/14712598.7.10.1597. [DOI] [PubMed] [Google Scholar]
- 29.Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science. 2001;293:876–80. doi: 10.1126/science.1062538. [DOI] [PubMed] [Google Scholar]
- 30.Gururajan M, Dasu T, Shahidain S, Jennings CD, Robertson DA, Rangnekar VM, et al. Spleen tyrosine kinase (Syk), a novel target of curcumin, is required for B lymphoma growth. J Immunol. 2007;178:111–21. doi: 10.4049/jimmunol.178.1.111. [DOI] [PubMed] [Google Scholar]
- 31.Seckinger P, Fougereau M. Activation of src family kinases in human pre-B cells by IL-7. J Immunol. 1994;153:97–109. [PubMed] [Google Scholar]
- 32.Taguchi T, Kiyokawa N, Sato N, Saito M, Fujimoto J. Characteristic expression of Hck in human B-cell precursors. Exp Hematol. 2000;28:349. doi: 10.1016/s0301-472x(99)00127-7. [DOI] [PubMed] [Google Scholar]
- 33.Johnson FM, Saigal B, Tran H, Donato NJ. Abrogation of signal transducer and activator of transcription 3 reactivation after Src kinase inhibition results in synergistic antitumor effects. Clin Cancer Res. 2007;13:4233–44. doi: 10.1158/1078-0432.CCR-06-2981. [DOI] [PubMed] [Google Scholar]
- 34.Hofmeister R, Khaled AR, Benbernou N, Rajnavolgyi E, Muegge K, Durum SK. Interleukin-7: physiological roles and mechanisms of action. Cytokine Growth Factor Rev. 1999;10:41–60. doi: 10.1016/s1359-6101(98)00025-2. [DOI] [PubMed] [Google Scholar]
- 35.Scupoli MT, Perbellini O, Krampera M, Vinante F, Cioffi F, Pizzolo G. Interleukin 7 requirement for survival of T-cell acute lymphoblastic leukemia and human thymocytes on bone marrow stroma. Haematologica. 2007;92:264–6. doi: 10.3324/haematol.10356. [DOI] [PubMed] [Google Scholar]
- 36.Scupoli MT, Vinante F, Krampera M, Vincenzi C, Nadali G, Zampieri F, et al. Thymic epithelial cells promote survival of human T-cell acute lymphoblastic leukemia blasts: the role of interleukin-7. Haematologica. 2003;88:1229–37. [PubMed] [Google Scholar]
- 37.Touw I, Pouwels K, van Agthoven T, van Gurp R, Budel L, Hoogerbrugge H, et al. Interleukin-7 is a growth factor of precursor B and T acute lymphoblastic leukemia. Blood. 1990;75:2097–101. [PubMed] [Google Scholar]
- 38.Baersch G, Mollers T, Hotte A, Dockhorn-Dworniczak B, Rube C, Ritter J, et al. Good engraftment of B-cell precursor ALL in NOD-SCID mice. Klin Padiatr. 1997;209:178–85. doi: 10.1055/s-2008-1043947. [DOI] [PubMed] [Google Scholar]
- 39.Consolini R, Pui CH, Behm FG, Raimondi SC, Campana D. In vitro cytotoxicity of docetaxel in childhood acute leukemias. J Clin Oncol. 1998;16:907–13. doi: 10.1200/JCO.1998.16.3.907. [DOI] [PubMed] [Google Scholar]
- 40.Ek O, Gaynon P, Zeren T, Chelstrom LM, Myers DE, Uckun FM. Treatment of human B-cell precursor leukemia in SCID mice by using a combination of the anti-CD19 immunotoxin B43-PAP with the standard chemotherapeutic drugs vincristine, methylprednisolone, and L-asparaginase. Leuk Lymphoma. 1998;31:143–9. doi: 10.3109/10428199809057594. [DOI] [PubMed] [Google Scholar]
- 41.Borgmann A, Baldy C, von Stackelberg A, Beyermann B, Fichtner I, Nurnberg P, et al. Childhood all blasts retain phenotypic and genotypic characteristics upon long-term serial passage in NOD/SCID mice. Pediatr Hematol Oncol. 2000;17:635–50. doi: 10.1080/08880010050211349. [DOI] [PubMed] [Google Scholar]
- 42.Liem NL, Papa RA, Milross CG, Schmid MA, Tajbakhsh M, Choi S, et al. Characterization of childhood acute lymphoblastic leukemia xenograft models for the preclinical evaluation of new therapies. Blood. 2004;103:3905–14. doi: 10.1182/blood-2003-08-2911. [DOI] [PubMed] [Google Scholar]
- 43.Kolb EA, Gorlick R, Houghton PJ, Morton CL, Lock RB, Tajbakhsh M, et al. Initial testing of dasatinib by the pediatric preclinical testing program. Pediatr Blood Cancer. 2008;50:1198–206. doi: 10.1002/pbc.21368. [DOI] [PubMed] [Google Scholar]
- 44.Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Ann Oncol. 2005;16:525–37. doi: 10.1093/annonc/mdi113. [DOI] [PubMed] [Google Scholar]
- 45.Houghton PJ, Huang S. mTOR as a target for cancer therapy. Curr Top Microbiol Immunol. 2004;279:339–59. doi: 10.1007/978-3-642-18930-2_20. [DOI] [PubMed] [Google Scholar]
- 46.Steelman LS, Pohnert SC, Shelton JG, Franklin RA, Bertrand FE, McCubrey JA. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189–218. doi: 10.1038/sj.leu.2403241. [DOI] [PubMed] [Google Scholar]
- 47.Amaravadi R, Thompson CB. The survival kinases Akt and Pim as potential pharmacological targets. J Clin Invest. 2005;115:2618–24. doi: 10.1172/JCI26273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–30. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 49.Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 50.Harrington LS, Findlay GM, Lamb RF. Restraining PI3K: mTOR signalling goes back to the membrane. Trends Biochem Sci. 2005;30:35–42. doi: 10.1016/j.tibs.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 51.Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098–101. doi: 10.1126/science.1106148. [DOI] [PubMed] [Google Scholar]
- 52.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, et al. Activation of Akt and eIF4E survival pathways by rapamycin-mediated mammalian target of rapamycin inhibition. Cancer Res. 2005;65:7052–8. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 53.O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Akcakanat A, Singh G, Hung MC, Meric-Bernstam F. Rapamycin regulates the phosphorylation of rictor. Biochem Biophys Res Commun. 2007;362:330–3. doi: 10.1016/j.bbrc.2007.07.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159–68. doi: 10.1016/j.molcel.2006.03.029. [DOI] [PubMed] [Google Scholar]
- 56.Zeng Z, Sarbassov dos D, Samudio IJ, Yee KW, Munsell MF, Ellen Jackson C, et al. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood. 2007;109:3509–12. doi: 10.1182/blood-2006-06-030833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Inoki K, Corradetti MN, Guan KL. Dysregulation of the TSC-mTOR pathway in human disease. Nat Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- 58.Hay N. The Akt-mTOR tango and its relevance to cancer. Cancer Cell. 2005;8:179–83. doi: 10.1016/j.ccr.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 59.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 60.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3 K pathway as an integrator of multiple inputs during tumorigenesis. Nat Rev Cancer. 2006;6:184–92. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 61.Sehgal SN, Baker H, Vezina C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo) 1975;28:727–32. doi: 10.7164/antibiotics.28.727. [DOI] [PubMed] [Google Scholar]
- 62.Easton JB, Houghton PJ. mTOR and cancer therapy. Oncogene. 2006;25:6436–46. doi: 10.1038/sj.onc.1209886. [DOI] [PubMed] [Google Scholar]
- 63.Sabatini DM. mTOR and cancer: insights into a complex relationship. Nat Rev Cancer. 2006;6:729–34. doi: 10.1038/nrc1974. [DOI] [PubMed] [Google Scholar]
- 64.Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, et al. Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med. 2007;356:2271–81. doi: 10.1056/NEJMoa066838. [DOI] [PubMed] [Google Scholar]
- 65.Majewski M, Korecka M, Kossev P, Li S, Goldman J, Moore J, et al. The immunosuppressive macro-lide RAD inhibits growth of human Epstein-Barr virus-transformed B lymphocytes in vitro and in vivo: a potential approach to prevention and treatment of posttransplant lymphoproliferative disorders. Proc Natl Acad Sci USA. 2000;97:4285–90. doi: 10.1073/pnas.080068597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gottschalk AR, Boise LH, Thompson CB, Quintans J. Identification of immunosuppressant-induced apoptosis in a murine B-cell line and its prevention by bcl-x but not bcl-2. Proc Natl Acad Sci USA. 1994;91:7350–4. doi: 10.1073/pnas.91.15.7350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Muthukkumar S, Ramesh TM, Bondada S. Rapamycin, a potent immunosuppressive drug, causes programmed cell death in B lymphoma cells. Transplantation. 1995;60:264–70. doi: 10.1097/00007890-199508000-00010. [DOI] [PubMed] [Google Scholar]
- 68.Ringshausen I, Peschel C, Decker T. Mammalian target of rapamycin (mTOR) inhibition in chronic lymphocytic B-cell leukemia: a new therapeutic option. Leuk Lymphoma. 2005;46:11–9. doi: 10.1080/10428190400005353. [DOI] [PubMed] [Google Scholar]
- 69.Brown VI, Fang J, Alcorn K, Barr R, Kim JM, Wasserman R, et al. Rapamycin is active against B-precursor leukemia in vitro and in vivo, an effect that is modulated by IL-7-mediated signaling. Proc Natl Acad Sci USA. 2003;100:15113–8. doi: 10.1073/pnas.2436348100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Brown VI, Hulitt J, Fish J, Sheen C, Bruno M, Xu Q, et al. Thymic stromal-derived lymphopoietin induces proliferation of pre-B leukemia and antagonizes mTOR inhibitors, suggesting a role for interleukin-7Ralpha signaling. Cancer Res. 2007;67:9963–70. doi: 10.1158/0008-5472.CAN-06-4704. [DOI] [PubMed] [Google Scholar]
- 71.Teachey DT, Obzut DA, Cooperman J, Fang J, Carroll M, Choi JK, et al. The mTOR inhibitor CCI-779 induces apoptosis and inhibits growth in preclinical models of primary adult human ALL. Blood. 2006;107:1149–55. doi: 10.1182/blood-2005-05-1935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Goodfellow PJ, Wells SA., Jr RET gene and its implications for cancer. J Natl Cancer Inst. 1995;87:1515–23. doi: 10.1093/jnci/87.20.1515. [DOI] [PubMed] [Google Scholar]
- 73.Wasserman R, Zeng XX, Hardy RR. The evolution of B precursor leukemia in the Emu-ret mouse. Blood. 1998;92:273–82. [PubMed] [Google Scholar]
- 74.Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul. 1984;22:27–55. doi: 10.1016/0065-2571(84)90007-4. [DOI] [PubMed] [Google Scholar]
- 75.Fan QW, Cheng CK, Nicolaides TP, Hackett CS, Knight ZA, Shokat KM, et al. A dual phosphoinositide-3-kinase alpha/mTOR inhibitor cooperates with blockade of epidermal growth factor receptor in PTEN-mutant glioma. Cancer Res. 2007;67:7960–5. doi: 10.1158/0008-5472.CAN-07-2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Raynaud FI, Eccles S, Clarke PA, Hayes A, Nutley B, Alix S, et al. Pharmacologic characterization of a potent inhibitor of class I phosphatidylinositide 3-kinases. Cancer Res. 2007;67:5840–50. doi: 10.1158/0008-5472.CAN-06-4615. [DOI] [PubMed] [Google Scholar]
- 77.Kang J, Der SD. Cytokine functions in the formative stages of a lymphocyte’s life. Curr Opin Immunol. 2004;16:180–90. doi: 10.1016/j.coi.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 78.Lin JX, Migone TS, Tsang M, Friedmann M, Weatherbee JA, Zhou L, et al. The role of shared receptor motifs and common Stat proteins in the generation of cytokine pleiotropy and redundancy by IL-2, IL-4, IL-7, IL-13, and IL-15. Immunity. 1995;2:331–9. doi: 10.1016/1074-7613(95)90141-8. [DOI] [PubMed] [Google Scholar]
- 79.Venkitaraman AR, Cowling RJ. Interleukin-7 induces the association of phosphatidylinositol 3-kinase with the alpha chain of the interleukin-7 receptor. Eur J Immunol. 1994;24:2168–74. doi: 10.1002/eji.1830240935. [DOI] [PubMed] [Google Scholar]
- 80.Palmer MJ, Mahajan VS, Trajman LC, Irvine DJ, Lauffenburger DA, Chen J. Interleukin-7 receptor signaling network: an integrated systems perspective. Cell Mol Immunol. 2008;5:79–89. doi: 10.1038/cmi.2008.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Pandey A, Ozaki K, Baumann H, Levin SD, Puel A, Farr AG, et al. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat Immunol. 2000;1:59–64. doi: 10.1038/76923. [DOI] [PubMed] [Google Scholar]
- 82.Isaksen DE, Baumann H, Trobridge PA, Farr AG, Levin SD, Ziegler SF. Requirement for stat5 in thymic stromal lymphopoietin-mediated signal transduction. J Immunol. 1999;163:5971–7. [PubMed] [Google Scholar]
- 83.Nagasawa T. Microenvironmental niches in the bone marrow required for B-cell development. Nat Rev Immunol. 2006;6:107–16. doi: 10.1038/nri1780. [DOI] [PubMed] [Google Scholar]
- 84.Mudry RE, Fortney JE, York T, Hall BM, Gibson LF. Stromal cells regulate survival of B-lineage leukemic cells during chemotherapy. Blood. 2000;96:1926–32. [PubMed] [Google Scholar]
- 85.Lagneaux L, Delforge A, Bron D, De Bruyn C, Stryckmans P. Chronic lymphocytic leukemic B cells but not normal B cells are rescued from apoptosis by contact with normal bone marrow stromal cells. Blood. 1998;91:2387–96. [PubMed] [Google Scholar]
- 86.Meads MB, Hazlehurst LA, Dalton WS. The bone marrow microenvironment as a tumor sanctuary and contributor to drug resistance. Clin Cancer Res. 2008;14:2519–26. doi: 10.1158/1078-0432.CCR-07-2223. [DOI] [PubMed] [Google Scholar]
- 87.Corcione A, Arduino N, Ferretti E, Pistorio A, Spinelli M, Ottonello L, et al. Chemokine receptor expression and function in childhood acute lymphoblastic leukemia of B-lineage. Leuk Res. 2006;30:365–72. doi: 10.1016/j.leukres.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 88.Juarez J, Baraz R, Gaundar S, Bradstock K, Bendall L. Interaction of interleukin-7 and interleukin-3 with the CXCL12-induced proliferation of B-cell progenitor acute lymphoblastic leukemia. Haematologica. 2007;92:450–9. doi: 10.3324/haematol.10621. [DOI] [PubMed] [Google Scholar]
- 89.Zeng Z, Samudio IJ, Munsell M, An J, Huang Z, Estey E, et al. Inhibition of CXCR4 with the novel RCP168 peptide overcomes stroma-mediated chemoresistance in chronic and acute leukemias. Mol Cancer Ther. 2006;5:3113–21. doi: 10.1158/1535-7163.MCT-06-0228. [DOI] [PubMed] [Google Scholar]
- 90.Bertrand FE, Eckfeldt CE, Fink JR, Lysholm AS, Pribyl JA, Shah N, et al. Microenvironmental influences on human B-cell development. Immunol Rev. 2000;175:175–86. [PubMed] [Google Scholar]
- 91.Bertrand FE, Spengemen JD, Shelton JG, McCubrey JA. Inhibition of PI3K, mTOR and MEK signaling pathways promotes rapid apoptosis in B-lineage ALL in the presence of stromal cell support. Leukemia. 2005;19:98–102. doi: 10.1038/sj.leu.2403560. [DOI] [PubMed] [Google Scholar]
- 92.Wang L, Fortney JE, Gibson LF. Stromal cell protection of B-lineage acute lymphoblastic leukemic cells during chemotherapy requires active Akt. Leuk Res. 2004;28:733–42. doi: 10.1016/j.leukres.2003.10.033. [DOI] [PubMed] [Google Scholar]
- 93.Fortney JE, Zhao W, Wenger SL, Gibson LF. Bone marrow stromal cells regulate caspase 3 activity in leukemic cells during chemotherapy. Leuk Res. 2001;25:901–7. doi: 10.1016/s0145-2126(01)00051-0. [DOI] [PubMed] [Google Scholar]
- 94.Kay NE, Shanafelt TD, Strege AK, Lee YK, Bone ND, Raza A. Bone biopsy derived marrow stromal elements rescue chronic lymphocytic leukemia B-cells from spontaneous and drug induced cell death and facilitates an “angiogenic switch”. Leuk Res. 2007;31:899–906. doi: 10.1016/j.leukres.2006.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sims JE, Williams DE, Morrissey PJ, Garka K, Foxworthe D, Price V, et al. Molecular cloning and biological characterization of a novel murine lymphoid growth factor. J Exp Med. 2000;192:671–80. doi: 10.1084/jem.192.5.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wei C, Zeff R, Goldschneider I. Murine pro-B cells require IL-7 and its receptor complex to up-regulate IL-7R alpha, terminal deoxynucleotidyltransferase, and c mu expression. J Immunol. 2000;164:1961–70. doi: 10.4049/jimmunol.164.4.1961. [DOI] [PubMed] [Google Scholar]
- 97.Levin SD, Koelling RM, Friend SL, Isaksen DE, Ziegler SF, Perlmutter RM, et al. Thymic stromal lymphopoietin: a cytokine that promotes the development of IgM+ B cells in vitro and signals via a novel mechanism. J Immunol. 1999;162:677–83. [PubMed] [Google Scholar]
- 98.Vosshenrich CA, Cumano A, Muller W, Di Santo JP, Vieira P. Thymic stromal-derived lymphopoietin distinguishes fetal from adult B cell development. Nat Immunol. 2003;4:773–9. doi: 10.1038/ni956. [DOI] [PubMed] [Google Scholar]
- 99.Karawajew L, Ruppert V, Wuchter C, Kosser A, Schrappe M, Dorken B, et al. Inhibition of in vitro spontaneous apoptosis by IL-7 correlates with bcl-2 up-regulation, cortical/mature immunophenotype, and better early cytoreduction of childhood T-cell acute lymphoblastic leukemia. Blood. 2000;96:297–306. [PubMed] [Google Scholar]
- 100.Wuchter C, Ruppert V, Schrappe M, Dorken B, Ludwig WD, Karawajew L. In vitro susceptibility to dexamethasone- and doxorubicin-induced apoptotic cell death in context of maturation stage, responsiveness to interleukin 7, and early cytoreduction in vivo in childhood T-cell acute lymphoblastic leukemia. Blood. 2002;99:4109–15. doi: 10.1182/blood.v99.11.4109. [DOI] [PubMed] [Google Scholar]
- 101.Renard N, Duvert V, Matthews DJ, Pages MP, Magaud JP, Manel AM, et al. Proliferation of MIELIKI a novel t(7;9) early pre-B acute lymphoblastic leukemia cell line is inhibited concomitantly by IL-4 and IL-7. Leukemia. 1995;9:1219–26. [PubMed] [Google Scholar]
- 102.Nishii K, Katayama N, Miwa H, Shikami M, Masuya M, Shiku H, et al. Survival of human leukaemic B-cell precursors is supported by stromal cells and cytokines: association with the expression of bcl-2 protein. Br J Haematol. 1999;105:701–10. doi: 10.1046/j.1365-2141.1999.01380.x. [DOI] [PubMed] [Google Scholar]
- 103.Teachey DT, Sheen C, Hall J, Ryan T, Brown VI, Fish J, et al. mTOR inhibitors are synergistic with methotrexate: an effective combination to treat acute lymphoblastic leukemia (ALL) Blood. 2008 June 10; doi: 10.1182/blood-2008-02-137141. Prepublished online. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Saydem G, Celikkaya H, Cole P, Bertino J, Erickan-Abali E. mTOR inhibition leads to increased sensitivity to methotrexate. AACR 98th Annual Meeting; 2005. [Google Scholar]
- 105.Serra M, Reverter-Branchat G, Maurici D, Benini S, Shen JN, Chano T, et al. Analysis of dihydrofolate reductase and reduced folate carrier gene status in relation to methotrexate resistance in osteosarcoma cells. Ann Oncol. 2004;15:151–60. doi: 10.1093/annonc/mdh004. [DOI] [PubMed] [Google Scholar]
- 106.Laakko T, Schwartz RC, Fraker PJ. IL-7-mediated protection of pro and pre-B cells from the adverse effects of corticosterone. Cell Immunol. 2002;220:39–50. doi: 10.1016/s0008-8749(03)00023-6. [DOI] [PubMed] [Google Scholar]
- 107.Shah OJ, Iniguez-Lluhi JA, Romanelli A, Kimball SR, Jefferson LS. The activated glucocorticoid receptor modulates presumptive autoregulation of ribosomal protein S6 protein kinase, p70 S6K. J Biol Chem. 2002;277:2525–33. doi: 10.1074/jbc.M105935200. [DOI] [PubMed] [Google Scholar]
- 108.Nagata S, Golstein P. The Fas death factor. Science. 1995;267:1449–56. doi: 10.1126/science.7533326. [DOI] [PubMed] [Google Scholar]
- 109.Bleesing JJ. Autoimmune lymphoproliferative syndrome: a genetic disorder of abnormal lymphocyte apoptosis. Immunol Allergy Clin North Am. 2002;22:339–49. [Google Scholar]
- 110.Sneller MC, Straus SE, Jaffe ES, Jaffe JS, Fleisher TA, Stetler-Stevenson M, et al. A novel lymphoproliferative/autoimmune syndrome resembling murine lpr/gld disease. J Clin Invest. 1992;90:334–41. doi: 10.1172/JCI115867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bleesing JJ, Brown MR, Novicio C, Guarraia D, Dale JK, Straus SE, et al. A composite picture of TcR alpha/beta(+) CD4(−)CD8(−) T Cells (alpha/beta-DNTCs) in humans with autoimmune lymphoproliferative syndrome. Clin Immunol. 2002;104:21–30. doi: 10.1006/clim.2002.5225. [DOI] [PubMed] [Google Scholar]
- 112.Takahashi T, Tanaka M, Brannan CI, Jenkins NA, Copeland NG, Suda T, et al. Generalized lymphoproliferative disease in mice, caused by a point mutation in the Fas ligand. Cell. 1994;76:969–76. doi: 10.1016/0092-8674(94)90375-1. [DOI] [PubMed] [Google Scholar]
- 113.Watanabe-Fukunaga R, Brannan CI, Copeland NG, Jenkins NA, Nagata S. Lymphoproliferation disorder in mice explained by defects in Fas antigen that mediates apoptosis. Nature. 1992;356:314–7. doi: 10.1038/356314a0. [DOI] [PubMed] [Google Scholar]
- 114.Fisher GH, Rosenberg FJ, Straus SE, Dale JK, Middleton LA, Lin AY, et al. Dominant interfering Fas gene mutations impair apoptosis in a human autoimmune lymphoproliferative syndrome. Cell. 1995;81:935–46. doi: 10.1016/0092-8674(95)90013-6. [DOI] [PubMed] [Google Scholar]
- 115.Rieux-Laucat F, Le Deist F, Hivroz C, Roberts IA, Debatin KM, Fischer A, et al. Mutations in Fas associated with human lymphoproliferative syndrome and autoimmunity. Science. 1995;268:1347–9. doi: 10.1126/science.7539157. [DOI] [PubMed] [Google Scholar]
- 116.Rieux-Laucat F, Le Deist F, Fischer A. Autoimmune lymphoproliferative syndromes: genetic defects of apoptosis pathways. Cell Death Differ. 2003;10:124–33. doi: 10.1038/sj.cdd.4401190. [DOI] [PubMed] [Google Scholar]
- 117.Jackson CE, Fischer RE, Hsu AP, Anderson SM, Choi Y, Wang J, et al. Autoimmune lymphoproliferative syndrome with defective Fas: genotype influences penetrance. Am J Hum Genet. 1999;64:1002–14. doi: 10.1086/302333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Straus SE, Sneller M, Lenardo MJ, Puck JM, Strober W. An inherited disorder of lymphocyte apoptosis: the autoimmune lymphoproliferative syndrome. Ann Intern Med. 1999;130:591–601. doi: 10.7326/0003-4819-130-7-199904060-00020. [DOI] [PubMed] [Google Scholar]
- 119.Sneller MC, Wang J, Dale JK, Strober W, Middelton LA, Choi Y, et al. Clincial, immunologic, and genetic features of an autoimmune lymphoproliferative syndrome associated with abnormal lymphocyte apoptosis. Blood. 1997;89:1341–8. [PubMed] [Google Scholar]
- 120.Straus SE, Jaffe ES, Puck JM, Dale JK, Elkon KB, Rosen-Wolff A, et al. The development of lymphomas in families with autoimmune lymphoproliferative syndrome with germline Fas mutations and defective lymphocyte apoptosis. Blood. 2001;98:194–200. doi: 10.1182/blood.v98.1.194. [DOI] [PubMed] [Google Scholar]
- 121.Bleesing JJ, Straus SE, Fleisher TA. Autoimmune lymphoproliferative syndrome. A human disorder of abnormal lymphocyte survival. Pediatr Clin North Am. 2000;47:1291–310. doi: 10.1016/s0031-3955(05)70272-8. [DOI] [PubMed] [Google Scholar]
- 122.Drappa J, Vaishnaw AK, Sullivan KE, Chu JL, Elkon KB. Fas gene mutations in the Canale-Smith syndrome, an inherited lymphoproliferative disorder associated with autoimmunity. N Engl J Med. 1996;335:1643–9. doi: 10.1056/NEJM199611283352204. [DOI] [PubMed] [Google Scholar]
- 123.Heelan BT, Tormey V, Amlot P, Payne E, Mehta A, Webster AD. Effect of anti-CD20 (rituximab) on resistant thrombocytopenia in autoimmune lymphoproliferative syndrome. Br J Haematol. 2002;118:1078–81. doi: 10.1046/j.1365-2141.2002.03753.x. [DOI] [PubMed] [Google Scholar]
- 124.Rao VK, Dugan F, Dale JK, Davis J, Tretler J, Hurley JK, et al. Use of mycophenolate mofetil for chronic, refractory immune cytopenias in children with autoimmune lymphoproliferative syndrome. Br J Haematol. 2005;129:534–8. doi: 10.1111/j.1365-2141.2005.05496.x. [DOI] [PubMed] [Google Scholar]
- 125.van der Werff Ten Bosch J, Schotte P, Ferster A, Azzi N, Boehler T, Laurey G, et al. Reversion of autoimmune lymphoproliferative syndrome with an antimalarial drug: preliminary results of a clinical cohort study and molecular observations. Br J Haematol. 2002;117:176–88. doi: 10.1046/j.1365-2141.2002.03357.x. [DOI] [PubMed] [Google Scholar]
- 126.Rao VK, Dowdell KC, Dale JK, Dugan F, Pesnicak L, Bi LL, et al. Pyrimethamine treatment does not ameliorate lymphoproliferation or autoimmune disease in MRL/lpr−/− mice or in patients with autoimmune lymphoproliferative syndrome. Am J Hematol. 2007;82:1049–55. doi: 10.1002/ajh.21007. [DOI] [PubMed] [Google Scholar]
- 127.Teachey DT, Obzut DA, Axsom K, Choi JK, Goldsmith KC, Hall J, et al. Rapamycin improves lymphoproliferative disease in murine autoimmune lymphoproliferative syndrome (ALPS) Blood. 2006;108:1965–71. doi: 10.1182/blood-2006-01-010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Teachey DT, Seif AE, Brown VI, Bruno M, Bunte RM, Chang YJ, et al. Targeting Notch signaling in autoimmune and lymphoproliferative disease. Blood. 2008;111:705–14. doi: 10.1182/blood-2007-05-087353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dowdell KC, Pesnicak L, Hoffmann V, Steadman KMR, Rao VK, et al. Valproic acid (VPA), a histone deacetylase (HDAC) inhibitor, diminishes lymphoproliferation in the fas deficient MRL/lpr−/− murine model of autoimmune lymphoproliferative syndrome (ALPS) Blood (ASH Annual Meeting Abstracts) 2006;108:2497. doi: 10.1016/j.exphem.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bobe P, Bonardelle D, Benihoud K, Opolon P, Chelbi-Alix MK. Arsenic trioxide: a promising novel therapeutic agent for lymphoproliferative and autoimmune syndromes in MRL/lpr mice. Blood. 2006;108:3967–75. doi: 10.1182/blood-2006-04-020610. [DOI] [PubMed] [Google Scholar]
- 131.Maillard I, Fang T, Pear WS. Regulation of lymphoid development, differentiation, and function by the Notch pathway. Annu Rev Immunol. 2005;23:945–74. doi: 10.1146/annurev.immunol.23.021704.115747. [DOI] [PubMed] [Google Scholar]
- 132.Hyde LA, McHugh NA, Chen J, Zhang Q, Manfra D, Nomeir AA, et al. Studies to investigate the in vivo therapeutic window of the gamma-secretase inhibitor N2-[(2S)-2-(3,5-difluorophenyl)-2-hydroxyethanoyl]-N1-[(7S)-5-methyl-6-oxo-6,7-dihydro-5H-dibenzo[b,d]azepin-7-yl]-L-alanina-mide (LY411,575) in the CRND8 mouse. J Pharmacol Exp Ther. 2006;319:1133–43. doi: 10.1124/jpet.106.111716. [DOI] [PubMed] [Google Scholar]
- 133.van Es JH, van Gijn ME, Riccio O, Van den Born M, Vooijs M, Begthel H, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–63. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 134.Best JD, Smith DW, Reilly MA, O’Donnell R, Lewis HD, Ellis S, et al. The novel gamma secretase inhibitor N-[cis-4-[(4-chlorophenyl)sulfonyl]-4-(2, 5-difluorophenyl)cyclohexyl]-1,1,1-trifluoromethane-sulfonamide (MRK-560) reduces amyloid plaque deposition without evidence of notch-related pathology in the Tg2576 mouse. J Pharmacol Exp Ther. 2007;320:552–8. doi: 10.1124/jpet.106.114330. [DOI] [PubMed] [Google Scholar]
- 135.Wong GT, Manfra D, Poulet FM, Zhang Q, Josien H, Bara T, et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J Biol Chem. 2004;279:12876–82. doi: 10.1074/jbc.M311652200. [DOI] [PubMed] [Google Scholar]
- 136.Akiko I, Yoichi M, Hiroshi S, Koji J. Inhibition of gamma-secretase mediated notch signaling suppresses experimental autoimmune uveoretinitis. Proc Jpn Soc Immunol. 2006;36:194. [Google Scholar]
- 137.Jurynczyk M, Jurewicz A, Bielecki B, Raine CS, Selmaj K. Overcoming failure to repair demyelination in EAE: gamma-secretase inhibition of Notch signaling. J Neurol Sci. 2008;265:5–11. doi: 10.1016/j.jns.2007.09.007. [DOI] [PubMed] [Google Scholar]
- 138.Demehri S, Liu Z, Lee J, Lin MH, Crosby SD, Roberts CJ, et al. Notch-deficient skin induces a lethal systemic B-lymphoproliferative disorder by secreting TSLP, a sentinel for epidermal integrity. PLoS Biol. 2008;6:e123. doi: 10.1371/journal.pbio.0060123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Tournoy J, Bossuyt X, Snellinx A, Regent M, Garmyn M, Serneels L, et al. Partial loss of presenilins causes seborrheic keratosis and autoimmune disease in mice. Hum Mol Genet. 2004;13:1321–31. doi: 10.1093/hmg/ddh151. [DOI] [PubMed] [Google Scholar]
- 140.Bunin N, Carston M, Wall D, Adams R, Casper J, Kamani N, et al. Unrelated marrow transplantation for children with acute lymphoblastic leukemia in second remission. Blood. 2002;99:3151–7. doi: 10.1182/blood.v99.9.3151. [DOI] [PubMed] [Google Scholar]
- 141.Horowitz MM, Gale RP, Sondel PM, Goldman JM, Kersey J, Kolb HJ, et al. Graft-versus-leukemia reactions after bone marrow transplantation. Blood. 1990;75:555–62. [PubMed] [Google Scholar]
- 142.Weiden PL, Sullivan KM, Flournoy N, Storb R, Thomas ED. Antileukemic effect of chronic graft-versus-host disease: contribution to improved survival after allogeneic marrow transplantation. N Engl J Med. 1981;304:1529–33. doi: 10.1056/NEJM198106183042507. [DOI] [PubMed] [Google Scholar]
- 143.Barbaric D, Corthals SL, Jastaniah WA, Asalanian S, Shimizu H, Reid GS, et al. Detection of WT1-specific T cells in paediatric acute lymphoblastic leukaemia patients in first remission. Br J Haematol. 2008;141:271–3. doi: 10.1111/j.1365-2141.2008.07001.x. [DOI] [PubMed] [Google Scholar]
- 144.Yotnda P, Garcia F, Peuchmaur M, Grandchamp B, Duval M, Lemonnier F, et al. Cytotoxic T cell response against the chimeric ETV6-AML1 protein in childhood acute lymphoblastic leukemia. J Clin Invest. 1998;102:455–62. doi: 10.1172/JCI3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Nijmeijer BA, Willemze R, Falkenburg JH. An animal model for human cellular immunotherapy: specific eradication of human acute lymphoblastic leukemia by cytotoxic T lymphocytes in NOD/scid mice. Blood. 2002;100:654–60. doi: 10.1182/blood.v100.2.654. [DOI] [PubMed] [Google Scholar]
- 146.Reid GS, Terrett L, Alessandri AJ, Grubb S, Stork L, Seibel N, et al. Altered patterns of T cell cytokine production induced by relapsed pre-B ALL cells. Leuk Res. 2003;27:1135–42. doi: 10.1016/s0145-2126(03)00106-1. [DOI] [PubMed] [Google Scholar]
- 147.Alessandri AJ, Reid GS, Bader SA, Massing BG, Sorensen PH, Schultz KR. ETV6 (TEL)-AML1 pre-B acute lymphoblastic leukaemia cells are associated with a distinct antigen-presenting phenotype. Br J Haematol. 2002;116:266–72. [PubMed] [Google Scholar]
- 148.Maecker B, Mougiakakos D, Zimmermann M, Behrens M, Hollander S, Schrauder A, et al. Dendritic cell deficiencies in pediatric acute lymphoblastic leukemia patients. Leukemia. 2006;20:645–9. doi: 10.1038/sj.leu.2404146. [DOI] [PubMed] [Google Scholar]
- 149.Reid GS, Bharya S, Klingemann HG, Schultz KR. Differential killing of pre-B acute lymphoblastic leukaemia cells by activated NK cells and the NK-92 ci cell line. Clin Exp Immunol. 2002;129:265–71. doi: 10.1046/j.1365-2249.2002.01919.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Romanski A, Bug G, Becker S, Kampfmann M, Seifried E, Hoelzer D, et al. Mechanisms of resistance to natural killer cell-mediated cytotoxicity in acute lymphoblastic leukemia. Exp Hematol. 2005;33:344–52. doi: 10.1016/j.exphem.2004.11.006. [DOI] [PubMed] [Google Scholar]
- 151.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–76. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 152.Krieg AM. CpG motifs in bacterial DNA and their immune effects. Annu Rev Immunol. 2002;20:709–60. doi: 10.1146/annurev.immunol.20.100301.064842. [DOI] [PubMed] [Google Scholar]
- 153.Fujii H, Trudeau JD, Teachey DT, Fish JD, Grupp SA, Schultz KR, et al. In vivo control of acute lymphoblastic leukemia by immunostimulatory CpG oligonucleotides. Blood. 2007;109:2008–13. doi: 10.1182/blood-2006-02-002055. [DOI] [PubMed] [Google Scholar]
- 154.Reid GS, She K, Terrett L, Food MR, Trudeau JD, Schultz KR. CpG stimulation of precursor B-lineage acute lymphoblastic leukemia induces a distinct change in costimulatory molecule expression and shifts allogeneic T cells toward a Th1 response. Blood. 2005;105:3641–7. doi: 10.1182/blood-2004-06-2468. [DOI] [PubMed] [Google Scholar]
- 155.Cardoso AA, Schultze JL, Boussiotis VA, Freeman GJ, Seamon MJ, Laszlo S, et al. Pre-B acute lymphoblastic leukemia cells may induce T-cell anergy to alloantigen. Blood. 1996;88:41–8. [PubMed] [Google Scholar]
- 156.Corthals SL, Wynne K, She K, Shimizu H, Curman D, Garbutt K, et al. Differential immune effects mediated by Toll-like receptors stimulation in precursor B-cell acute lymphoblastic leukaemia. Br J Haematol. 2006;132:452–8. doi: 10.1111/j.1365-2141.2005.05893.x. [DOI] [PubMed] [Google Scholar]
- 157.Montagna D, Maccario R, Locatelli F, Montini E, Pagani S, Bonetti F, et al. Emergence of antitumor cytolytic T cells is associated with maintenance of hematologic remission in children with acute myeloid leukemia. Blood. 2006;108:3843–50. doi: 10.1182/blood-2006-05-021535. [DOI] [PubMed] [Google Scholar]
- 158.Miller RW, Young JL, Jr, Novakovic B. Childhood cancer. Cancer. 1995;75:395–405. doi: 10.1002/1097-0142(19950101)75:1+<395::aid-cncr2820751321>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]